
Evolutionary Relationships of Unclassified Coronaviruses in Canadian Bat Species
 , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Sampling
2.3. Bat Tissue Harvesting and RNA Extraction
2.4. Pan-Coronavirus for Screening of Bat Specimens
2.5. Next-Generation Sequencing
2.6. Phylogenetic Tree Construction
2.7. Data Availability
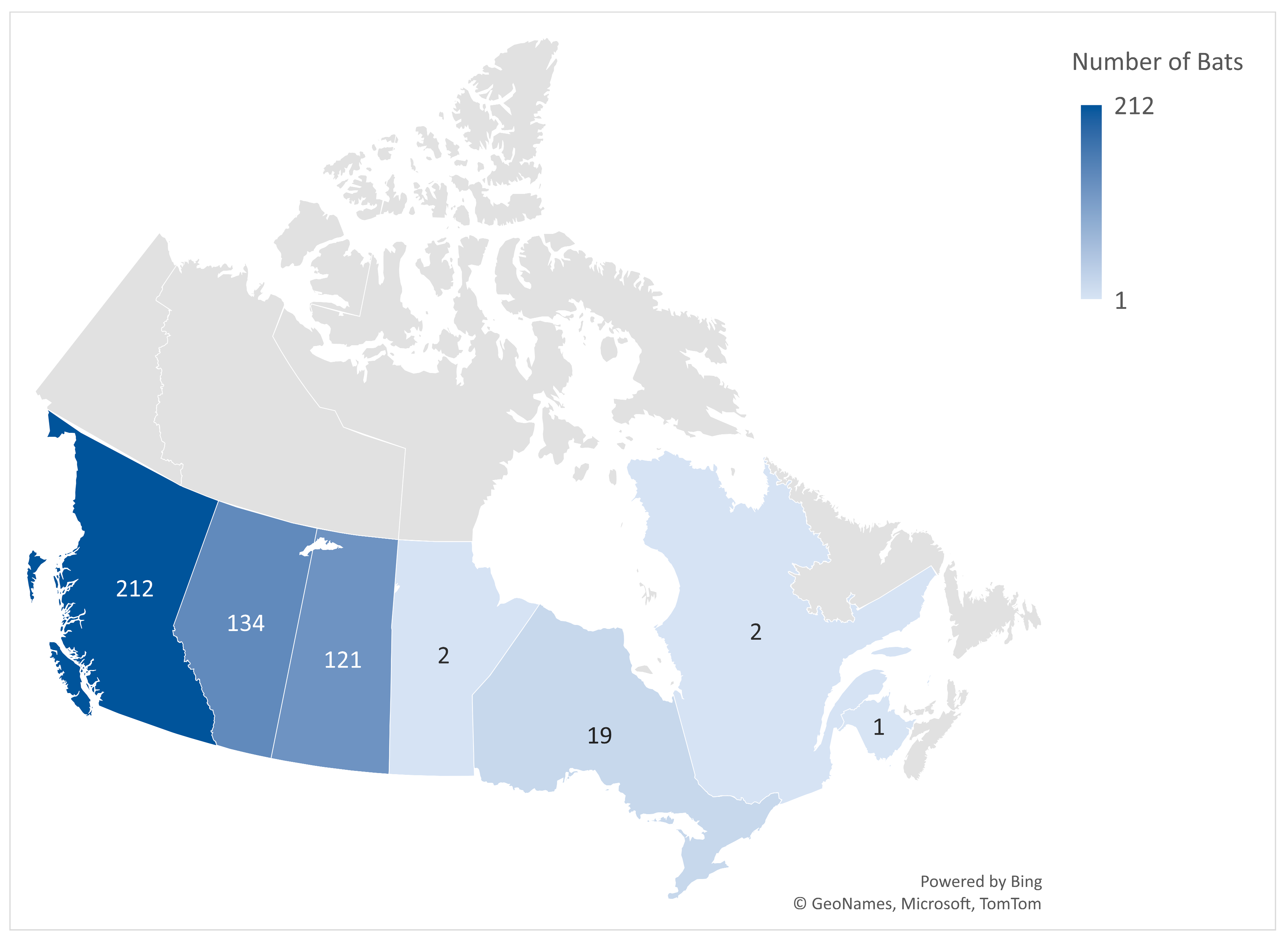
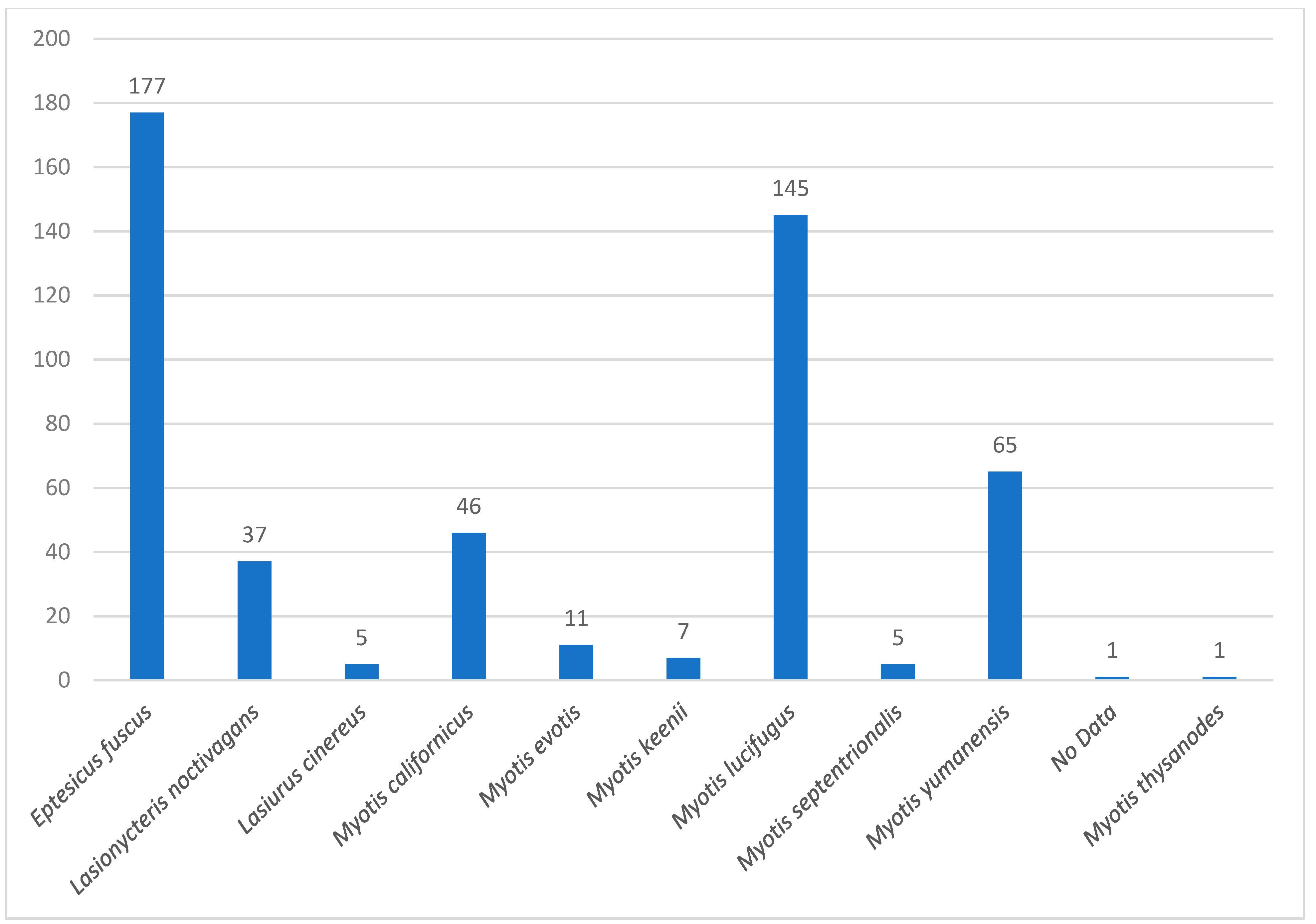
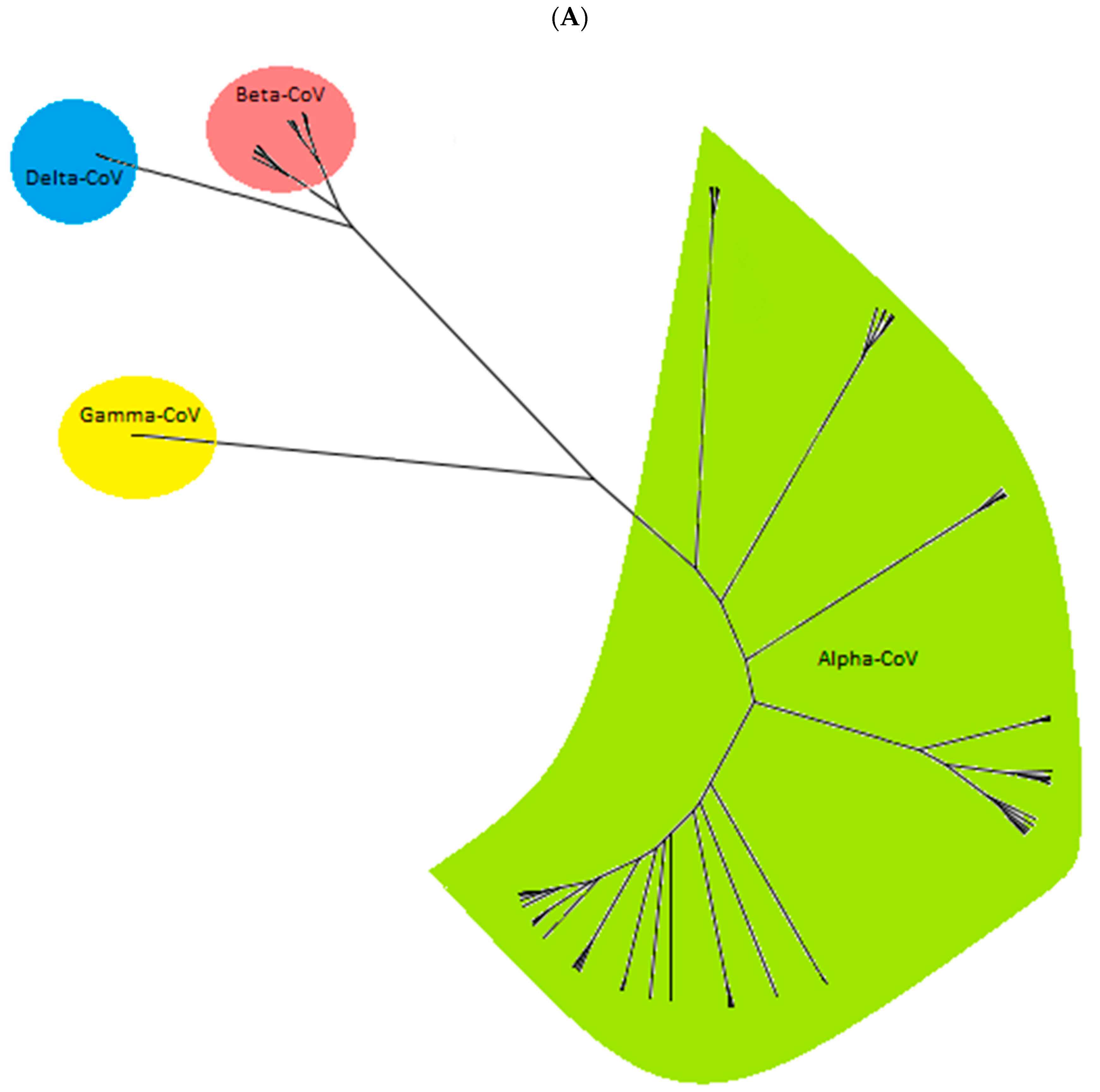
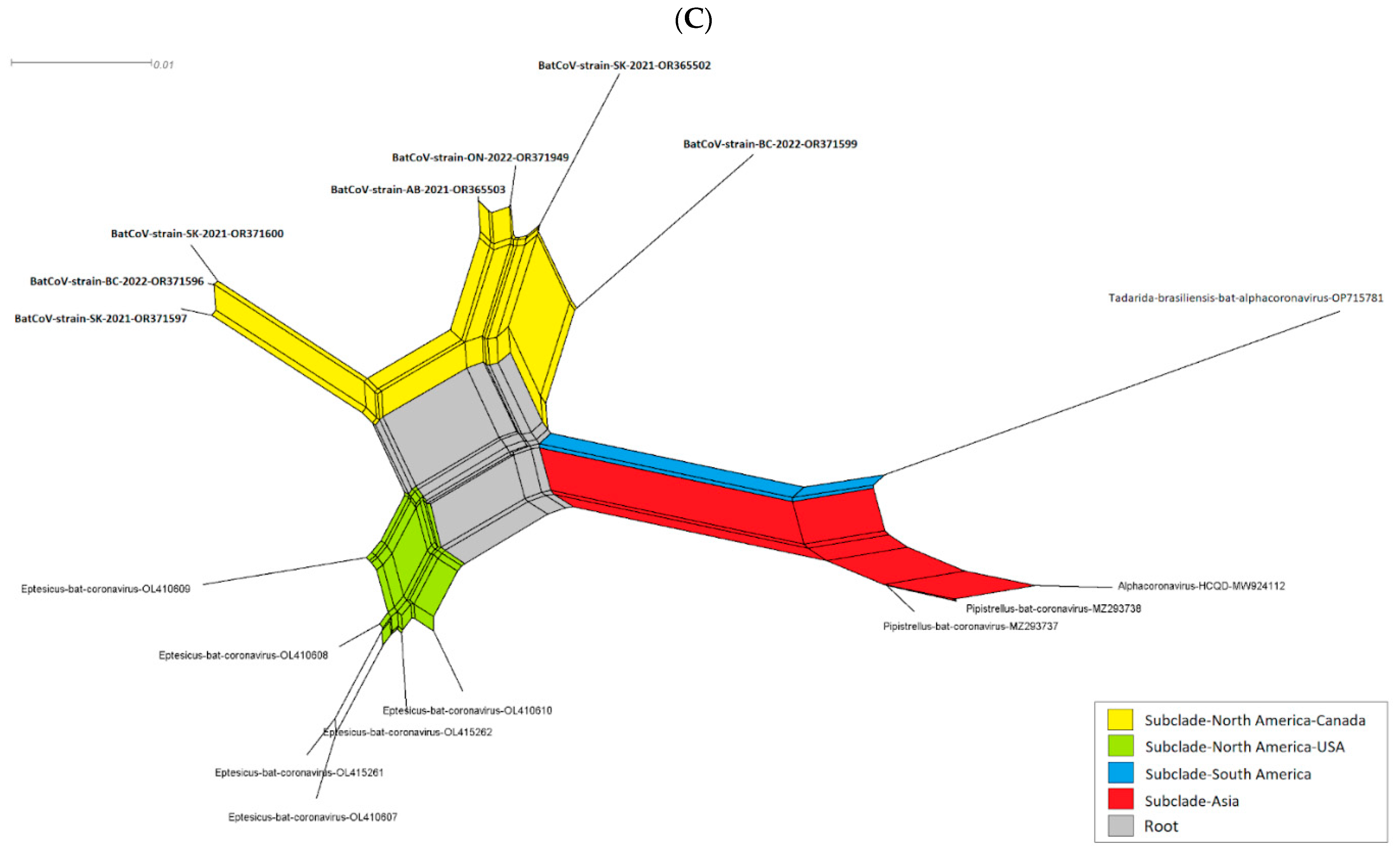
3. Results
3.1. Prevalence of CoVs in Bat Species
3.2. Evolutionary Relationship of Bat CoVs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shahhosseini, N.; Babuadze, G.; Wong, G.; Kobinger, G.P. Mutation signatures and