
Structural Conservation and Functional Diversity of the Poxvirus Immune Evasion (PIE) Domain Superfamily

Abstract
:1. Introduction
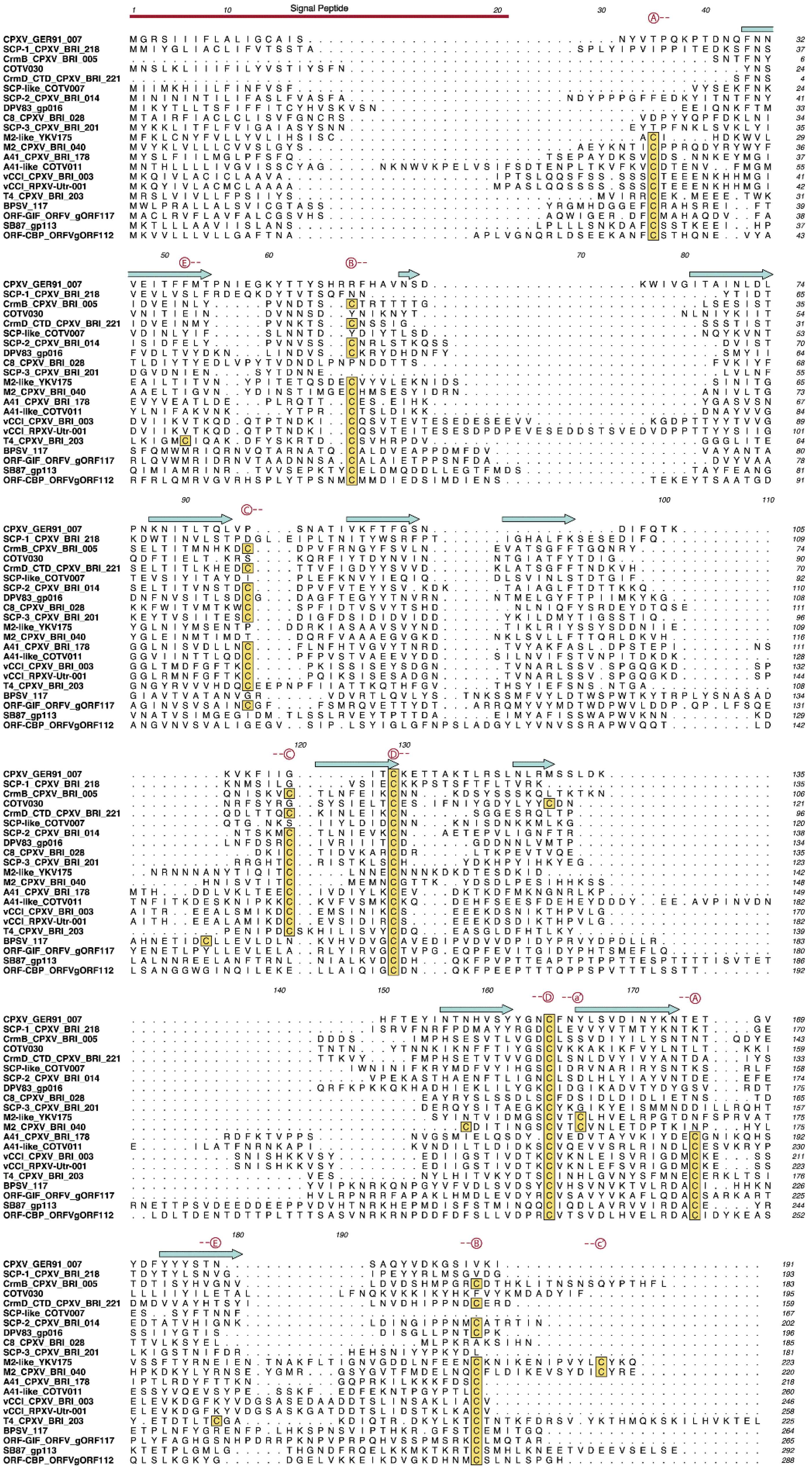
2. PIE Domains of Known Structure
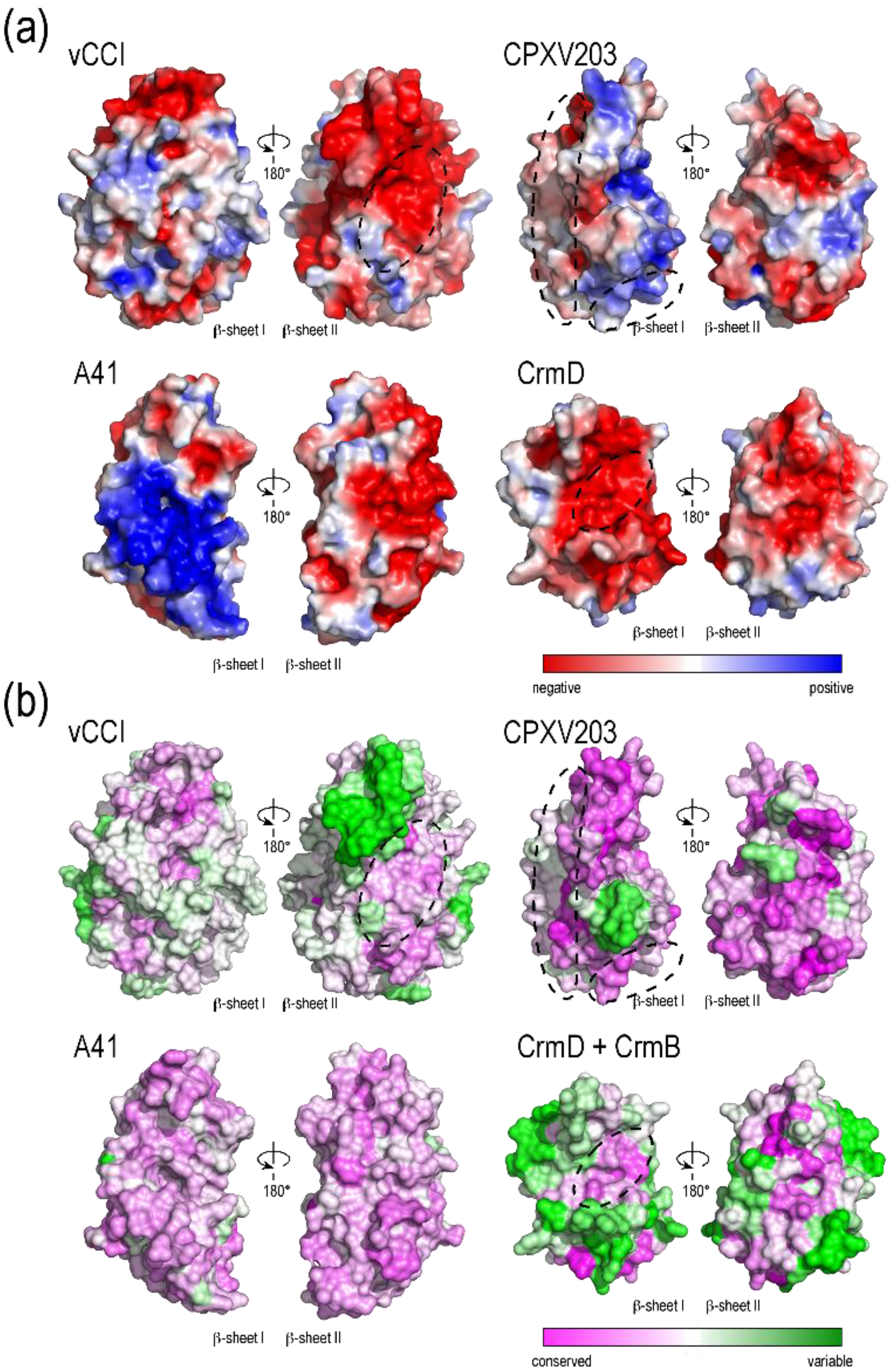
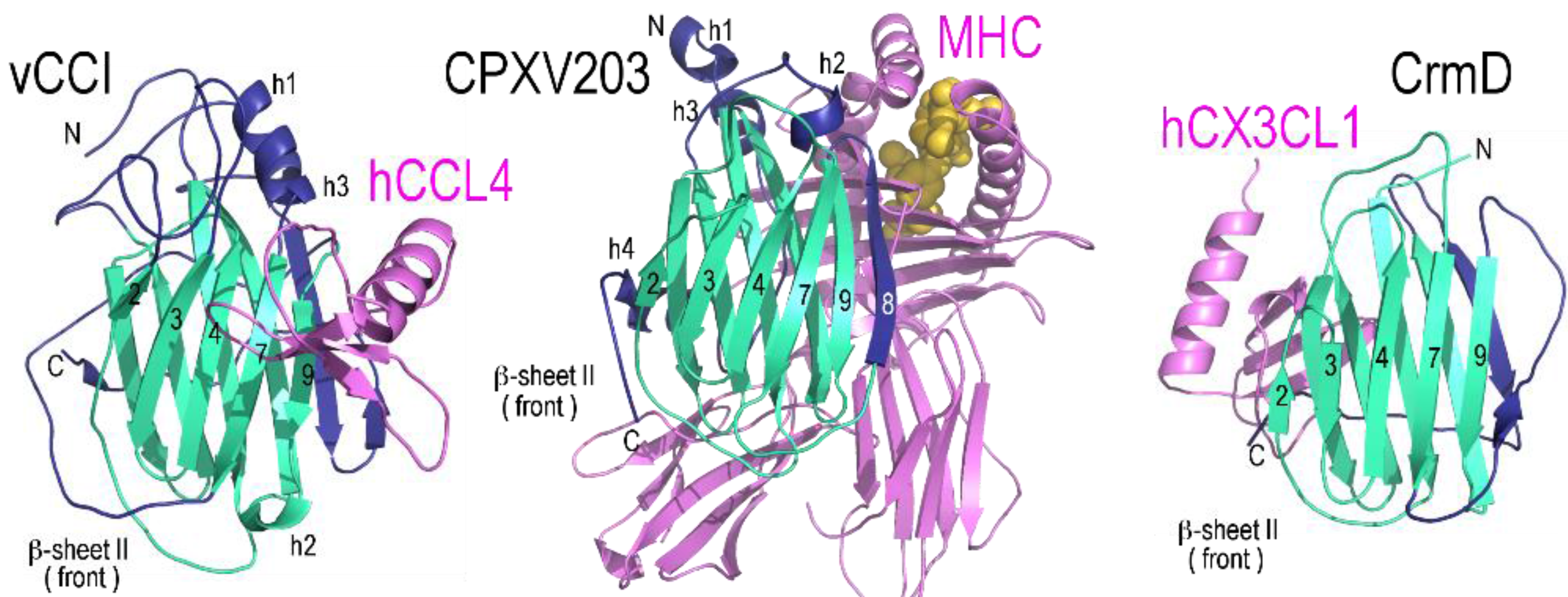
2.1. vCCI
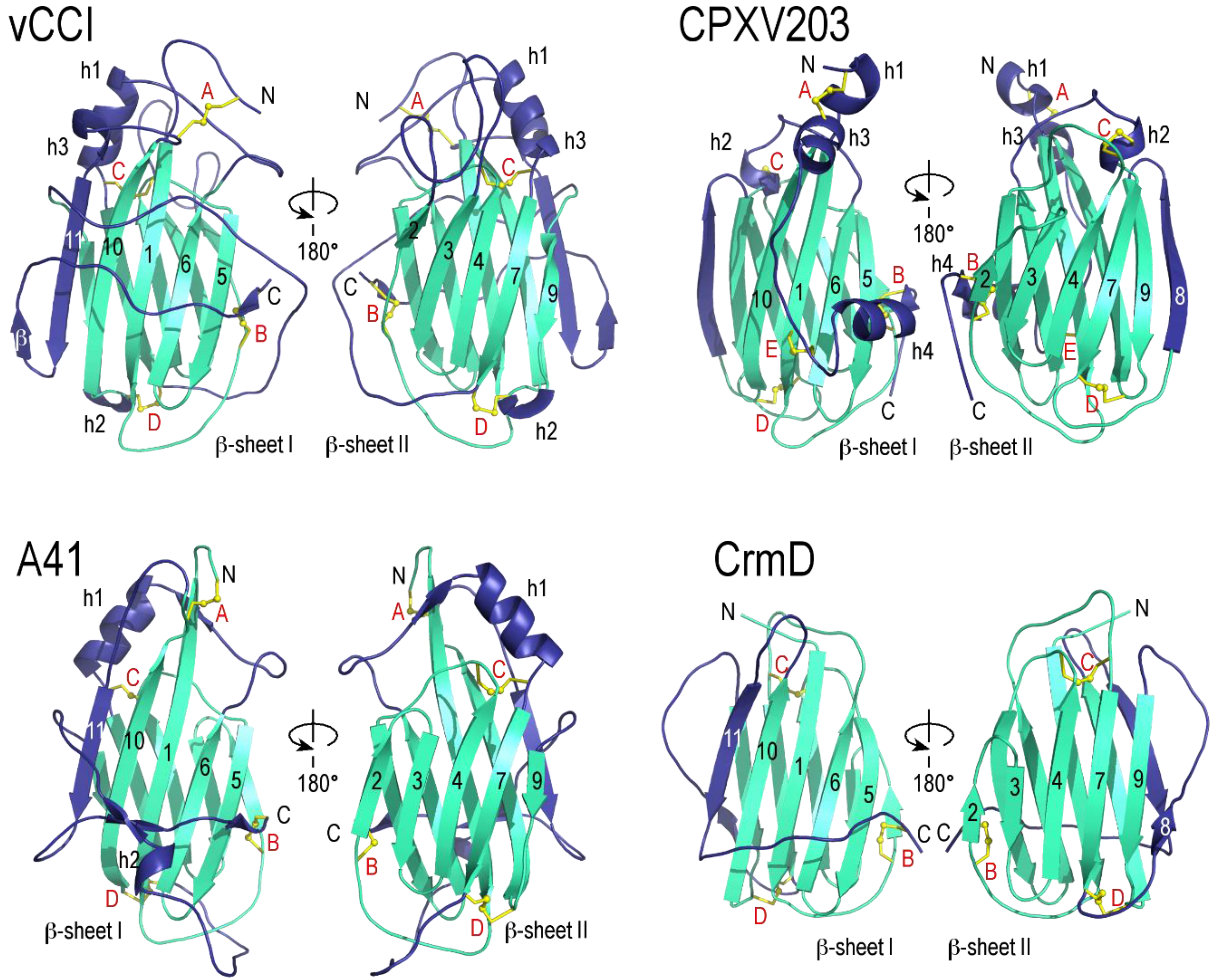
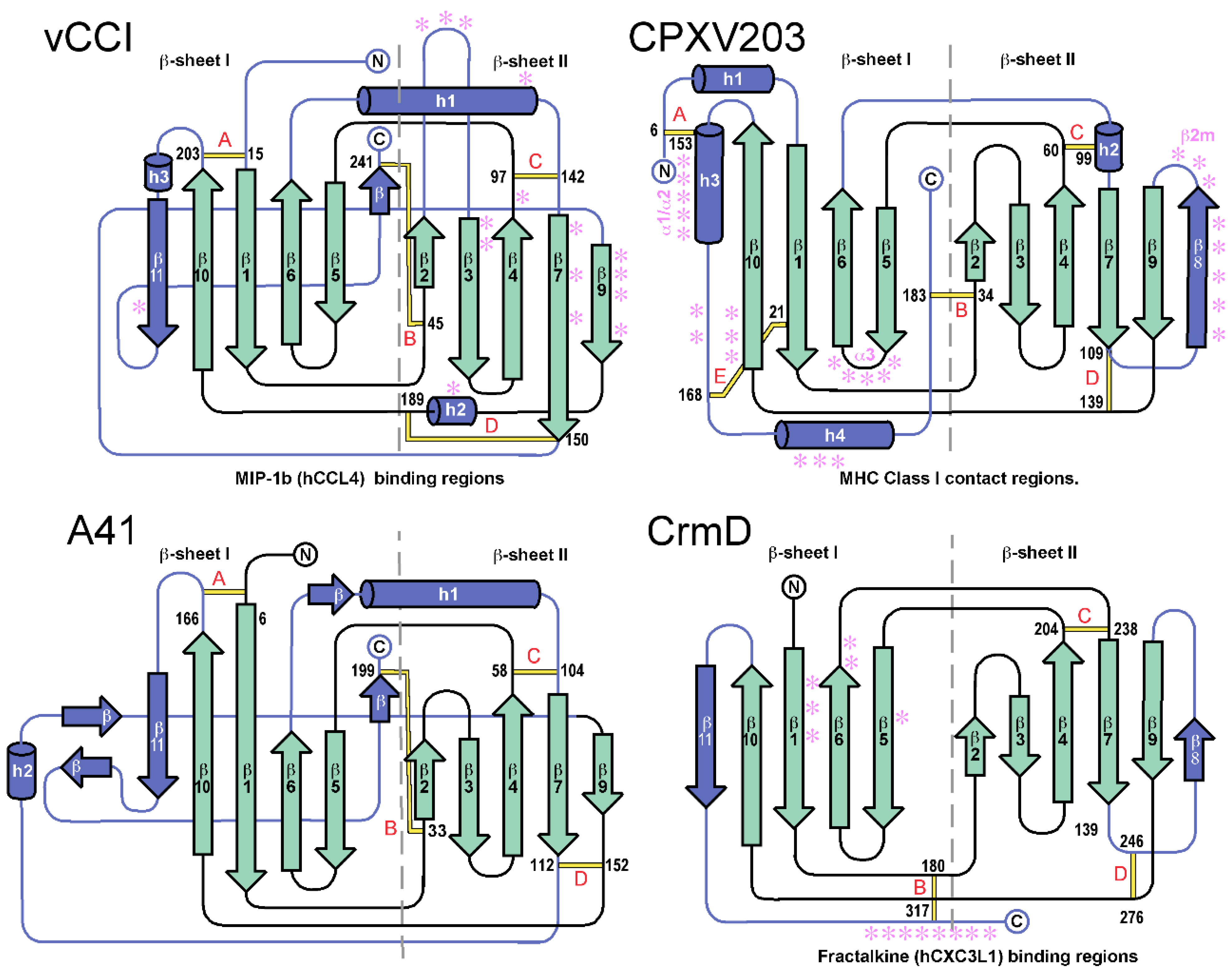
2.2. A41
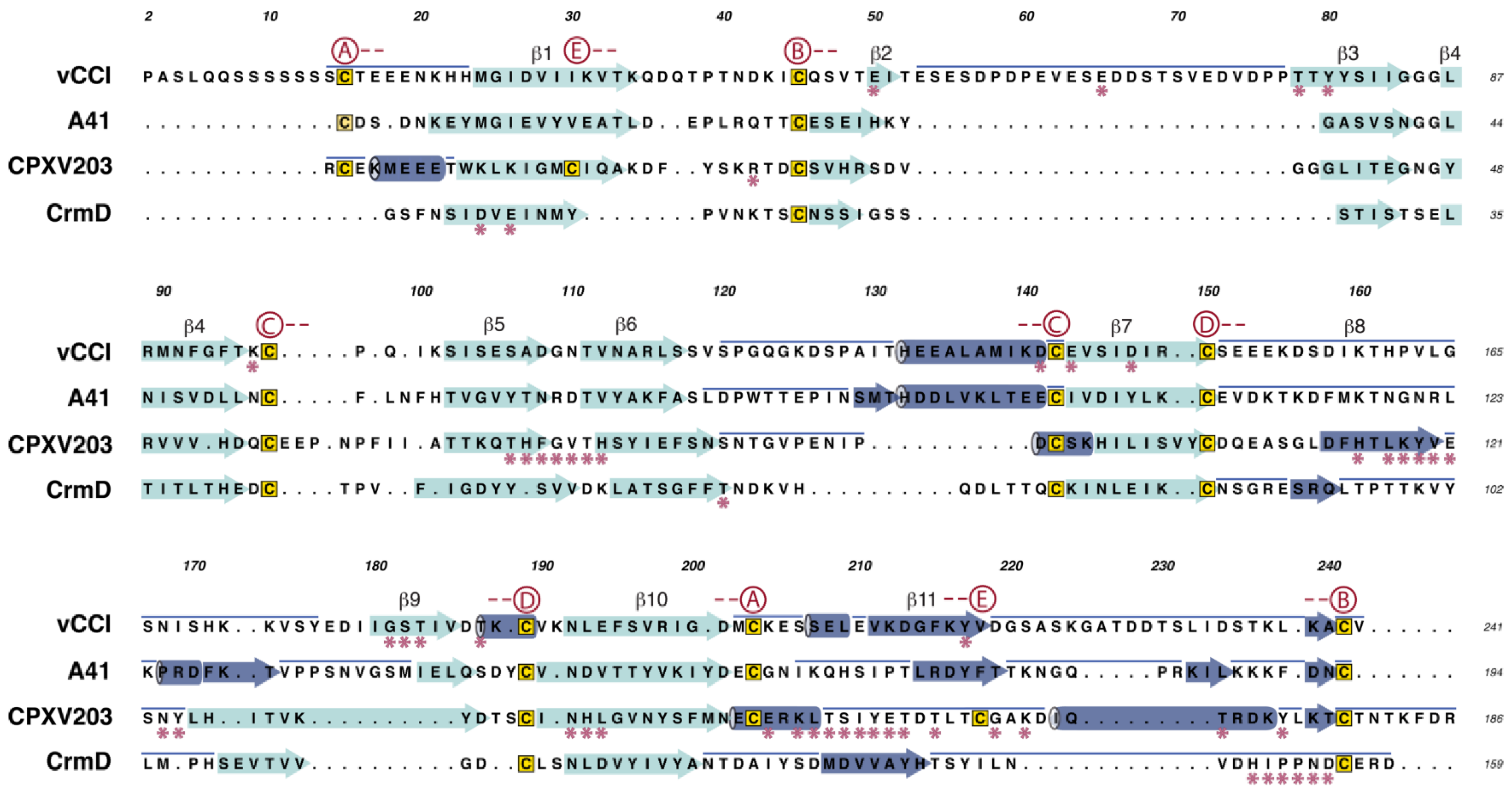

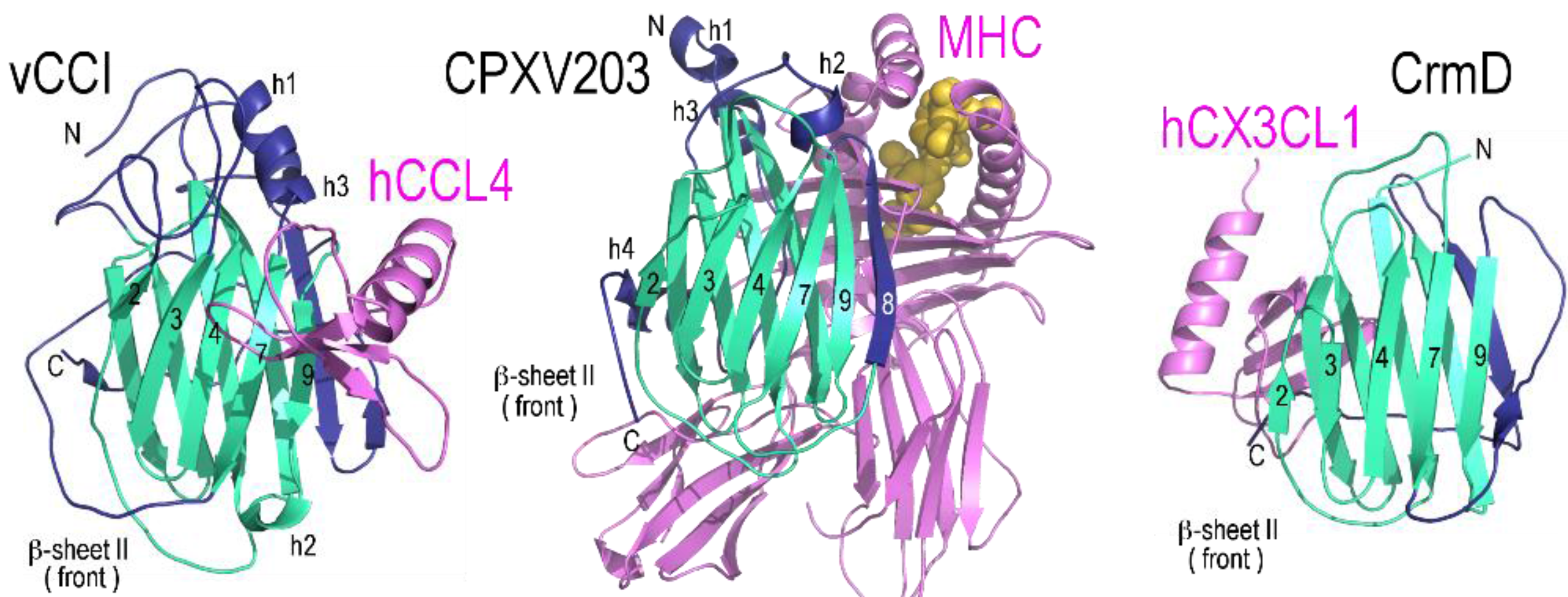
2.3. CrmD C-Terminal Domain
2.4. CPXV203
3. Putative PIE Domains
3.1. ORF-GIF Family
3.2. ORF-CBP Family
3.3. Search for Additional PIE Domains

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genus | Species | Strain | Abbreviation | GenBank # |
|---|---|---|---|---|
| Orthopoxvirus | Variola virus | Brazil 1966 (v66-39 São Paulo) | VARV BRZ66 | DQ441419 |
| Congo 1970 v70-46 Kinshasa | VARV CNG70 | DQ437583 | ||
| Garcia-1966 | VARV GAR | Y16780 | ||
| Guinea 1969 (005) | VARV GUI69 | DQ441426 | ||
| India 1964 7124 Vellore | VARV IND64 | DQ437585 | ||
| Sierra Leone 1969 (V68-258) | VARV SLN68 | DQ441437 | ||
| Monkeypox virus | Sierra Leone V70 | MPXV SL | AY741551 | |
| Zaire-96-I-16 | MPXV Z96 | AF380138 | ||
| Camelpox virus | M-96 | CMLV M96 | AF438165 | |
| Taterapox virus | Dahomey 1968 | TATV | NC_008291 | |
| Horsepox virus | MNR-76 | HPXV | DQ792504 | |
| Cowpox virus | GRI-90 | CPXV GRI | X94355 | |
| Brighton Red | CPXV BRI | AF482758 | ||
| Germany 91-3 | CPXV GER | DQ437593 | ||
| Ectromelia virus | Moscow | ECTV MOS | AF012825 | |
| Vaccinia virus | Western Reserve | VACV WR | NC_006998 | |
| Unclassified | Yoka poxvirus | DakArB 4268 | YOKA | NC_015960 |
| Leporipoxvirus | Myxoma virus | Lausanne | MYXV LAU | AF170726 |
| Yatapoxvirus | Yaba monkey tumor virus | YLD | YMTV YLD | AJ293568 |
| Unclassified | Cotia virus | SPAn232 | COTV | NC_016924 |
| Cervidpoxvirus | Deerpox virus | W-848-83 | DPXV W83 | AY689436 |
| Capripoxvirus | Lumpy skin disease virus | Neethling 2490 | LSDV 2490 | AF325528 |
| Sheeppox virus | A | SPPV A | AY077833 | |
| Goatpox virus | G20-LKV | GTP G20 | AY077836 | |
| Pellor | GTP PEL | NC_004003 | ||
| Suipoxvirus | Swinepox virus | 17077-99 | SWPV 99 | AF410153 |
| Parapoxvirus | Bovine papular stomatitis virus | BV-AR02 | BPSV | NC_005337 |
| Orf virus | OV-SA00 | ORF | NC_005336 | |
| Pseudocowpox virus | VR634 | PCPV | NC_013804 | |
| Parapoxvirus red deer | HL953 | SB87 | NC_025963 | |
| Avipoxvirus | Fowlpox virus | FCV | FWPV FCV | AF198100 |
| Canarypox virus | Wheatley C93 | CNPV WC93 | NC_005309 | |
| Molluscipoxvirus | Molluscum contagiosum virus | Subtype 1 | MOCV SB1 | MCU60315 |
| Genus | Strain | vCCI | CrmB | SCP-2 | C8 | M2 | A41 | SCP-3 | T4 | SCP-1 | CrmD | COTV030 | CPXV-007 | SCP-like | M2-like | A41-like | ORF-CBP | SB87-113 | DPXV-016 | BPSV-117 | ORF-GIF |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Orthopoxvirus | VARV BRZ66 | 1 | 1 | 1 | 1 | ||||||||||||||||
| VARV CNG70 | 1 | 1 | 1 | 1 | |||||||||||||||||
| VARV GAR | 1 | 1 | 1 | 1 | |||||||||||||||||
| VARV GUI69 | 1 | 1 | 1 | 1 | |||||||||||||||||
| VARV IND64 | 1 | 1 | 1 | 1 | |||||||||||||||||
| VARV SLN68 | 1 | 1 | 1 | 1 | |||||||||||||||||
| MPXV SL | 2 | 2 | 1 | 1 | 1 | 1 | |||||||||||||||
