
Receptor Binding for the Entry Mechanisms of SARS-CoV-2: Insights from the Original Strain and Emerging Variants

 ,
,  , , and
, , and
Abstract
1. Introduction
2. Entry Receptors for Coronaviruses
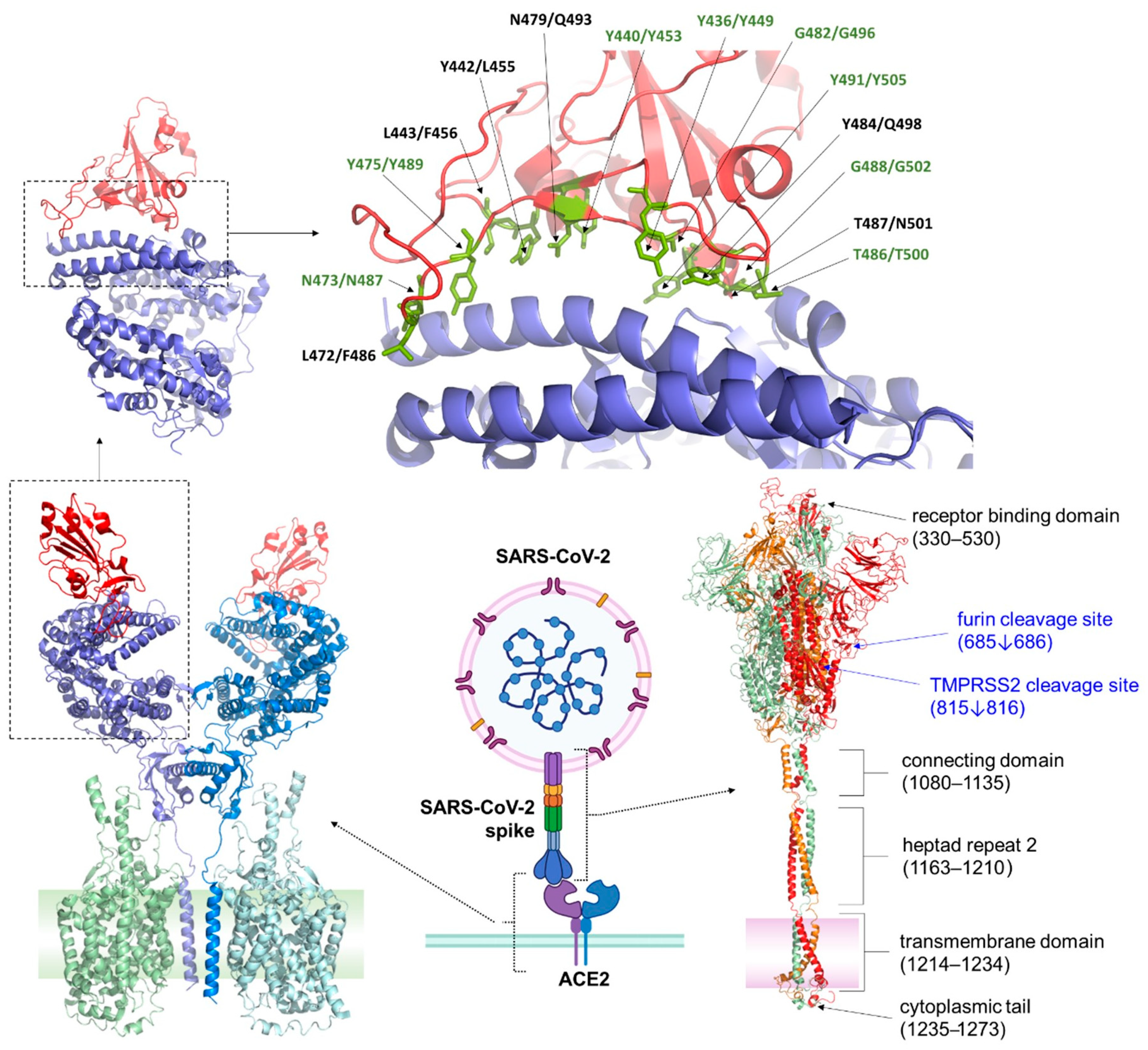
2.1. ACE2 (Angiotensin-Converting Enzyme 2)
2.2. Transmembrane Protease, Serine 2
2.3. TMPSS2-Independent Pathways
3. Role of Other Receptors
3.1. Dipeptidyl Peptidase 4 (DPP4)
3.2. Neuropilin-1 (NRP1)
3.3. Aminopeptidase N (APN)
3.4. Glucose-Regulated Protein 78 (GRP78)
3.5. Carcinoembryonic Antigen-Related Cell Adhesion Molecule 1 (CEACAM1)
4. Other Potential Receptors
5. Receptor-Mediated Endocytotic Pathways
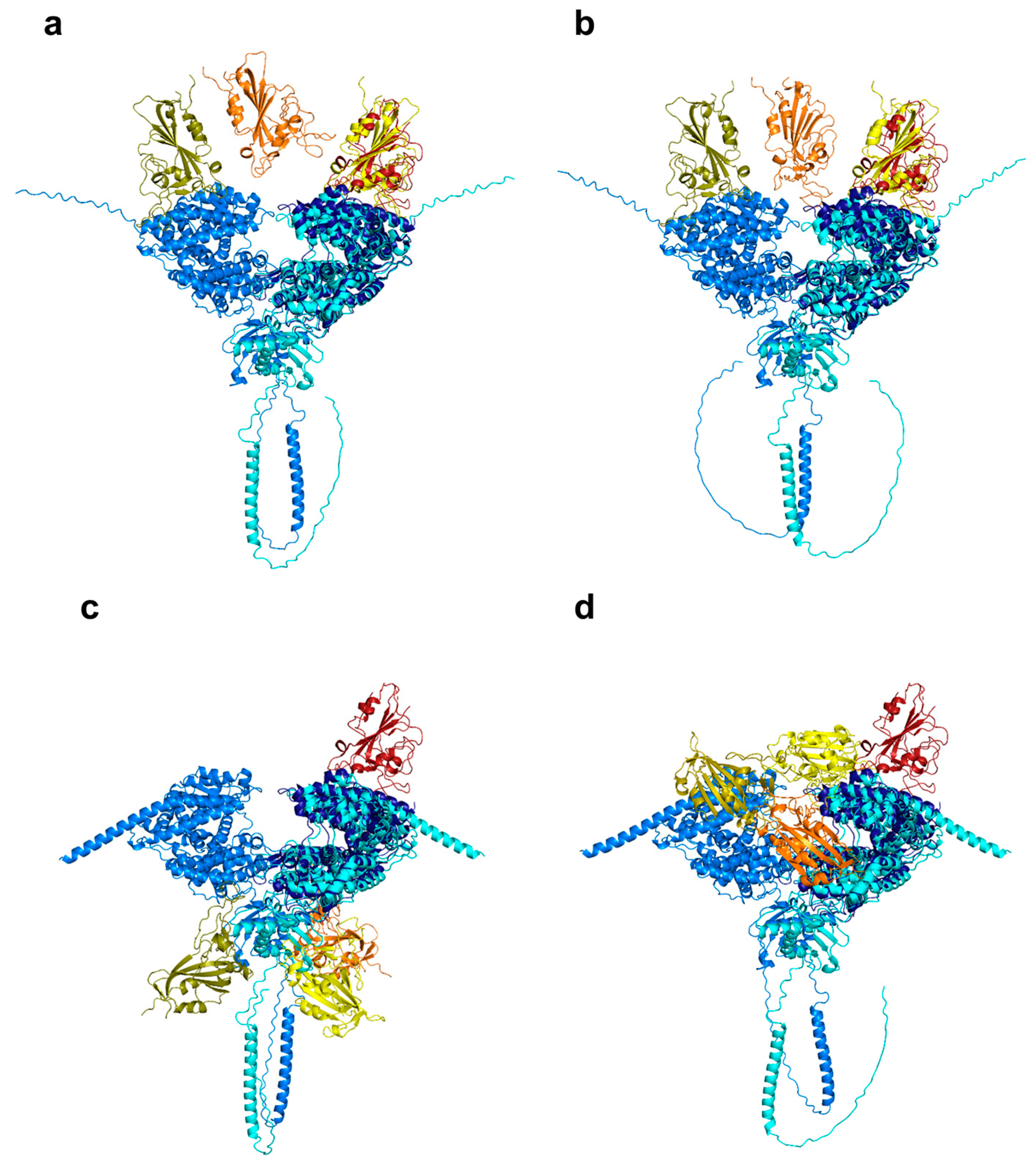
6. Mutations in Omicron Subvariants: Implications for Receptor Binding and Viral Entry
7. Evidence of Shift in Receptor Utilization from the Changing Clinical Spectrum of Infection
8. Therapeutic Implications
9. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
List of Abbreviations
References
- Peiris, J.S.M. Coronaviruses. In Medical Microbiology, 18th ed.; Churchill Livingstone: London, UK, 2012. [Google Scholar] [CrossRef]
- Yang, Y.; Peng, F.; Wang, R.; Yange, M.; Guan, K.; Jiang, T.; Xu, G.; Sun, J.; Chang, C. The deadly coronaviruses: The 2003 sars pandemic and the 2020 novel coronavirus epidemic in China. J. Autoimmun. 2020, 109, 102434. [Google Scholar] [CrossRef] [PubMed]
- Gorbalenya, A.E.; Enjuanes, L.; Ziebuhr, J.; Snijder, E.J. Nidovirales: Evolving the largest rna virus genome. Virus Res. 2006, 117, 17–37. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Lee, Y.; Michelow, I.C.; Choe, Y.J. Global seasonality of human coronaviruses: A systematic review. Open Forum Infect. Dis. 2020, 7, ofaa443. [Google Scholar] [CrossRef] [PubMed]
- Pfefferle, S.; Oppong, S.; Drexler, J.F.; Gloza-Rausch, F.; Ipsen, A.; Seebens, A.; Muller, M.A.; Annan, A.; Vallo, P.; Adu-Sarkodie, Y.; et al. Distant relatives of severe acute respiratory syndrome coronavirus and close relatives of human coronavirus 229e in bats, ghana. Emerg. Infect. Dis. 2009, 15, 1377–1384. [Google Scholar] [CrossRef]
- Vijgen, L.; Keyaerts, E.; Moes, E.; Thoelen, I.; Wollants, E.; Lemey, P.; Vandamme, A.M.; Van Ranst, M. Complete genomic sequence of human coronavirus OC43: Molecular clock analysis suggests a relatively recent zoonotic coronavirus transmission event. J. Virol. 2005, 79, 1595–1604. [Google Scholar] [CrossRef]
- Milewska, A.; Zarebski, M.; Nowak, P.; Stozek, K.; Potempa, J.; Pyrc, K. Human coronavirus nl63 utilizes heparan sulfate proteoglycans for attachment to target cells. J. Virol. 2014, 88, 13221–13230. [Google Scholar] [CrossRef]
- Barlan, A.; Zhao, J.; Sarkar, M.K.; Li, K.; McCray, P.B., Jr.; Perlman, S.; Gallagher, T. Receptor variation and susceptibility to middle east respiratory syndrome coronavirus infection. J. Virol. 2014, 88, 4953–4961. [Google Scholar] [CrossRef]
- Faramarzi, A.; Norouzi, S.; Dehdarirad, H.; Aghlmand, S.; Yusefzadeh, H.; Javan-Noughabi, J. The global economic burden of COVID-19 disease: A comprehensive systematic review and meta-analysis. Syst. Rev. 2024, 13, 68. [Google Scholar] [CrossRef]
- Kirtipal, N.; Bharadwaj, S.; Kang, S.G. From sars to SARS-CoV-2, insights on structure, pathogenicity and immunity aspects of pandemic human coronaviruses. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2020, 85, 104502. [Google Scholar]
- Li, W.; Shi, Z.; Yu, M.; Ren, W.; Smith, C.; Epstein, J.H.; Wang, H.; Crameri, G.; Hu, Z.; Zhang, H.; et al. Bats are natural reservoirs of SARS-like coronaviruses. Science 2005, 310, 676–679. [Google Scholar] [CrossRef]
- Wang, L.F.; Eaton, B.T. Bats, civets and the emergence of sars. Curr. Top. Microbiol. Immunol. 2007, 315, 325–344. [Google Scholar] [PubMed]
- de Groot, R.J.; Baker, S.C.; Baric, R.S.; Brown, C.S.; Drosten, C.; Enjuanes, L.; Fouchier, R.A.; Galiano, M.; Gorbalenya, A.E.; Memish, Z.A.; et al. Middle east respiratory syndrome coronavirus (MERS-CoV): Announcement of the coronavirus study group. J. Virol. 2013, 87, 7790–7792. [Google Scholar] [CrossRef]
- Wrobel, A.G.; Benton, D.J.; Xu, P.; Roustan, C.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. SARS-CoV-2 and bat RaTG13 spike glycoprotein structures inform on virus evolution and furin-cleavage effects. Nat. Struct. Mol. Biol. 2020, 27, 763–767. [Google Scholar] [CrossRef] [PubMed]
- Drake, J.W.; Holland, J.J. Mutation rates among RNA viruses. Proc. Natl. Acad. Sci. USA 1999, 96, 13910–13913. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Garcia-Crespo, C.; Lobo-Vega, R.; Perales, C. Mutation rates, mutation frequencies, and proofreading-repair activities in RNA virus genetics. Viruses 2021, 13, 1882. [Google Scholar] [CrossRef]
- Graham, R.L.; Baric, R.S. Recombination, reservoirs, and the modular spike: Mechanisms of coronavirus cross-species transmission. J. Virol. 2010, 84, 3134–3146. [Google Scholar] [CrossRef]
- Everest, H.; Stevenson-Leggett, P.; Bailey, D.; Bickerton, E.; Keep, S. Known cellular and receptor interactions of animal and human coronaviruses: A review. Viruses 2022, 14, 351. [Google Scholar] [CrossRef]
- Markov, P.V.; Ghafari, M.; Beer, M.; Lythgoe, K.; Simmonds, P.; Stilianakis, N.I.; Katzourakis, A. The evolution of SARS-CoV-2. Nat. Rev. Microbiol. 2023, 21, 361–379. [Google Scholar] [CrossRef]
- Belouzard, S.; Millet, J.K.; Licitra, B.N.; Whittaker, G.R. Mechanisms of coronavirus cell entry mediated by the viral spike protein. Viruses 2012, 4, 1011–1033. [Google Scholar] [CrossRef]
- Brian, D.A.; Baric, R.S. Coronavirus genome structure and replication. Curr. Top. Microbiol. Immunol. 2005, 287, 1–30. [Google Scholar]
- EA, J.A.; Jones, I.M. Membrane binding proteins of coronaviruses. Future Virol. 2019, 14, 275–286. [Google Scholar]
- Edinger, T.O.; Pohl, M.O.; Stertz, S. Entry of influenza a virus: Host factors and antiviral targets. J. Gen. Virol. 2014, 95, 263–277. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Lv, F.; Li, Z.; Zhao, C.; Wang, X.; Zhu, P.; Zhou, X. Cross-species susceptibility of emerging variants of SARS-CoV-2 spike. Genes 2024, 15, 1321. [Google Scholar] [CrossRef] [PubMed]
- Nassar, A.; Ibrahim, I.M.; Amin, F.G.; Magdy, M.; Elgharib, A.M.; Azzam, E.B.; Nasser, F.; Yousry, K.; Shamkh, I.M.; Mahdy, S.M.; et al. A review of human coronaviruses’ receptors: The host-cell targets for the crown bearing viruses. Molecules 2021, 26, 6455. [Google Scholar] [CrossRef]