in silico docking of novel SARS-CoV-2 variants of concern. Microorganisms 2021, 9, 926. [Google Scholar] [CrossRef] [PubMed]
- Drosten, C.; Günther, S.; Preiser, W.; Van Der Werf, S.; Brodt, H.-R.; Becker, S.; Rabenau, H.; Panning, M.; Kolesnikova, L.; Fouchier, R.A. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1967–1976. [Google Scholar] [CrossRef] [PubMed]
- Zumla, A.; Hui, D.S.; Perlman, S. Middle East respiratory syndrome. Lancet 2015, 386, 995–1007. [Google Scholar] [CrossRef] [PubMed]
- Memish, Z.A.; Mishra, N.; Olival, K.J.; Fagbo, S.F.; Kapoor, V.; Epstein, J.H.; AlHakeem, R.; Durosinloun, A.; Al Asmari, M.; Islam, A. Middle East respiratory syndrome coronavirus in bats, Saudi Arabia. Emerg. Infect. Dis. 2013, 19, 1819. [Google Scholar] [CrossRef] [PubMed]
- Shahhosseini, N.; Wong, G.; Kobinger, G.P.; Chinikar, S. SARS-CoV-2 spillover transmission due to recombination event. Gene Rep. 2021, 23, 101045. [Google Scholar] [CrossRef]
- Wertheim, J.O.; Chu, D.K.; Peiris, J.S.; Kosakovsky Pond, S.L.; Poon, L.L. A case for the ancient origin of coronaviruses. J. Virol. 2013, 87, 7039–7045. [Google Scholar] [CrossRef]
- Hernández-Aguilar, I.; Lorenzo, C.; Santos-Moreno, A.; Naranjo, E.J.; Navarrete-Gutiérrez, D. Coronaviruses in bats: A review for the Americas. Viruses 2021, 13, 1226. [Google Scholar] [CrossRef]