| MPXV Z96 | 2 | 2 | 1 | 1 | 1 | 1 | 1 | ||||||||||||||
| CMLV M96 | 2 | 2 | 1 | 1 | 1 | 0 | 1 | 1 | |||||||||||||
| TATV | 0/0 | 0/0 | 0 | 1 | 1 | 1 | 1 | 1 | |||||||||||||
| HPXV | 2 | 2 | 0 | 1 | 1 | 1 | 1 | 0 | 1 | ||||||||||||
| CPXV GRI | 2 | 2 | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | |||||||||||
| CPXV BRI | 2 | 2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | |||||||||||
| CPXV GER | 2 | 2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | ||||||||||
| ECTV MOS | 2 | 0/0 | 1 | 1 | 1 | 2 | |||||||||||||||
| VACV WR | 0/0 | 0/0 | 0/0 | 1 | 1 | 1 | 1 | 0 | 1 | ||||||||||||
| Unclassified | YOKA | 1 | 2 | 1 | |||||||||||||||||
| Leporipoxvirus | MYXV LAU | 2 | 2 | 1 | 2 | ||||||||||||||||
| Yatapoxvirus | YMTV YLD | 1 | 1 | ||||||||||||||||||
| Unclassified | COTV | 1 | 1 | 1 | 2 | 2 | |||||||||||||||
| Cervidpoxvirus | DPXV W83 | 1 | 2 | 1 | 1 | ||||||||||||||||
| Caporipoxvirus | LSDV 2490 | 2 | |||||||||||||||||||
| SPPV A | 2 | ||||||||||||||||||||
| GTP G20 | 2 | ||||||||||||||||||||
| GTP PEL | 2 | ||||||||||||||||||||
| Parapoxvirus | BPSV | 1 | 1 | ||||||||||||||||||
| ORF | 1 | 1 | |||||||||||||||||||
| SB87 | 2 | 1 | 1 | ||||||||||||||||||
| PCPV | 1 | 1 | |||||||||||||||||||
| Ligands | Chemokines | Chemokines | Chemokines | Unknown | Unknown | CK/GAGs | Chemokines | MHC class I | Chemokines | Chemokines | Unknown | Unknown | Unknown | Unknown | Unknown | Chemokines | Unknown | Unknown | Unknown | GM-CSF, IL-2 | |
| Gene | Length (aa) | Mol wt (Da) | pI | N-linked | Cysteines | Disulfides |
|---|---|---|---|---|---|---|
| vCCI (CPXV-BRI-003) | 226 | 24487 | 4.6 | 0 | 8 | A B C D |
| CrmB-CTD (CPXV-BRI-005) | 183 | 20567 | 4.8 | 2 | 6 | _ B C D |
| SCP-2 (CPXV-BRI-014) | 181 | 20208 | 4.5 | 5 | 6 | _ B C D |
| C8 (CPXV-BRI-028) | 164 | 19242 | 4.4 | 2 | 4 | _ _ C D |
| M2 (CPXV-BRI-040) | 203 | 23302 | 5.1 | 4 | 9 | A' B C' D |
| A41 (CPXV-BRI-178) | 202 | 22899 | 5.2 | 1 | 8 | A B C D |
| SCP-3 (CPXV-BRI-201) | 160 | 18793 | 5.3 | 0 | 4 | _ _ C D |
| T4 (CPXV-BRI-203) | 209 | 24027 | 6.8 | 1 | 10 | A B C D E |
| SCP-1 (CPXV-BRI-218) | 175 | 20133 | 5.0 | 2 | 2 | _ _ _ D |
| CrmD-CTD (CPXV-BRI-221) | 159 | 17648 | 4.5 | 1 | 6 | _ B C D |
| COTV030 | 174 | 20738 | 9.2 | 3 | 3 | _ _ _ D |
| CPXV_GER91_007 | 174 | 20070 | 9.1 | 2 | 2 | _ _ _ D |
| SCP-like (COTV007) | 151 | 17839 | 5.8 | 3 | 2 | _ _ _ D |
| M2-like (YKV175) | 203 | 23019 | 4.5 | 3 | 8 | A' B C' D |
| A41-like(COTV011) | 241 | 27586 | 4.7 | 1 | 8 | A B C D |
| ORF-CBP (ORF-112) | 272 | 29780 | 4.6 | 4 | 6 | A B _ D |
| SB87-gp113 | 276 | 31204 | 4.6 | 4 | 6 | A B _ D |
| DPXV-016 | 172 | 19713 | 5.3 | 1 | 6 | _ B C D |
| BPSV-117 | 245 | 27765 | 6.1 | 4 | 7 | A B _ D |
| ORF-GIF (ORFVgORF117) | 246 | 28111 | 6.2 | 4 | 7 | A B _ D |

3.4. PIEs of Unknown Function
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lefkowitz, E.J.; Wang, C.; Upton, C. Poxviruses: Past, Present and Future. Virus Res. 2006, 117, 105–118. [Google Scholar] [CrossRef]
- Upton, C.; Slack, S.; Hunter, A.L.; Ehlers, A.; Roper, R.L. Poxvirus Orthologous Clusters: Toward Defining the Minimum Essential Poxvirus Genome. J. Virol. 2003, 77, 7590–7600. [Google Scholar] [CrossRef] [PubMed]