- Hulswit, R.J.G.; Lang, Y.; Bakkers, M.J.G.; Li, W.; Li, Z.; Schouten, A.; Ophorst, B.; van Kuppeveld, F.J.M.; Boons, G.J.; Bosch, B.J.; et al. Human coronaviruses OC43 and HKU1 bind to 9-O-acetylated sialic acids via a conserved receptor-binding site in spike protein domain A. Proc. Natl. Acad. Sci. USA 2019, 116, 2681–2690. [Google Scholar] [CrossRef]
- Hofmann, H.; Pyrc, K.; van der Hoek, L.; Geier, M.; Berkhout, B.; Pohlmann, S. Human coronavirus NL63 employs the severe acute respiratory syndrome coronavirus receptor for cellular entry. Proc. Natl. Acad. Sci. USA 2005, 102, 7988–7993. [Google Scholar] [CrossRef]
- Saunders, N.; Fernandez, I.; Planchais, C.; Michel, V.; Rajah, M.M.; Baquero Salazar, E.; Postal, J.; Porrot, F.; Guivel-Benhassine, F.; Blanc, C.; et al. TMPRSS2 is a functional receptor for human coronavirus HKU1. Nature 2023, 624, 207–214. [Google Scholar] [CrossRef]
- Wentworth, D.E.; Holmes, K.V. Molecular determinants of species specificity in the coronavirus receptor aminopeptidase N (CD13): Influence of N-linked glycosylation. J. Virol. 2001, 75, 9741–9752. [Google Scholar] [CrossRef]
- Li, W.; Zhang, C.; Sui, J.; Kuhn, J.H.; Moore, M.J.; Luo, S.; Wong, S.K.; Huang, I.C.; Xu, K.; Vasilieva, N.; et al. Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J. 2005, 24, 1634–1643. [Google Scholar] [CrossRef]
- Raj, V.S.; Mou, H.; Smits, S.L.; Dekkers, D.H.; Muller, M.A.; Dijkman, R.; Muth, D.; Demmers, J.A.; Zaki, A.; Fouchier, R.A.; et al. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature 2013, 495, 251–254. [Google Scholar] [CrossRef]
- Clausen, T.M.; Sandoval, D.R.; Spliid, C.B.; Pihl, J.; Perrett, H.R.; Painter, C.D.; Narayanan, A.; Majowicz, S.A.; Kwong, E.M.; McVicar, R.N.; et al. SARS-CoV-2 infection depends on cellular heparan sulfate and ACE2. Cell 2020, 183, 1043–1057.e15. [Google Scholar] [CrossRef] [PubMed]
- Valero-Rello, A.; Sanjuan, R. Enveloped viruses show increased propensity to cross-species transmission and zoonosis. Proc. Natl. Acad. Sci. USA 2022, 119, e2215600119. [Google Scholar] [CrossRef]
- Valero-Rello, A.; Baeza-Delgado, C.; Andreu-Moreno, I.; Sanjuan, R. Cellular receptors for mammalian viruses. PLoS Pathog. 2024, 20, e1012021. [Google Scholar] [CrossRef]
- Donoghue, M.; Hsieh, F.; Baronas, E.; Godbout, K.; Gosselin, M.; Stagliano, N.; Donovan, M.; Woolf, B.; Robison, K.; Jeyaseelan, R.; et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ. Res. 2000, 87, E1–E9. [Google Scholar] [CrossRef] [PubMed]
- Menach, E.; Hashida, Y.; Yasukawa, K.; Inouye, K. Effects of conversion of the zinc-binding motif sequence of thermolysin, HEXXH, to that of dipeptidyl peptidase III, HEXXXH, on the activity and stability of thermolysin. Biosci. Biotechnol. Biochem. 2013, 77, 1901–1906. [Google Scholar] [CrossRef]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef]
- Patel, S.K.; Velkoska, E.; Burrell, L.M. Emerging markers in cardiovascular disease: Where does angiotensin-converting enzyme 2 fit in? Clin. Exp. Pharmacol. Physiol. 2013, 40, 551–559. [Google Scholar] [CrossRef]
- Li, Y.; Zhou, W.; Yang, L.; You, R. Physiological and pathological regulation of ACE2, the SARS-CoV-2 receptor. Pharmacol. Res. 2020, 157, 104833. [Google Scholar] [CrossRef] [PubMed]
- Lambert, D.W.; Yarski, M.; Warner, F.J.; Thornhill, P.; Parkin, E.T.; Smith, A.I.; Hooper, N.M.; Turner, A.J. Tumor necrosis factor-alpha convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2). J. Biol. Chem. 2005, 280, 30113–30119. [Google Scholar] [CrossRef]
- Zoufaly, A.; Poglitsch, M.; Aberle, J.H.; Hoepler, W.; Seitz, T.; Traugott, M.; Grieb, A.; Pawelka, E.; Laferl, H.; Wenisch, C.; et al. Human recombinant soluble ACE2 in severe COVID-19. Lancet Respir. Med. 2020, 8, 1154–1158. [Google Scholar] [CrossRef]
- Hikmet, F.; Mear, L.; Edvinsson, A.; Micke, P.; Uhlen, M.; Lindskog, C. The protein expression profile of ACE2 in human tissues. Mol. Syst. Biol. 2020, 16, e9610. [Google Scholar] [CrossRef] [PubMed]
- Hamming, I.; Timens, W.; Bulthuis, M.L.; Lely, A.T.; Navis, G.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for sars coronavirus. A first step in understanding sars pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef]
- Crackower, M.A.; Sarao, R.; Oudit, G.Y.; Yagil, C.; Kozieradzki, I.; Scanga, S.E.; Oliveira-dos-Santos, A.J.; da Costa, J.; Zhang, L.; Pei, Y.; et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature 2002, 417, 822–828. [Google Scholar] [CrossRef] [PubMed]
- Chua, R.L.; Lukassen, S.; Trump, S.; Hennig, B.P.; Wendisch, D.; Pott, F.; Debnath, O.; Thurmann, L.; Kurth, F.; Volker, M.T.; et al. COVID-19 severity correlates with airway epithelium-immune cell interactions identified by single-cell analysis. Nat. Biotechnol. 2020, 38, 970–979. [Google Scholar] [CrossRef]
- Blume, C.; Jackson, C.L.; Spalluto, C.M.; Legebeke, J.; Nazlamova, L.; Conforti, F.; Perotin, J.M.; Frank, M.; Butler, J.; Crispin, M.; et al. A novel ACE2 isoform is expressed in human respiratory epithelia and is upregulated in response to interferons and RNA respiratory virus infection. Nat. Genet. 2021, 53, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef]
- Letko, M.; Marzi, A.; Munster, V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol. 2020, 5, 562–569. [Google Scholar] [CrossRef]
- Ma, X.; Lu, M.; Gorman, J.; Terry, D.S.; Hong, X.; Zhou, Z.; Zhao, H.; Altman, R.B.; Arthos, J.; Blanchard, S.C.; et al. HIV-1 env trimer opens through an asymmetric intermediate in which individual protomers adopt distinct conformations. eLife 2018, 7, e34271. [Google Scholar] [CrossRef]
- Cervantes, M.; Hess, T.; Morbioli, G.G.; Sengar, A.; Kasson, P.M. The ACE2 receptor accelerates but is not biochemically required for SARS-CoV-2 membrane fusion. Chem. Sci. 2023, 14, 6997–7004. [Google Scholar] [CrossRef]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef]
- Yu, S.; Zheng, X.; Zhou, B.; Li, J.; Chen, M.; Deng, R.; Wong, G.; Lavillette, D.; Meng, G. SARS-CoV-2 spike engagement of ACE2 primes s2′ site cleavage and fusion initiation. Proc. Natl. Acad. Sci. USA 2022, 119, e2111199119. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Lan, Q.; Su, S.; Wang, X.; Xu, W.; Liu, Z.; Zhu, Y.; Wang, Q.; Lu, L.; Jiang, S. The role of furin cleavage site in SARS-CoV-2 spike protein-mediated membrane fusion in the presence or absence of trypsin. Signal Transduct. Target. Ther. 2020, 5, 92. [Google Scholar] [CrossRef] [PubMed]