- Gubser, C.; Hue, S.; Kellam, P.; Smith, G.L. Poxvirus Genomes: A Phylogenetic Analysis. J. Gen. Virol. 2004, 85, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.S.; Ilyinskii, P.; Philippon, V.; Gritz, L.; Yafal, A.G.; Zinnack, K.; Beaudry, K.R.; Manson, K.H.; Lifton, M.A.; Kuroda, M.J.; et al. Role of Genes That Modulate Host Immune Responses in the Immunogenicity and Pathogenicity of Vaccinia Virus. J. Virol. 2005, 79, 6554–6559. [Google Scholar] [CrossRef] [PubMed]
- Werden, S.J.; Rahman, M.M.; McFadden, G. Poxvirus Host Range Genes. Adv. Virus Res. 2008, 71, 135–171. [Google Scholar] [PubMed]
- Hughes, A.L.; Irausquin, S.; Friedman, R. The Evolutionary Biology of Poxviruses. Infect. Genet. Evol. 2010, 10, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Odom, M.R.; Hendrickson, R.C.; Lefkowitz, E.J. Poxvirus Protein Evolution: Family Wide Assessment of Possible Horizontal Gene Transfer Events. Virus Res. 2009, 144, 233–249. [Google Scholar] [CrossRef] [PubMed]
- Bratke, K.A.; McLysaght, A.; Rothenburg, S. A Survey of Host Range Genes in Poxvirus Genomes. Infect. Genet. Evol. 2013, 14, 406–425. [Google Scholar] [CrossRef] [PubMed]
- Haller, S.L.; Peng, C.; McFadden, G.; Rothenburg, S. Poxviruses and the Evolution of Host Range and Virulence. Infect. Genet. Evol. 2014, 21, 15–40. [Google Scholar] [CrossRef] [PubMed]
- Moss, B.; Winters, E.; Cooper, N. Instability and Reiteration of DNA Sequences within the Vaccinia Virus Genome. Proc. Natl. Acad. Sci. USA 1981, 78, 1614–1618. [Google Scholar] [CrossRef] [PubMed]
- Elde, N.C.; Child, S.J.; Eickbush, M.T.; Kitzman, J.O.; Rogers, K.S.; Shendure, J.; Geballe, A.P.; Malik, H.S. Poxviruses Deploy Genomic Accordions to Adapt Rapidly against Host Antiviral Defenses. Cell 2012, 150, 831–841. [Google Scholar] [CrossRef] [PubMed]
- Sonnberg, S.; Fleming, S.B.; Mercer, A.A. Phylogenetic Analysis of the Large Family of Poxvirus Ankyrin-Repeat Proteins Reveals Orthologue Groups within and across Chordopoxvirus Genera. J. Gen. Virol. 2011, 92, 2596–2607. [Google Scholar] [CrossRef] [PubMed]
- Silverman, G.A.; Bird, P.I.; Carrell, R.W.; Church, F.C.; Coughlin, P.B.; Gettins, P.G.; Irving, J.A.; Lomas, D.A.; Luke, C.J.; Moyer, R.W.; et al. The Serpins Are an Expanding Superfamily of Structurally Similar but Functionally Diverse Proteins. Evolution, Mechanism of Inhibition, Novel Functions, and a Revised Nomenclature. J. Biol. Chem. 2001, 276, 33293–33296. [Google Scholar]
- Liu, J.; Rothenburg, S.; McFadden, G. The Poxvirus C7l Host Range Factor Superfamily. Curr. Opin. Virol. 2012, 2, 764–772. [Google Scholar] [CrossRef] [PubMed]
- Shchelkunov, S.; Totmenin, A.; Kolosova, I. Species-Specific Differences in Organization of Orthopoxvirus Kelch-Like Proteins. Virus Genes 2002, 24, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Bahar, M.W.; Graham, S.C.; Chen, R.A.; Cooray, S.; Smith, G.L.; Stuart, D.I.; Grimes, J.M. How Vaccinia Virus Has Evolved to Subvert the Host Immune Response. J. Struct. Biol. 2011, 175, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, J.M.; Esteban, M. A Poxvirus Bcl-2-Like Gene Family Involved in Regulation of Host Immune Response: Sequence Similarity and Evolutionary History. Virol. J. 2010, 7, e59. [Google Scholar] [CrossRef] [PubMed]