- Almehdi, A.M.; Khoder, G.; Alchakee, A.S.; Alsayyid, A.T.; Sarg, N.H.; Soliman, S.S.M. SARS-CoV-2 spike protein: Pathogenesis, vaccines, and potential therapies. Infection 2021, 49, 855–876. [Google Scholar] [CrossRef]
- Ismail, A.M.; Elfiky, A.A. SARS-CoV-2 spike behavior in situ: A Cryo-EM images for a better understanding of the COVID-19 pandemic. Signal Transduct. Target. Ther. 2020, 5, 252. [Google Scholar] [CrossRef]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 2020, 183, 1735. [Google Scholar] [CrossRef]
- Casalino, L.; Gaieb, Z.; Goldsmith, J.A.; Hjorth, C.K.; Dommer, A.C.; Harbison, A.M.; Fogarty, C.A.; Barros, E.P.; Taylor, B.C.; McLellan, J.S.; et al. Beyond shielding: The roles of glycans in the SARS-CoV-2 spike protein. ACS Cent. Sci. 2020, 6, 1722–1734. [Google Scholar] [CrossRef]
- Liu, H.; Wei, P.; Kappler, J.W.; Marrack, P.; Zhang, G. SARS-CoV-2 variants of concern and variants of interest receptor binding domain mutations and virus infectivity. Front. Immunol. 2022, 13, 825256. [Google Scholar]
- Cherian, S.; Potdar, V.; Jadhav, S.; Yadav, P.; Gupta, N.; Das, M.; Rakshit, P.; Singh, S.; Abraham, P.; Panda, S.; et al. SARS-CoV-2 spike mutations, L452R, T478K, E484Q and P681R, in the second wave of COVID-19 in Maharashtra, India. Microorganisms 2021, 9, 1542. [Google Scholar] [CrossRef]
- Le, K.; Kannappan, S.; Kim, T.; Lee, J.H.; Lee, H.R.; Kim, K.K. Structural understanding of SARS-CoV-2 virus entry to host cells. Front. Mol. Biosci. 2023, 10, 1288686. [Google Scholar] [CrossRef] [PubMed]
- Cele, S.; Karim, F.; Lustig, G.; San, J.E.; Hermanus, T.; Tegally, H.; Snyman, J.; Moyo-Gwete, T.; Wilkinson, E.; Bernstein, M.; et al. SARS-CoV-2 prolonged infection during advanced HIV disease evolves extensive immune escape. Cell Host Microbe 2022, 30, 154–162.e5. [Google Scholar] [CrossRef] [PubMed]
- Hattab, D.; Amer, M.F.A.; Al-Alami, Z.M.; Bakhtiar, A. SARS-CoV-2 journey: From alpha variant to omicron and its sub-variants. Infection 2024, 52, 767–786. [Google Scholar] [CrossRef]
- Tzou, P.L.; Tao, K.; Pond, S.L.K.; Shafer, R.W. Coronavirus resistance database (CoV-RDB): SARS-CoV-2 susceptibility to monoclonal antibodies, convalescent plasma, and plasma from vaccinated persons. PLoS ONE 2022, 17, e0261045. [Google Scholar] [CrossRef]
- Niu, S.; Wang, J.; Bai, B.; Wu, L.; Zheng, A.; Chen, Q.; Du, P.; Han, P.; Zhang, Y.; Jia, Y.; et al. Molecular basis of cross-species ACE2 interactions with SARS-CoV-2-like viruses of pangolin origin. EMBO J. 2021, 40, e107786. [Google Scholar] [CrossRef] [PubMed]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef]
- Monteiro da Silva, G.; Cui, J.Y.; Dalgarno, D.C.; Lisi, G.P.; Rubenstein, B.M. High-throughput prediction of protein conformational distributions with subsampled AlphaFold2. Nat. Commun. 2024, 15, 2464. [Google Scholar] [CrossRef]
- Singh, A.; Copeland, M.M.; Kundrotas, P.J.; Vakser, I.A. Gramm web server for protein docking. Methods Mol. Biol. 2024, 2714, 101–112. [Google Scholar]
- Fraser, B.J.; Beldar, S.; Seitova, A.; Hutchinson, A.; Mannar, D.; Li, Y.; Kwon, D.; Tan, R.; Wilson, R.P.; Leopold, K.; et al. Structure and activity of human TMPRSS2 protease implicated in SARS-CoV-2 activation. Nat. Chem. Biol. 2022, 18, 963–971. [Google Scholar] [CrossRef]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARA-CoV. Nat. Commun. 2020, 11, 1620. [Google Scholar] [CrossRef] [PubMed]
- Piva, F.; Sabanovic, B.; Cecati, M.; Giulietti, M. Expression and co-expression analyses of TMPRSS2, a key element in COVID-19. Eur. J. Clin. Microbiol. Infect. Dis. Off. Publ. Eur. Soc. Clin. Microbiol. 2021, 40, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Limburg, H.; Harbig, A.; Bestle, D.; Stein, D.A.; Moulton, H.M.; Jaeger, J.; Janga, H.; Hardes, K.; Koepke, J.; Schulte, L.; et al. TMPRSS2 is the major activating protease of influenza a virus in primary human airway cells and influenza B virus in human type II pneumocytes. J. Virol. 2019, 93, e00649-19. [Google Scholar] [CrossRef]
- Shulla, A.; Heald-Sargent, T.; Subramanya, G.; Zhao, J.; Perlman, S.; Gallagher, T. A transmembrane serine protease is linked to the severe acute respiratory syndrome coronavirus receptor and activates virus entry. J. Virol. 2011, 85, 873–882. [Google Scholar] [CrossRef]
- Shirato, K.; Kawase, M.; Matsuyama, S. Wild-type human coronaviruses prefer cell-surface tmprss2 to endosomal cathepsins for cell entry. Virology 2018, 517, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, X.; Zhang, X.; Zhao, Z.; Lu, Y.; Pu, D.; Zhang, Z.; Chen, J.; Wang, Y.; Li, M.; et al. Tmprss2 and glycan receptors synergistically facilitate coronavirus entry. Cell 2024, 187, 4261–4271.e17. [Google Scholar] [CrossRef]
- Lu, G.; Wang, Q.; Gao, G.F. Bat-to-human: Spike features determining ‘host jump’ of coronaviruses SARS-CoV, MERS-CoV, and beyond. Trends Microbiol. 2015, 23, 468–478. [Google Scholar] [CrossRef]
- Ou, T.; Mou, H.; Zhang, L.; Ojha, A.; Choe, H.; Farzan, M. Hydroxychloroquine-mediated inhibition of SARS-CoV-2 entry is attenuated by TMPRSS2. PLoS Pathog. 2021, 17, e1009212. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 cell entry depends on ACE2 and tmprss2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, J.; Johnson, B.A.; Xia, H.; Ku, Z.; Schindewolf, C.; Widen, S.G.; An, Z.; Weaver, S.C.; Menachery, V.D.; et al. Delta spike P681R mutation enhances SARS-CoV-2 fitness over alpha variant. Cell Rep. 2022, 39, 110829. [Google Scholar] [CrossRef]
- Furusawa, Y.; Kiso, M.; Iida, S.; Uraki, R.; Hirata, Y.; Imai, M.; Suzuki, T.; Yamayoshi, S.; Kawaoka, Y. In SARS-CoV-2 delta variants, spike-P681R and D950N promote membrane fusion, spike-P681R enhances spike cleavage, but neither substitution affects pathogenicity in hamsters. EBioMedicine 2023, 91, 104561. [Google Scholar] [CrossRef] [PubMed]
- Park, S.B.; Khan, M.; Chiliveri, S.C.; Hu, X.; Irvin, P.; Leek, M.; Grieshaber, A.; Hu, Z.; Jang, E.S.; Bax, A.; et al. SARS-CoV-2 omicron variants harbor spike protein mutations responsible for their attenuated fusogenic phenotype. Commun. Biol. 2023, 6, 556. [Google Scholar] [CrossRef] [PubMed]