- Carfi, A.; Smith, C.A.; Smolak, P.J.; McGrew, J.; Wiley, D.C. Structure of a Soluble Secreted Chemokine Inhibitor vCCI (P35) from Cowpox Virus. Proc. Natl. Acad. Sci. USA 1999, 96, 12379–12383. [Google Scholar] [CrossRef] [PubMed]
- Arnold, P.L.; Fremont, D.H. Structural Determinants of Chemokine Binding by an Ectromelia Virus-Encoded Decoy Receptor. J. Virol. 2006, 80, 7439–7449. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Derider, M.; McCornack, M.A.; Jao, S.C.; Isern, N.; Ness, T.; Moyer, R.; LiWang, P.J. Solution Structure of the Complex between Poxvirus-Encoded Cc Chemokine Inhibitor vCCI and Human Mip-1beta. Proc. Natl. Acad. Sci. USA 2006, 103, 13985–13990. [Google Scholar] [CrossRef] [PubMed]
- Epperson, M.L.; Lee, C.A.; Fremont, D.H. Subversion of Cytokine Networks by Virally Encoded Decoy Receptors. Immunol. Rev. 2012, 250, 199–215. [Google Scholar] [CrossRef] [PubMed]
- Bahar, M.W.; Kenyon, J.C.; Putz, M.M.; Abrescia, N.G.; Pease, J.E.; Wise, E.L.; Stuart, D.I.; Smith, G.L.; Grimes, J.M. Structure and Function of A41, a Vaccinia Virus Chemokine Binding Protein. PLoS Pathog. 2008, 4, e5. [Google Scholar] [CrossRef] [PubMed]
- Antonets, D.V.; Nepomnyashchikh, T.S.; Shchelkunov, S.N. Secret Domain of Variola Virus CrmB Protein Can Be a Member of Poxviral Type Ii Chemokine-Binding Proteins Family. BMC Res. Notes 2010, 3, e271. [Google Scholar] [CrossRef] [PubMed]
- Alejo, A.; Ruiz-Arguello, M.B.; Ho, Y.; Smith, V.P.; Saraiva, M.; Alcami, A. A Chemokine-Binding Domain in the Tumor Necrosis Factor Receptor from Variola (Smallpox) Virus. Proc. Natl. Acad. Sci. USA 2006, 103, 5995–6000. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Lu, Q.; Wei, H.; Wang, D.; Chen, D.; He, G.; Huang, L.; Wang, H.; Wang, X. Structural Basis of Chemokine Sequestration by CrmD, a Poxvirus-Encoded Tumor Necrosis Factor Receptor. PLoS Pathog. 2011, 7, e1002162. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, A.; Hammarlund, E.; Slifka, M.K.; Fruh, K. Cowpox Virus Evades CTL Recognition and Inhibits the Intracellular Transport of MHC Class I Molecules. J. Immunol. 2007, 178, 1654–1661. [Google Scholar] [CrossRef] [PubMed]
- Byun, M.; Wang, X.; Pak, M.; Hansen, T.H.; Yokoyama, W.M. Cowpox Virus Exploits the Endoplasmic Reticulum Retention Pathway to Inhibit MHC Class I Transport to the Cell Surface. Cell Host Microbe 2007, 2, 306–315. [Google Scholar] [CrossRef]
- McCoy, W.H.T.; Wang, X.; Yokoyama, W.M.; Hansen, T.H.; Fremont, D.H. Structural Mechanism of ER Retrieval of MHC Class I by Cowpox. PLoS Biol. 2012, 10, e1001432. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.H.; Gaffney, D.F.; Subak-Sharpe, J.H.; Stow, N.D. DNA Sequence of the Gene Encoding a Major Secreted Protein of Vaccinia Virus, Strain Lister. J. Gen. Virol. 1990, 71, 2013–2021. [Google Scholar] [CrossRef] [PubMed]
- Seet, B.T.; Singh, R.; Paavola, C.; Lau, E.K.; Handel, T.M.; McFadden, G. Molecular Determinants for CC-Chemokine Recognition by a Poxvirus CC-Chemokine Inhibitor. Proc. Natl. Acad. Sci. USA 2001, 98, 9008–9013. [Google Scholar] [CrossRef] [PubMed]
- Beck, C.G.; Studer, C.; Zuber, J.F.; Demange, B.J.; Manning, U.; Urfer, R. The Viral Cc Chemokine-Binding Protein vCCI Inhibits Monocyte Chemoattractant Protein-1 Activity by Masking Its CCR2B-Binding Site. J. Biol. Chem. 2001, 276, 43270–43276. [Google Scholar] [CrossRef] [PubMed]
- Alcami, A.; Symons, J.A.; Collins, P.D.; Williams, T.J.; Smith, G.L. Blockade of Chemokine Activity by a Soluble Chemokine Binding Protein from Vaccinia Virus. J. Immunol. 1998, 160, 624–633. [Google Scholar] [PubMed]
- Baggiolini, M. Chemokines and Leukocyte Traffic. Nature 1998, 392, 565–568. [Google Scholar] [CrossRef] [PubMed]
- Zlotnik, A.; Yoshie, O. Chemokines: A New Classification System and Their Role in Immunity. Immunity 2000, 12, 121–127. [Google Scholar] [CrossRef]
- Graham, K.A.; Lalani, A.S.; Macen, J.L.; Ness, T.L.; Barry, M.; Liu, L.Y.; Lucas, A.; Clark-Lewis, I.; Moyer, R.W.; McFadden, G. The T1/35kDa Family of Poxvirus-Secreted Proteins Bind Chemokines and Modulate Leukocyte Influx into Virus-Infected Tissues. Virology 1997, 229, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; Smith, T.D.; Smolak, P.J.; Friend, D.; Hagen, H.; Gerhart, M.; Park, L.; Pickup, D.J.; Torrance, D.; Mohler, K.; et al. Poxvirus Genomes Encode a Secreted, Soluble Protein That Preferentially Inhibits Beta Chemokine Activity yet Lacks Sequence Homology to Known Chemokine Receptors. Virology 1997, 236, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Lalani, A.S.; Ness, T.L.; Singh, R.; Harrison, J.K.; Seet, B.T.; Kelvin, D.J.; McFadden, G.; Moyer, R.W. Functional Comparisons among Members of the Poxvirus T1/35kDa Family of Soluble CC-Chemokine Inhibitor Glycoproteins. Virology 1998, 250, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Burns, J.M.; Dairaghi, D.J.; Deitz, M.; Tsang, M.; Schall, T.J. Comprehensive Mapping of Poxvirus vCCI Chemokine-Binding Protein. Expanded Range of Ligand Interactions and Unusual Dissociation Kinetics. J. Biol. Chem. 2002, 277, 2785–2789. [Google Scholar] [CrossRef] [PubMed]
- White, G.E.; McNeill, E.; Christou, I.; Channon, K.M.; Greaves, D.R. Site-Directed Mutagenesis of the CC Chemokine Binding Protein 35k-Fc Reveals Residues Essential for Activity and Mutations That Increase the Potency of CC Chemokine Blockade. Mol. Pharmacol. 2011, 80, 328–336. [Google Scholar]
- Lalani, A.S.; Masters, J.; Graham, K.; Liu, L.; Lucas, A.; McFadden, G. Role of the Myxoma Virus Soluble CC-Chemokine Inhibitor Glycoprotein, M-T1, During Myxoma Virus Pathogenesis. Virology 1999, 256, 233–245. [Google Scholar]
- Bursill, C.A.; Cai, S.; Channon, K.M.; Greaves, D.R. Adenoviral-Mediated Delivery of a Viral Chemokine Binding Protein Blocks CC-Chemokine Activity in Vitro and in Vivo. Immunobiology 2003, 207, 187–196. [Google Scholar]
- Reading, P.C.; Symons, J.A.; Smith, G.L. A Soluble Chemokine-Binding Protein from Vaccinia Virus Reduces Virus Virulence and the Inflammatory Response to Infection. J. Immunol. 2003, 170, 1435–1442. [Google Scholar]
- Dabbagh, K.; Xiao, Y.; Smith, C.; Stepick-Biek, P.; Kim, S.G.; Lamm, W.J.; Liggitt, D.H.; Lewis, D.B. Local Blockade of Allergic Airway Hyperreactivity and Inflammation by the Poxvirus-Derived Pan-CC-Chemokine Inhibitor vCCI. J. Immunol. 2000, 165, 3418–3422. [Google Scholar]
- Buatois, V.; Fagete, S.; Magistrelli, G.; Chatel, L.; Fischer, N.; Kosco-Vilbois, M.H.; Ferlin, W.G. Pan-Cc Chemokine Neutralization Restricts Splenocyte Egress and Reduces Inflammation in a Model of Arthritis. J. Immunol. 2010, 185, 2544–2554. [Google Scholar]