- Afrin, S.Z.; Sathi, F.A.; Nooruzzaman, M.; Parvin, R. Molecular insights into the SARS-CoV-2 omicron variant from bangladesh suggest diverse and continuous evolution. Virology 2023, 587, 109882. [Google Scholar] [CrossRef] [PubMed]
- Escalera, A.; Gonzalez-Reiche, A.S.; Aslam, S.; Mena, I.; Laporte, M.; Pearl, R.L.; Fossati, A.; Rathnasinghe, R.; Alshammary, H.; van de Guchte, A.; et al. Mutations in SARS-CoV-2 variants of concern link to increased spike cleavage and virus transmission. Cell Host Microbe 2022, 30, 373–387.e7. [Google Scholar] [CrossRef]
- Takeda, M. Proteolytic activation of SARS-CoV-2 spike protein. Microbiol. Immunol. 2022, 66, 15–23. [Google Scholar] [CrossRef]
- Chakraborty, C.; Saha, A.; Bhattacharya, M.; Dhama, K.; Agoramoorthy, G. Natural selection of the D614G mutation in SARS-CoV-2 Omicron (B.1.1.529) variant and its subvariants. Mol. Therapy Nucleic Acids 2023, 31, 437–439. [Google Scholar] [CrossRef]
- Gellenoncourt, S.; Saunders, N.; Robinot, R.; Auguste, L.; Rajah, M.M.; Kervevan, J.; Jeger-Madiot, R.; Staropoli, I.; Planchais, C.; Mouquet, H.; et al. The spike-stabilizing D614G mutation interacts with S1/S2 cleavage site mutations to promote the infectious potential of SARS-CoV-2 variants. J. Virol. 2022, 96, e0130122. [Google Scholar] [CrossRef]
- Laporte, M.; Raeymaekers, V.; Van Berwaer, R.; Vandeput, J.; Marchand-Casas, I.; Thibaut, H.J.; Van Looveren, D.; Martens, K.; Hoffmann, M.; Maes, P.; et al. The SARS-CoV-2 and other human coronavirus spike proteins are fine-tuned towards temperature and proteases of the human airways. PLoS Pathog. 2021, 17, e1009500. [Google Scholar] [CrossRef]
- Scheepers, C.; Everatt, J.; Amoako, D.G.; Tegally, H.; Wibmer, C.K.; Mnguni, A.; Ismail, A.; Mahlangu, B.; Lambson, B.E.; Martin, D.P.; et al. Emergence and phenotypic characterization of the global SARS-CoV-2 C.1.2 lineage. Nat. Commun. 2022, 13, 1976. [Google Scholar] [CrossRef]
- Simmons, G.; Gosalia, D.N.; Rennekamp, A.J.; Reeves, J.D.; Diamond, S.L.; Bates, P. Inhibitors of cathepsin l prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl. Acad. Sci. USA 2005, 102, 11876–11881. [Google Scholar] [CrossRef]
- Maciewicz, R.A.; Etherington, D.J.; Kos, J.; Turk, V. Collagenolytic cathepsins of rabbit spleen—A kinetic-analysis of collagen degradation and inhibition by chicken cystatin. Collagen Relat. Res. 1987, 7, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Reilly, J.J.; Mason, R.W.; Chen, P.; Joseph, L.J.; Sukhatme, V.P.; Yee, R.; Chapman, H.A. Synthesis and processing of cathepsin-L, an elastase, by human alveolar macrophages. Biochem. J. 1989, 257, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.M.; Zhu, Y.; Zhang, L.; Zhong, G.; Tai, L.; Liu, S.; Yin, G.; Lu, J.; He, Q.; Li, M.J.; et al. Novel cleavage sites identified in SARS-CoV-2 spike protein reveal mechanism for cathepsin L-facilitated viral infection and treatment strategies. Cell Discov. 2022, 8, 53. [Google Scholar] [CrossRef]
- Park, J.E.; Li, K.; Barlan, A.; Fehr, A.R.; Perlman, S.; McCray, P.B., Jr.; Gallagher, T. Proteolytic processing of middle east respiratory syndrome coronavirus spikes expands virus tropism. Proc. Natl. Acad. Sci. USA 2016, 113, 12262–12267. [Google Scholar] [CrossRef] [PubMed]
- Bosch, B.J.; Bartelink, W.; Rottier, P.J. Cathepsin l functionally cleaves the severe acute respiratory syndrome coronavirus class I fusion protein upstream of rather than adjacent to the fusion peptide. J. Virol. 2008, 82, 8887–8890. [Google Scholar] [CrossRef]
- Bestle, D.; Heindl, M.R.; Limburg, H.; Van Lam van, T.; Pilgram, O.; Moulton, H.; Stein, D.A.; Hardes, K.; Eickmann, M.; Dolnik, O.; et al. TMPRSS2 and furin are both essential for proteolytic activation of SARS-CoV-2 in human airway cells. Life Sci. Alliance 2020, 3, e202000786. [Google Scholar] [CrossRef]
- Sasaki, M.; Uemura, K.; Sato, A.; Toba, S.; Sanaki, T.; Maenaka, K.; Hall, W.W.; Orba, Y.; Sawa, H. SARS-CoV-2 variants with mutations at the S1/S2 cleavage site are generated in vitro during propagation in TMPRSS2-deficient cells. PLoS Pathog. 2021, 17, e1009233. [Google Scholar] [CrossRef]
- Sarker, J.; Das, P.; Sarker, S.; Roy, A.K.; Momen, A. A review on expression, pathological roles, and inhibition of TMPRSS2, the serine protease responsible for SARS-CoV-2 spike protein activation. Scientifica 2021, 2021, 2706789. [Google Scholar] [CrossRef]
- Suzuki, R.; Yamasoba, D.; Kimura, I.; Wang, L.; Kishimoto, M.; Ito, J.; Morioka, Y.; Nao, N.; Nasser, H.; Uriu, K.; et al. Attenuated fusogenicity and pathogenicity of SARS-CoV-2 Omicron variant. Nature 2022, 603, 700–705. [Google Scholar] [CrossRef]
- Zhao, H.; Lu, L.; Peng, Z.; Chen, L.L.; Meng, X.; Zhang, C.; Ip, J.D.; Chan, W.M.; Chu, A.W.; Chan, K.H.; et al. SARS-CoV-2 omicron variant shows less efficient replication and fusion activity when compared with delta variant in TMPRSS2-expressed cells. Emerg. Microbes Infect. 2022, 11, 277–283. [Google Scholar] [CrossRef]
- Iwata-Yoshikawa, N.; Kakizaki, M.; Shiwa-Sudo, N.; Okura, T.; Tahara, M.; Fukushi, S.; Maeda, K.; Kawase, M.; Asanuma, H.; Tomita, Y.; et al. Essential role of TMPRSS2 in SARS-CoV-2 infection in murine airways. Nat. Commun. 2022, 13, 6100. [Google Scholar] [CrossRef] [PubMed]
- Meyerholz, D.K.; Lambertz, A.M.; McCray, P.B., Jr. Dipeptidyl peptidase 4 distribution in the human respiratory tract: Implications for the middle east respiratory syndrome. Am. J. Pathol. 2016, 186, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Sebastian-Martin, A.; Sanchez, B.G.; Mora-Rodriguez, J.M.; Bort, A.; Diaz-Laviada, I. Role of dipeptidyl peptidase-4 (DPP4) on COVID-19 physiopathology. Biomedicines 2022, 10, 2026. [Google Scholar] [CrossRef]
- Yang, Y.; Du, L.; Liu, C.; Wang, L.; Ma, C.; Tang, J.; Baric, R.S.; Jiang, S.; Li, F. Receptor usage and cell entry of bat coronavirus hku4 provide insight into bat-to-human transmission of mers coronavirus. Proc. Natl. Acad. Sci. USA 2014, 111, 12516–12521. [Google Scholar] [CrossRef]
- Roy, A.N.; Gupta, A.M.; Banerjee, D.; Chakrabarti, J.; Raghavendra, P.B. Unraveling dpp4 receptor interactions with SARS-CoV-2 variants and mers-cov: Insights into pulmonary disorders via immunoinformatics and molecular dynamics. Viruses 2023, 15, 2056. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Z.; Yang, L.; Lian, X.; Xie, Y.; Li, S.; Xin, S.; Cao, P.; Lu, J. The Mers-CoV receptor DPP4 as a candidate binding target of the SARS-CoV-2 spike. iScience 2020, 23, 101160. [Google Scholar] [CrossRef] [PubMed]