- Liu, L.; Dai, E.; Miller, L.; Seet, B.; Lalani, A.; Macauley, C.; Li, X.; Virgin, H.W.; Bunce, C.; Turner, P.; et al. Viral Chemokine-Binding Proteins Inhibit Inflammatory Responses and Aortic Allograft Transplant Vasculopathy in Rat Models. Transplantation 2004, 77, 1652–1660. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.M.; Messauodi, I.; Estep, R.D.; Orzechowska, B.; Wong, S.W. Monkeypox virus viral chemokine inhibitor (MPV vCCI), a potent inhibitor of rhesus macrophage inflammatory protein-1. Cytokine 2008, 43, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Schrodinger, L.L.C. The Pymol Molecular Graphics System, Version 1.6.0.0. Available online: https://www.pymol.org (accessed on 21 March 2015).
- Seet, B.T.; Barrett, J.; Robichaud, J.; Shilton, B.; Singh, R.; McFadden, G. Glycosaminoglycan Binding Properties of the Myxoma Virus CC-Chemokine Inhibitor, M-T1. J. Biol. Chem. 2001, 276, 30504–30513. [Google Scholar] [CrossRef] [PubMed]
- Ng, A.; Tscharke, D.C.; Reading, P.C.; Smith, G.L. The Vaccinia Virus A41L Protein Is a Soluble 30 kDa Glycoprotein That Affects Virus Virulence. J. Gen. Virol. 2001, 82, 2095–2105. [Google Scholar]
- Clark, R.H.; Kenyon, J.C.; Bartlett, N.W.; Tscharke, D.C.; Smith, G.L. Deletion of Gene A41L Enhances Vaccinia Virus Immunogenicity and Vaccine Efficacy. J. Gen. Virol. 2006, 87, 29–38. [Google Scholar]
- Ruiz-Arguello, M.B.; Smith, V.P.; Campanella, G.S.; Baleux, F.; Arenzana-Seisdedos, F.; Luster, A.D.; Alcami, A. An Ectromelia Virus Protein That Interacts with Chemokines through Their Glycosaminoglycan Binding Domain. J. Virol. 2008, 82, 917–926. [Google Scholar]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of Nanosystems: Application to Microtubules and the Ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar]
- Graham, S.C.; Bahar, M.W.; Abrescia, N.G.; Smith, G.L.; Stuart, D.I.; Grimes, J.M. Structure of CrmE, a Virus-Encoded Tumour Necrosis Factor Receptor. J. Mol. Biol. 2007, 372, 660–671. [Google Scholar]
- Loparev, V.N.; Parsons, J.M.; Knight, J.C.; Panus, J.F.; Ray, C.A.; Buller, R.M.; Pickup, D.J.; Esposito, J.J. A Third Distinct Tumor Necrosis Factor Receptor of Orthopoxviruses. Proc. Natl. Acad. Sci. USA 1998, 95, 3786–3791. [Google Scholar]
- Smith, C.A.; Davis, T.; Wignall, J.M.; Din, W.S.; Farrah, T.; Upton, C.; McFadden, G.; Goodwin, R.G. T2 Open Reading Frame from the Shope Fibroma Virus Encodes a Soluble Form of the TNF Receptor. Biochem. Biophys. Res. Commun. 1991, 176, 335–342. [Google Scholar]
- Upton, C.; Macen, J.L.; Schreiber, M.; McFadden, G. Myxoma Virus Expresses a Secreted Protein with Homology to the Tumor Necrosis Factor Receptor Gene Family That Contributes to Viral Virulence. Virology 1991, 184, 370–382. [Google Scholar]
- Alejo, A.; Pontejo, S.M.; Alcami, A. Poxviral TNFRs: Properties and Role in Viral Pathogenesis. Adv. Exp. Med. Biol. 2011, 691, 203–210. [Google Scholar]
- Kelley, L.A.; Sternberg, M.J. Protein Structure Prediction on the Web: A Case Study Using the Phyre Server. Nat. Protoc. 2009, 4, 363–371. [Google Scholar]
- Byun, M.; Verweij, M.C.; Pickup, D.J.; Wiertz, E.J.; Hansen, T.H.; Yokoyama, W.M. Two Mechanistically Distinct Immune Evasion Proteins of Cowpox Virus Combine to Avoid Antiviral CD8 T Cells. Cell Host Microbe 2009, 6, 422–432. [Google Scholar]
- Lin, J.; Eggensperger, S.; Hank, S.; Wycisk, A.I.; Wieneke, R.; Mayerhofer, P.U.; Tampe, R. A Negative Feedback Modulator of Antigen Processing Evolved from a Frameshift in the Cowpox Virus Genome. PLoS Pathog. 2014, 10, e1004554. [Google Scholar]
- Luteijn, R.D.; Hoelen, H.; Kruse, E.; van Leeuwen, W.F.; Grootens, J.; Horst, D.; Koorengevel, M.; Drijfhout, J.W.; Kremmer, E.; Fruh, K.; et al. Cowpox Virus Protein CPXV012 Eludes CTLs by Blocking ATP Binding to Tap. J. Immunol. 2014, 193, 1578–1589. [Google Scholar] [CrossRef] [PubMed]
- McCoy, W.H.T.; Wang, X.; Yokoyama, W.M.; Hansen, T.H.; Fremont, D.H. Cowpox Virus Employs a Two-Pronged Strategy to Outflank MHCI Antigen Presentation. Mol. Immunol. 2013, 55, 156–158. [Google Scholar]
- Pelham, H.R. The Dynamic Organisation of the Secretory Pathway. Cell Struct. Funct. 1996, 21, 413–419. [Google Scholar]
- Hnatiuk, S.; Barry, M.; Zeng, W.; Liu, L.; Lucas, A.; Percy, D.; McFadden, G. Role of the C-Terminal RDEL Motif of the Myxoma Virus M-T4 Protein in Terms of Apoptosis Regulation and Viral Pathogenesis. Virology 1999, 263, 290–306. [Google Scholar]
- Deane, D.; McInnes, C.J.; Percival, A.; Wood, A.; Thomson, J.; Lear, A.; Gilray, J.; Fleming, S.; Mercer, A.; Haig, D. Orf Virus Encodes a Novel Secreted Protein Inhibitor of Granulocyte-Macrophage Colony-Stimulating Factor and Interleukin-2. J. Virol. 2000, 74, 1313–1320. [Google Scholar] [CrossRef] [PubMed]
- Rozwarski, D.A.; Gronenborn, A.M.; Clore, G.M.; Bazan, J.F.; Bohm, A.; Wlodawer, A.; Hatada, M.; Karplus, P.A. Structural Comparisons among the Short-Chain Helical Cytokines. Structure 1994, 2, 159–173. [Google Scholar]
- Deane, D.; Ueda, N.; Wise, L.M.; Wood, A.R.; Percival, A.; Jepson, C.; Inglis, N.F.; Fleming, S.B.; Mercer, A.A.; McInnes, C.J. Conservation and Variation of the Parapoxvirus GM-CSF-Inhibitory Factor (Gif) Proteins. J. Gen. Virol. 2009, 90, 970–977. [Google Scholar]
- Seet, B.T.; McCaughan, C.A.; Handel, T.M.; Mercer, A.; Brunetti, C.; McFadden, G.; Fleming, S.B. Analysis of an Orf Virus Chemokine-Binding Protein: Shifting Ligand Specificities among a Family of Poxvirus Viroceptors. Proc. Natl. Acad. Sci. USA 2003, 100, 15137–15142. [Google Scholar]
- Lee, S.; Chu, H.X.; Kim, H.A.; Real, N.C.; Sharif, S.; Fleming, S.B.; Mercer, A.A.; Wise, L.M.; Drummond, G.R.; Sobey, C.G. Effect of a Broad-Specificity Chemokine-Binding Protein on Brain Leukocyte Infiltration and Infarct Development. Stroke 2015, 46, 537–544. [Google Scholar]
- Lateef, Z.; Baird, M.A.; Wise, L.M.; Mercer, A.A.; Fleming, S.B. Orf Virus-Encoded Chemokine-Binding Protein Is a Potent Inhibitor of Inflammatory Monocyte Recruitment in a Mouse Skin Model. J. Gen. Virol. 2009, 90, 1477–1482. [Google Scholar]
- Lateef, Z.; Baird, M.A.; Wise, L.M.; Young, S.; Mercer, A.A.; Fleming, S.B. The Chemokine-Binding Protein Encoded by the Poxvirus Orf Virus Inhibits Recruitment of Dendritic Cells to Sites of Skin Inflammation and Migration to Peripheral Lymph Nodes. Cell. Microbiol. 2010, 12, 665–676. [Google Scholar]