- Nyland, J.E.; Raja-Khan, N.T.; Bettermann, K.; Haouzi, P.A.; Leslie, D.L.; Kraschnewski, J.L.; Parent, L.J.; Grigson, P.S. Diabetes, drug treatment, and mortality in COVID-19: A multinational retrospective cohort study. Diabetes 2021, 70, 2903–2916. [Google Scholar] [CrossRef]
- Abbasi, F.; Adatorwovor, R.; Davarpanah, M.A.; Mansoori, Y.; Hajiani, M.; Azodi, F.; Sefidbakht, S.; Davoudi, S.; Rezaei, F.; Mohammadmoradi, S.; et al. A randomized trial of sitagliptin and spironolactone with combination therapy in hospitalized adults with COVID-19. J. Endocr. Soc. 2022, 6, bvac017. [Google Scholar] [CrossRef]
- Martinez, T.E.; Mayilsamy, K.; Mohapatra, S.S.; Mohapatra, S. Modulation of paracellular permeability in SARS-CoV-2 blood-to-brain transcytosis. Viruses 2024, 16, 785. [Google Scholar] [CrossRef]
- Pellet-Many, C.; Frankel, P.; Jia, H.; Zachary, I. Neuropilins: Structure, function and role in disease. Biochem. J. 2008, 411, 211–226. [Google Scholar] [CrossRef]
- Daly, J.L.; Simonetti, B.; Klein, K.; Chen, K.E.; Williamson, M.K.; Anton-Plagaro, C.; Shoemark, D.K.; Simon-Gracia, L.; Bauer, M.; Hollandi, R.; et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. Science 2020, 370, 861–865. [Google Scholar] [CrossRef] [PubMed]
- Hou, D.; Cao, W.; Kim, S.; Cui, X.; Ziarnik, M.; Im, W.; Zhang, X.F. Biophysical investigation of interactions between SARS-CoV-2 spike protein and neuropilin-1. Protein Sci. A Publ. Protein Soc. 2023, 32, e4773. [Google Scholar] [CrossRef] [PubMed]
- Kong, W.; Montano, M.; Corley, M.J.; Helmy, E.; Kobayashi, H.; Kinisu, M.; Suryawanshi, R.; Luo, X.; Royer, L.A.; Roan, N.R.; et al. Neuropilin-1 mediates SARS-CoV-2 infection of astrocytes in brain organoids, inducing inflammation leading to dysfunction and death of neurons. mBio 2022, 13, e0230822. [Google Scholar] [CrossRef]
- Nguyen, H.L.; Hieu, H.K.; Nguyen, T.Q.; Nhung, N.T.A.; Li, M.S. Neuropilin-1 protein may serve as a receptor for SARS-CoV-2 infection: Evidence from molecular dynamics simulations. J. Phys. Chem. B 2024, 128, 7141–7147. [Google Scholar] [CrossRef]
- Morgan, R.L.; Behbahani-Nejad, N.; Endres, J.; Amin, M.A.; Lepore, N.J.; Du, Y.; Urquhart, A.; Chung, K.C.; Fox, D.A. Localization, shedding, regulation and function of aminopeptidase N/CD13 on fibroblast like synoviocytes. PLoS ONE 2016, 11, e0162008. [Google Scholar] [CrossRef]
- Delmas, B.; Gelfi, J.; Sjostrom, H.; Noren, O.; Laude, H. Further characterization of aminopeptidase-N as a receptor for coronaviruses. Adv. Exp. Med. Biol. 1993, 342, 293–298. [Google Scholar]
- Tresnan, D.B.; Holmes, K.V. Feline aminopeptidase N is a receptor for all group I coronaviruses. Coronaviruses Arter. 1998, 440, 69–75. [Google Scholar]
- Chen, L.; Lin, Y.L.; Peng, G.; Li, F. Structural basis for multifunctional roles of mammalian aminopeptidase n. Proc. Natl. Acad. Sci. USA 2012, 109, 17966–17971. [Google Scholar] [CrossRef]
- Qi, F.; Qian, S.; Zhang, S.; Zhang, Z. Single cell RNA sequencing of 13 human tissues identify cell types and receptors of human coronaviruses. Biochem. Biophys. Res. Commun. 2020, 526, 135–140. [Google Scholar] [CrossRef]
- Devarakonda, C.K.V.; Meredith, E.; Ghosh, M.; Shapiro, L.H. Coronavirus receptors as immune modulators. J. Immunol. 2021, 206, 923–929. [Google Scholar] [CrossRef]
- Alves, M.; Mahnke, L.C.; Macedo, T.C.; Silva, T.; Carvalho Junior, L.B. The enzymes in COVID-19: A review. Biochimie 2022, 197, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Gopal, U.; Pizzo, S.V. Cell surface GRP78 signaling: An emerging role as a transcriptional modulator in cancer. J. Cell. Physiol. 2021, 236, 2352–2363. [Google Scholar] [CrossRef]
- Lee, A.S. The ER chaperone and signaling regulator GRP78/BiP as a monitor of endoplasmic reticulum stress. Methods 2005, 35, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Toyoda, S.; Fukuhara, A.; Shimomura, I. GRP78, a novel host factor for SARS-CoV-2: The emerging roles in COVID-19 related to metabolic risk factors. Biomedicines 2022, 10, 1995. [Google Scholar] [CrossRef] [PubMed]
- Carlos, A.J.; Ha, D.P.; Yeh, D.W.; Van Krieken, R.; Tseng, C.C.; Zhang, P.; Gill, P.; Machida, K.; Lee, A.S. The chaperone GRP78 is a host auxiliary factor for SARS-CoV-2 and GRP78 depleting antibody blocks viral entry and infection. J. Biol. Chem. 2021, 296, 100759. [Google Scholar] [CrossRef]
- Han, B.; Lv, Y.; Moser, D.; Zhou, X.; Woehrle, T.; Han, L.; Osterman, A.; Rudelius, M.; Chouker, A.; Lei, P. ACE2-independent SARS-CoV-2 virus entry through cell surface GRP78 on monocytes—Evidence from a translational clinical and experimental approach. EBioMedicine 2023, 98, 104869. [Google Scholar] [CrossRef]
- Chen, Z.; Chen, L.; Qiao, S.W.; Nagaishi, T.; Blumberg, R.S. Carcinoembryonic antigen-related cell adhesion molecule 1 inhibits proximal TCR signaling by targeting ZAP-70. J. Immunol. 2008, 180, 6085–6093. [Google Scholar] [CrossRef]
- Dveksler, G.S.; Pensiero, M.N.; Cardellichio, C.B.; Williams, R.K.; Jiang, G.S.; Holmes, K.V.; Dieffenbach, C.W. Cloning of the mouse hepatitis virus (MHV) receptor: Expression in human and hamster cell lines confers susceptibility to MHV. J. Virol. 1991, 65, 6881–6891. [Google Scholar] [CrossRef]
- Peng, G.; Sun, D.; Rajashankar, K.R.; Qian, Z.; Holmes, K.V.; Li, F. Crystal structure of mouse coronavirus receptor-binding domain complexed with its murine receptor. Proc. Natl. Acad. Sci. USA 2011, 108, 10696–10701. [Google Scholar] [CrossRef]
- Saheb Sharif-Askari, N.; Saheb Sharif-Askari, F.; Mdkhana, B.; Al Heialy, S.; Alsafar, H.S.; Hamoudi, R.; Hamid, Q.; Halwani, R. Enhanced expression of immune checkpoint receptors during SARS-CoV-2 viral infection. Mol. Ther. Methods Clin. Dev. 2021, 20, 109–121. [Google Scholar] [CrossRef]
- Elgundi, Z.; Papanicolaou, M.; Major, G.; Cox, T.R.; Melrose, J.; Whitelock, J.M.; Farrugia, B.L. Cancer metastasis: The role of the extracellular matrix and the heparan sulfate proteoglycan perlecan. Front. Oncol. 2019, 9, 1482. [Google Scholar] [CrossRef]
- Zhang, Q.; Pavlinov, I.; Ye, Y.; Zheng, W. Therapeutic development targeting host heparan sulfate proteoglycan in SARS-CoV-2 infection. Front. Med. 2024, 11, 1364657. [Google Scholar]