- Hemmerich, S.; Paavola, C.; Bloom, A.; Bhakta, S.; Freedman, R.; Grunberger, D.; Krstenansky, J.; Lee, S.; McCarley, D.; Mulkins, M.; et al. Identification of Residues in the Monocyte Chemotactic Protein-1 That Contact the MCP-1 Receptor, CCR2. Biochemistry 1999, 38, 13013–13025. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Clements, J.; Eddy, S.R. Hmmer Web Server: Interactive Sequence Similarity Searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar]
- Kelley, L.A.; Mexulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web protal for protein modeling prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar]
- Phyre2. Available online: http://www.sbg.bio.ic.ac.uk/phyre2 (accessed on 21 March 2015).
- Hendrickson, R.C.; Wang, C.; Hatcher, E.L.; Lefkowitz, E.J. Orthopoxvirus Genome Evolution: The Role of Gene Loss. Viruses 2010, 2, 1933–1967. [Google Scholar]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X Version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, M.R.; Gasteiger, E.; Bairoch, A.; Sanchez, J.C.; Williams, K.L.; Appel, R.D.; Hochstrasser, D.F. Protein Identification and Analysis Tools in the Expasy Server. Methods Mol. Biol. 1999, 112, 531–552. [Google Scholar]
- NetNGlyc 1.0 Server. Available online: http://www.cbs.dtu.dk/services/NetNGlyc/ (accessed on 21 March 2015).
- Altekar, G.; Dwarkadas, S.; Huelsenbeck, J.P.; Ronquist, F. Parallel Metropolis Coupled Markov Chain Monte Carlo for Bayesian Phylogenetic Inference. Bioinformatics 2004, 20, 407–415. [Google Scholar]
- Gedey, R.; Jin, X.L.; Hinthong, O.; Shisler, J.L. Poxviral Regulation of the Host Nf-Kappab Response: The Vaccinia Virus M2L Protein Inhibits Induction of NF-KappaB Activation Via an ERK2 Pathway in Virus-Infected Human Embryonic Kidney Cells. J. Virol. 2006, 80, 8676–8685. [Google Scholar] [CrossRef] [PubMed]
- Halaby, D.M.; Poupon, A.; Mornon, J. The Immunoglobulin Fold Family: Sequence Analysis and 3D Structure Comparisons. Protein Eng. 1999, 12, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Bork, P.; Holm, L.; Sander, C. The Immunoglobulin Fold. Structural Classification, Sequence Patterns and Common Core. J. Mol. Biol. 1994, 242, 309–320. [Google Scholar] [CrossRef]
- Gewurz, B.E.; Gaudet, R.; Tortorella, D.; Wang, E.W.; Ploegh, H.L. Virus Subversion of Immunity: A Structural Perspective. Curr. Opin. Immunol. 2001, 13, 442–450. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nelson, C.A.; Epperson, M.L.; Singh, S.; Elliott, J.I.; Fremont, D.H. Structural Conservation and Functional Diversity of the Poxvirus Immune Evasion (PIE) Domain Superfamily. Viruses 2015, 7, 4873-4893. https://doi.org/10.3390/v7092848
Nelson CA, Epperson ML, Singh S, Elliott JI, Fremont DH. Structural Conservation and Functional Diversity of the Poxvirus Immune Evasion (PIE) Domain Superfamily. Viruses. 2015; 7(9):4873-4893. https://doi.org/10.3390/v7092848
Chicago/Turabian StyleNelson, Christopher A., Megan L. Epperson, Sukrit Singh, Jabari I. Elliott, and Daved H. Fremont. 2015. "Structural Conservation and Functional Diversity of the Poxvirus Immune Evasion (PIE) Domain Superfamily" Viruses 7, no. 9: 4873-4893. https://doi.org/10.3390/v7092848
APA StyleNelson, C. A., Epperson, M. L., Singh, S., Elliott, J. I., & Fremont, D. H. (2015). Structural Conservation and Functional Diversity of the Poxvirus Immune Evasion (PIE) Domain Superfamily. Viruses, 7(9), 4873-4893. https://doi.org/10.3390/v7092848