- Yue, J.; Jin, W.; Yang, H.; Faulkner, J.; Song, X.; Qiu, H.; Teng, M.; Azadi, P.; Zhang, F.; Linhardt, R.J.; et al. Heparan sulfate facilitates spike protein-mediated SARS-CoV-2 host cell invasion and contributes to increased infection of SARS-CoV-2 G614 mutant and in lung cancer. Front. Mol. Biosci. 2021, 8, 649575. [Google Scholar] [CrossRef]
- Kim, S.Y.; Jin, W.; Sood, A.; Montgomery, D.W.; Grant, O.C.; Fuster, M.M.; Fu, L.; Dordick, J.S.; Woods, R.J.; Zhang, F.; et al. Characterization of heparin and severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) spike glycoprotein binding interactions. Antivir. Res. 2020, 181, 104873. [Google Scholar] [CrossRef] [PubMed]
- Connell, B.J.; Lortat-Jacob, H. Human immunodeficiency virus and heparan sulfate: From attachment to entry inhibition. Front. Immunol. 2013, 4, 385. [Google Scholar] [CrossRef]
- Fenizia, C.; Galbiati, S.; Vanetti, C.; Vago, R.; Clerici, M.; Tacchetti, C.; Daniele, T. SARS-CoV-2 entry: At the crossroads of CD147 and ACE2. Cells 2021, 10, 1434. [Google Scholar] [CrossRef]
- Shilts, J.; Crozier, T.W.M.; Greenwood, E.J.D.; Lehner, P.J.; Wright, G.J. No evidence for basigin/CD147 as a direct SARS-CoV-2 spike binding receptor. Sci. Rep. 2021, 11, 413. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.Q.; Wang, K.; Wang, X.Y.; Cui, H.Y.; Zhao, Y.; Zhu, P.; Chen, Z.N. SARS-CoV-2 pseudovirus enters the host cells through spike protein-CD147 in an Arf6-dependent manner. Emerg. Microbes Infect. 2022, 11, 1135–1144. [Google Scholar] [CrossRef]
- Geijtenbeek, T.B.H.; van Kooyk, Y. Pathogens target DC-SIGN to influence their fate—DC-SIGN functions as a pathogen receptor with broad specificity. APMIS 2003, 111, 698–714. [Google Scholar] [CrossRef]
- Jeffers, S.A.; Tusell, S.M.; Gillim-Ross, L.; Hemmila, E.M.; Achenbach, J.E.; Babcock, G.J.; Thomas, W.D.; Thackray, L.B.; Young, M.D.; Mason, R.J.; et al. CD209L (L-SIGN) is a receptor for severe acute respiratory syndrome coronavirus. Proc. Natl. Acad. Sci. USA 2004, 101, 15748–15753. [Google Scholar] [CrossRef]
- Choudhury, A.; Mukherjee, S. In silico studies on the comparative characterization of the interactions of SARS-CoV-2 spike glycoprotein with ace-2 receptor homologs and human tlrs. J. Med. Virol. 2020, 92, 2105–2113. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Liu, X.; Ou, H.; Li, X.; Liu, R.; Lv, X.; Xiao, S.; Hu, M.; Liang, T.; Chen, T.; et al. The histamine receptor H1 acts as an alternative receptor for SARS-CoV-2. mBio 2024, 15, e0108824. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. Drug Approvals and Databases. Available online: https://www.fda.gov/drugs/development-approval-process-drugs/drug-approvals-and-databases (accessed on 20 February 2025).
- Mak, T.W.; Saunders, M.E. The immune response basic and clinical principles preface. In Immune Response: Basic and Clinical Principles; Academic Press: New York, NY, USA, 2006; Volume VII. [Google Scholar]
- Chaudhary, N.; Gomez, G.A.; Howes, M.T.; Lo, H.P.; McMahon, K.A.; Rae, J.A.; Schieber, N.L.; Hill, M.M.; Gaus, K.; Yap, A.S.; et al. Endocytic crosstalk: Cavins, caveolins, and caveolae regulate clathrin-independent endocytosis. PLoS Biol. 2014, 12, e1001832. [Google Scholar] [CrossRef]
- Smith, J.L.; Campos, S.K.; Wandinger-Ness, A.; Ozbun, M.A. Caveolin-1-dependent infectious entry of human papillomavirus Type 31 in human keratinocytes proceeds to the endosomal pathway for pH-dependent uncoating. J. Virol. 2008, 82, 9505–9512. [Google Scholar] [CrossRef]
- Norkin, L.C.; Anderson, H.A.; Wolfrom, S.A.; Oppenheim, A. Caveolar endocytosis of simian virus 40 is followed by brefeldin A-sensitive transport to the endoplasmic reticulum, where the virus disassembles. J. Virol. 2002, 76, 5156–5166. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.T.; Kiessling, V.; Simmons, J.A.; White, J.M.; Tamm, L.K. HIV gp41-mediated membrane fusion occurs at edges of cholesterol-rich lipid domains. Nat. Chem. Biol. 2015, 11, 424–431. [Google Scholar] [CrossRef]
- Li, Y.J.; Chen, C.Y.; Yang, J.H.; Chiu, Y.F. Modulating cholesterol-rich lipid rafts to disrupt influenza a virus infection. Front. Immunol. 2022, 13, 982264. [Google Scholar] [CrossRef]
- Kaksonen, M.; Roux, A. Mechanisms of clathrin-mediated endocytosis. Nat. reviews. Mol. Cell Biol. 2018, 19, 313–326. [Google Scholar] [CrossRef]
- van der Schaar, H.M.; Rust, M.J.; Chen, C.; van der Ende-Metselaar, H.; Wilschut, J.; Zhuang, X.; Smit, J.M. Dissecting the cell entry pathway of dengue virus by single-particle tracking in living cells. PLoS Pathog. 2008, 4, e1000244. [Google Scholar] [CrossRef]
- Meier, O.; Greber, U.F. Adenovirus endocytosis. J. Gene Med. 2004, 6 (Suppl. S1), S152–S163. [Google Scholar] [CrossRef]
- Bayati, A.; Kumar, R.; Francis, V.; McPherson, P.S. SARS-CoV-2 infects cells after viral entry via clathrin-mediated endocytosis. J. Biol. Chem. 2021, 296, 100306. [Google Scholar] [CrossRef] [PubMed]
- Gorshkov, K.; Susumu, K.; Chen, J.; Xu, M.; Pradhan, M.; Zhu, W.; Hu, X.; Breger, J.C.; Wolak, M.; Oh, E. Quantum dot-conjugated SARS-CoV-2 spike pseudo-virions enable tracking of angiotensin converting enzyme 2 binding and endocytosis. ACS Nano 2020, 14, 12234–12247. [Google Scholar] [CrossRef]
- Ponka, P.; Lok, C.N. The transferrin receptor: Role in health and disease. Int. J. Biochem. Cell Biol. 1999, 31, 1111–1137. [Google Scholar] [CrossRef] [PubMed]
- Mazel-Sanchez, B.; Niu, C.; Williams, N.; Bachmann, M.; Choltus, H.; Silva, F.; Serre-Beinier, V.; Karenovics, W.; Iwaszkiewicz, J.; Zoete, V.; et al. Influenza a virus exploits transferrin receptor recycling to enter host cells. Proc. Natl. Acad. Sci. USA 2023, 120, e2214936120. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.S.L.; Murphy, W.J.; Wang, D.; O’Brien, S.J.; Parrish, C.R. Canine and feline parvoviruses can use human or feline transferrin receptors to bind, enter, and infect cells. J. Virol. 2001, 75, 3896–3902. [Google Scholar] [CrossRef]
- Liao, Z.; Wang, C.; Tang, X.; Yang, M.; Duan, Z.; Liu, L.; Lu, S.; Ma, L.; Cheng, R.; Wang, G.; et al. Human transferrin receptor can mediate SARS-CoV-2 infection. Proc. Natl. Acad. Sci. USA 2024, 121, e2317026121. [Google Scholar] [CrossRef]
- Zhang, W.; Shi, K.; Geng, Q.; Ye, G.; Aihara, H.; Li, F. Structural basis for mouse receptor recognition by SARS-CoV-2 omicron variant. Proc. Natl. Acad. Sci. USA 2022, 119, e2206509119. [Google Scholar] [CrossRef]
- Wu, Q.; Wu, H.; Hu, Y.; Zheng, X.; Chang, F.; Liu, Y.; Pan, Z.; Wang, Q.; Tang, F.; Qian, J.; et al. Immune evasion of omicron variants JN.1, KP.2, and KP.3 to the polyclonal and monoclonal antibodies from COVID-19 convalescents and vaccine recipients. Antivir. Res. 2025, 235, 106092. [Google Scholar] [CrossRef]
- Zheng, B.; Xiao, Y.; Tong, B.; Mao, Y.; Ge, R.; Tian, F.; Dong, X.; Zheng, P. S373P mutation stabilizes the receptor-binding domain of the spike protein in omicron and promotes binding. JACS Au 2023, 3, 1902–1910. [Google Scholar] [CrossRef]
- Yin, W.; Xu, Y.; Xu, P.; Cao, X.; Wu, C.; Gu, C.; He, X.; Wang, X.; Huang, S.; Yuan, Q.; et al. Structures of the omicron spike trimer with ACE2 and an anti-omicron antibody. Science 2022, 375, 1048–1053. [Google Scholar] [CrossRef]
- Geng, Q.; Shi, K.; Ye, G.; Zhang, W.; Aihara, H.; Li, F. Structural basis for human receptor recognition by SARS-CoV-2 omicron variant ba.1. J. Virol. 2022, 96, e0024922. [Google Scholar] [CrossRef] [PubMed]
- Wickenhagen, A.; Flagg, M.; Port, J.R.; Yinda, C.K.; Goldin, K.; Gallogly, S.; Schulz, J.E.; Lutterman, T.; Williamson, B.N.; Kaiser, F.; et al. Evolution of omicron lineage towards increased fitness in the upper respiratory tract in the absence of severe lung pathology. Nat. Commun. 2025, 16, 594. [Google Scholar] [CrossRef]
- Hoffmann, M.; Hofmann-Winkler, H.; Kruger, N.; Kempf, A.; Nehlmeier, I.; Graichen, L.; Arora, P.; Sidarovich, A.; Moldenhauer, A.S.; Winkler, M.S.; et al. SARS-CoV-2 variant B.1.617 is resistant to bamlanivimab and evades antibodies induced by infection and vaccination. Cell Rep. 2021, 36, 109415. [Google Scholar] [CrossRef] [PubMed]
- Willett, B.J.; Grove, J.; MacLean, O.A.; Wilkie, C.; De Lorenzo, G.; Furnon, W.; Cantoni, D.; Scott, S.; Logan, N.; Ashraf, S.; et al. SARS-CoV-2 omicron is an immune escape variant with an altered cell entry pathway. Nat. Microbiol. 2022, 7, 1161–1179. [Google Scholar] [CrossRef]
- Halfmann, P.J.; Iida, S.; Iwatsuki-Horimoto, K.; Maemura, T.; Kiso, M.; Scheaffer, S.M.; Darling, T.L.; Joshi, A.; Loeber, S.; Singh, G.; et al. SARS-CoV-2 omicron virus causes attenuated disease in mice and hamsters. Nature 2022, 603, 687–692. [Google Scholar] [CrossRef]
- Kaku, Y.; Uriu, K.; Kosugi, Y.; Okumura, K.; Yamasoba, D.; Uwamino, Y.; Kuramochi, J.; Sadamasu, K.; Yoshimura, K.; Asakura, H.; et al. Virological characteristics of the SARS-CoV-2 KP.2 variant. Lancet. Infect. Dis. 2024, 24, e416. [Google Scholar] [CrossRef]
- Gangavarapu, K.; Latif, A.A.; Mullen, J.L.; Alkuzweny, M.; Hufbauer, E.; Tsueng, G.; Haag, E.; Zeller, M.; Aceves, C.M.; Zaiets, K.; et al. Outbreak.Info genomic reports: Scalable and dynamic surveillance of SARS-CoV-2 variants and mutations. Nat. Methods 2023, 20, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Tsueng, G.; Mullen, J.L.; Alkuzweny, M.; Cano, M.; Rush, B.; Haag, E.; Lin, J.; Welzel, D.J.; Zhou, X.; Qian, Z.; et al. Outbreak.Info research library: A standardized, searchable platform to discover and explore COVID-19 resources. Nat. Methods 2023, 20, 536–540. [Google Scholar] [CrossRef]
- Shu, Y.; McCauley, J. Gisaid: Global initiative on sharing all influenza data—From vision to reality. Euro Surveill. 2017, 22, 30494. [Google Scholar] [CrossRef]
- Schulze, H.; Bayer, W. Changes in symptoms experienced by SARS-CoV-2-infected individuals—From the first wave to the Omicron variant. Front. Virol. 2022, 2, 880707. [Google Scholar] [CrossRef]
- Hu, Z.; Huang, X.; Zhang, J.; Fu, S.; Ding, D.; Tao, Z. Differences in clinical characteristics between Delta variant and wild-type SARS-CoV-2 infected patients. Front. Med. 2021, 8, 792135. [Google Scholar]
- Thi Khanh, H.N.; Cornelissen, L.; Castanares-Zapatero, D.; De Pauw, R.; Van Cauteren, D.; Demarest, S.; Drieskens, S.; Devleesschauwer, B.; De Ridder, K.; Charafeddine, R.; et al. Association between SARS-CoV-2 variants and post COVID-19 condition: Findings from a longitudinal cohort study in the belgian adult population. BMC Infect. Dis. 2023, 23, 774. [Google Scholar] [CrossRef] [PubMed]
- von Bartheld, C.S.; Wang, L. Prevalence of olfactory dysfunction with the omicron variant of SARS-CoV-2: A systematic review and meta-analysis. Cells 2023, 12, 430. [Google Scholar] [CrossRef] [PubMed]
- Torabi, S.H.; Riahi, S.M.; Ebrahimzadeh, A.; Salmani, F. Changes in symptoms and characteristics of COVID-19 patients across different variants: Two years study using neural network analysis. BMC Infect. Dis. 2023, 23, 838. [Google Scholar] [CrossRef]
- Fyles, M.; Vihta, K.D.; Sudre, C.H.; Long, H.; Das, R.; Jay, C.; Wingfield, T.; Cumming, F.; Green, W.; Hadjipantelis, P.; et al. Diversity of symptom phenotypes in SARS-CoV-2 community infections observed in multiple large datasets. Sci. Rep. 2023, 13, 21705. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. FDA Approves and Authorizes Updated mRNA COVID-19 Vaccines to Better Protect Against Currently Circulating Variants. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-and-authorizes-updated-mrna-COVID-19-vaccines-better-protect-against-currently (accessed on 22 August 2024).
- Cox, M.; Peacock, T.P.; Harvey, W.T.; Hughes, J.; Wright, D.W.; Consortium, C.-G.U.; Willett, B.J.; Thomson, E.; Gupta, R.K.; Peacock, S.J.; et al. SARS-CoV-2 variant evasion of monoclonal antibodies based on in vitro studies. Nat. Rev. Microbiol. 2023, 21, 112–124. [Google Scholar] [CrossRef]
- Gidari, A.; Sabbatini, S.; Bastianelli, S.; Pierucci, S.; Busti, C.; Svizzeretto, E.; Tommasi, A.; Pallotto, C.; Schiaroli, E.; Francisci, D. Tixagevimab/cilgavimab: Still a valid prophylaxis against COVID-19 new variants? Viruses 2024, 16, 354. [Google Scholar] [CrossRef]
- Focosi, D.; Casadevall, A. A critical analysis of the use of cilgavimab plus tixagevimab monoclonal antibody cocktail (evusheld) for COVID-19 prophylaxis and treatment. Viruses 2022, 14, 1999. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. FDA Announces Evusheld Is Not Currently Authorized for Emergency Use in the U.S. Available online: https://www.fda.gov/drugs/drug-safety-and-availability/fda-announces-evusheld-not-currently-authorized-emergency-use-us (accessed on 16 March 2025).
- U.S. Food and Drug Administration. Fact Sheet for Healthcare Providers: Emergency Use Authorization of Pemgarda (Pemivibart). Invivyd, Inc. Available online: https://www.fda.gov/media/177067/download (accessed on 16 March 2025).
- Voss, W.N.; Mallory, M.A.; Byrne, P.O.; Marchioni, J.M.; Knudson, S.A.; Powers, J.M.; Leist, S.R.; Dadonaite, B.; Townsend, D.R.; Kain, J.; et al. Hybrid immunity to SARS-CoV-2 arises from serological recall of IgG antibodies distinctly imprinted by infection or vaccination. Cell Rep. Med. 2024, 5, 101668. [Google Scholar] [CrossRef]
- Nagy, B., Jr.; Fejes, Z.; Szentkereszty, Z.; Suto, R.; Varkonyi, I.; Ajzner, E.; Kappelmayer, J.; Papp, Z.; Toth, A.; Fagyas, M. A dramatic rise in serum ACE2 activity in a critically ill COVID-19 patient. Int. J. Infect. Dis. 2021, 103, 412–414. [Google Scholar] [CrossRef]
- Urano, E.; Itoh, Y.; Suzuki, T.; Sasaki, T.; Kishikawa, J.I.; Akamatsu, K.; Higuchi, Y.; Sakai, Y.; Okamura, T.; Mitoma, S.; et al. An inhaled ACE2 decoy confers protection against SARS-CoV-2 infection in preclinical models. Sci. Transl. Med. 2023, 15, eadi2623. [Google Scholar] [CrossRef] [PubMed]
- Havranek, B.; Lindsey, G.W.; Higuchi, Y.; Itoh, Y.; Suzuki, T.; Okamoto, T.; Hoshino, A.; Procko, E.; Islam, S.M. A computationally designed ACE2 decoy has broad efficacy against SARS-CoV-2 omicron variants and related viruses in vitro and in vivo. Commun. Biol. 2023, 6, 513. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Yao, W.; Li, Y.; Ma, D.; Zhang, Z.; Wang, H.; Tang, X.; Wang, Y.; Li, C.; Cheng, D.; et al. Broadly effective ACE2 decoy proteins protect mice from lethal SARS-CoV-2 infection. Microbiol. Spectr. 2023, 11, e0110023. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Virus | Genus | Primary Receptors | Year of Outbreak | Disease Caused |
|---|---|---|---|---|
| HCoV-229E | Alphacoronavirus | APN (Aminopeptidase N) [29] | Identified in 1960s | Common cold, respiratory infections |
| HCoV-OC43 | Betacoronavirus | 9-O-acetylsialic acids as receptor [26] | Identified in 1960s | Common cold, bronchitis, pneumonia |
| SARS-CoV-1 | Betacoronavirus | ACE2 [30] | 2002–2003 | Severe acute respiratory syndrome (SARS) |
| HCoV-NL63 | Alphacoronavirus | ACE2 [27] | 2004 | Common cold, croup, bronchiolitis |
| HCoV-HKU1 | Betacoronavirus | 9-O-acetylsialic acids as receptor [26] | 2005 | Common cold, pneumonia |
| MERS-CoV | Betacoronavirus | DPP4 (CD26) [31] | 2012 | Middle East respiratory syndrome (MERS) |
| SARS-CoV-2 | Betacoronavirus | ACE2 [32] | 2019–2020 | Coronavirus disease of 2019 (COVID-19) |
| Pango Lineage | Variant | Key Mutations in RBD |
|---|---|---|
| B.1.1.7 | Alpha | N501Y |
| B.1.351 | Beta | K417N, E484K, N501Y |
| P.1 | Gamma | K417T, E484K, N501Y |
| B.1.617.2 | Delta | L452R, T478K |
| B.1.427/B.1.429 | Epsilon | L452R |
| P.2 | Zeta | E484K |
| B.1.525 | Eta | E484K |
| P.3 | Theta | E484K, N501Y |
| B.1.526 | Iota | E484K |
| B.1.617.1 | Kappa | L452R, E484Q |
| C.37 | Lambda | L452Q, F490S |
| B.1.621 | Mu | R346K, E484K, N501Y |
| Variant | Lineage | Key Spike Mutations in the RBD | Cumulative Prevalence Worldwide * |
|---|---|---|---|
| KP.2 | Descendant of BA.4 lineage of the Omicron variant | I332V, G339H, R346T/K, K356T, S371F, S373P, S375F, T376A, R403K, D405N, R408S, K417N, N440K, V445H, G446S, N450D, L452W/R, L455S, F456L, N460K, S477N, T478K, N481K, del483/483, E484K, F486P/V, Q493R, Q498R, N501Y, Y505H | 2% as of October 2024 |
| KP.3 | Evolved from the BA.2.12.1 subvariant | I332V, R346K, K356T, S371F/L, S373P, S375F, T376A, G339H, R403K, D405N, R408S, K417N, N440K, V445H, G446S, N450D, L452W/R, E554K, L455S, F456L, N460K, S477N, T478K, N481K, V483del, E484K, F486P/V, Q493E/R, Q498R, N501Y, Y505H | 27% as of December 2024 |
| LB.1 | Descendant of the BA.1 Omicron subvariant | I332V, G339H, R346T, K356T, S371F, S373P, S375F, T376A, R403K, D405N, R408S, K417N, N440K, V445H, G446S, N450D, L452W, L455S, F456L, N460K, S477N, T478K, N481K, del483/483, E484K, F486P, Q498R, N501Y, Y505H | 3% as of December 2024 |
| BA.2.86 | Descendant of BA.2 Omicron variant | G339H, K356T, S371F, S373P, S375F, T376A, R403K, D405N, R408S, K417N, N440K, V445, G446S, N450D, L452W, N460K, S477N, T478K, N481K, V483Δ, E484K, F486P, Q498R, N501Y, Y505H | 27% worldwide as of October 2024 |
| XBB.1.5 | Recombination between BA.2.10.1 and BA.2.75 subvariants | G339H, R346T, L368I, S371F/L, S373P, S375F, S477N, T376A, D405N, R408S, S413R, K417N, N440K, V445P, G446S, L452R, N460K, T478R, E484A, F486P, F490S, S494P, Q498R, N501Y, Y505H | 8% as of October 2024 |
| JN.1 | Subvariant of Omicron variant BA.2.86 | G339D, K356T, S371F, S373P, S375F, T376A, R403K, D405N, R408S, K417N, N440K, G446S, L452W/R, N450D, L455S, N460K, F486S, V445H, Y505H | 27% as of December 2024 |
| XEC | Recombinant variant combining genetic material from the KS.1.1 and KP.3.3 subvariants | I332V, G339H, R346K, K356T, S371F/L, S373P, S375F, T376A, R403K, D405N, R408S, K417N, N440K, V445H, G446S, N450D, L452W/R, L455S, F456L, N460K, S477N, T478K, N481K, del483/483, E484K, F486P/V, Q493E/R, Q498R, N501Y, Y505H | 1% as of October 2024 |
| Common Symptoms | Prominent Symptoms | |
|---|---|---|
| Original Strain (Wuhan) | Fever, cough, fatigue, shortness of breath, muscle aches | Anosmia, ageusia, pneumonia, ARDS |
| Alpha (B.1.1.7) | Fever, headache, vertigo, nasal congestion, throat pain, dyspnea, nausea, diarrhea | Anosmia, ageusia, delerium, depression, pneumonia, ARDS |
| Beta (B.1.351) | Fever, headache, vertigo, nasal congestion, throat pain, dyspnea, nausea, diarrhea | Anosmia, ageusia |
| Delta (B.1.617.2) | Fever, fatigue, muscle ache, diaphoresis, nasal congestion, dyspnea, diarrhea | Headache, throat pain, vomiting, diarrhea, chest pain, pneumonia, ARDS |
| Omicron (B.1.1.529) | Fever, cough, fatigue, nasal congestion, muscle aches, | Throat pain, vomiting, diarrhea, vertigo |
| Omicron Subvariants (BA.4/BA.5/XEC/JN.1) | Rhinorrhea, cough, fatigue, headache, fever, muscle aches | Sore throat, nasal congestion |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahdi, M.; Kiarie, I.W.; Mótyán, J.A.; Hoffka, G.; Al-Muffti, A.S.; Tóth, A.; Tőzsér, J. Receptor Binding for the Entry Mechanisms of SARS-CoV-2: Insights from the Original Strain and Emerging Variants. Viruses 2025, 17, 691. https://doi.org/10.3390/v17050691
Mahdi M, Kiarie IW, Mótyán JA, Hoffka G, Al-Muffti AS, Tóth A, Tőzsér J. Receptor Binding for the Entry Mechanisms of SARS-CoV-2: Insights from the Original Strain and Emerging Variants. Viruses. 2025; 17(5):691. https://doi.org/10.3390/v17050691
Chicago/Turabian StyleMahdi, Mohamed, Irene Wanjiru Kiarie, János András Mótyán, Gyula Hoffka, Aya Shamal Al-Muffti, Attila Tóth, and József Tőzsér. 2025. "Receptor Binding for the Entry Mechanisms of SARS-CoV-2: Insights from the Original Strain and Emerging Variants" Viruses 17, no. 5: 691. https://doi.org/10.3390/v17050691
APA StyleMahdi, M., Kiarie, I. W., Mótyán, J. A., Hoffka, G., Al-Muffti, A. S., Tóth, A., & Tőzsér, J. (2025). Receptor Binding for the Entry Mechanisms of SARS-CoV-2: Insights from the Original Strain and Emerging Variants. Viruses, 17(5), 691. https://doi.org/10.3390/v17050691