- Woo, P.C.; Lau, S.K.; Lam, C.S.; Lau, C.C.; Tsang, A.K.; Lau, J.H.; Bai, R.; Teng, J.L.; Tsang, C.C.; Wang, M. Discovery of seven novel Mammalian and avian coronaviruses in the genus deltacoronavirus supports bat coronaviruses as the gene source of alphacoronavirus and betacoronavirus and avian coronaviruses as the gene source of gammacoronavirus and deltacoronavirus. J. Virol. 2012, 86, 3995–4008. [Google Scholar]
- Ma, Y.; Zhang, Y.; Liang, X.; Lou, F.; Oglesbee, M.; Krakowka, S.; Li, J. Origin, evolution, and virulence of porcine deltacoronaviruses in the United States. mBio 2015, 6, e00064. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Q.; Guo, D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020, 92, 418–423. [Google Scholar] [CrossRef]
- Cui, J.; Li, F.; Shi, Z.-L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Grunewald, M.; Perlman, S. Coronaviruses: An updated overview of their replication and pathogenesis. Coronaviruses Methods Protoc. 2020, 2203, 1–29. [Google Scholar] [CrossRef]
- Corman, V.M.; Muth, D.; Niemeyer, D.; Drosten, C. Hosts and sources of endemic human coronaviruses. Adv. Virus Res. 2018, 100, 163–188. [Google Scholar] [PubMed]
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.-L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Forni, D.; Cagliani, R.; Clerici, M.; Sironi, M. Molecular evolution of human coronavirus genomes. Trends Microbiol. 2017, 25, 35–48. [Google Scholar] [CrossRef]
- Wang, Q.; Vlasova, A.N.; Kenney, S.P.; Saif, L.J. Emerging and re-emerging coronaviruses in pigs. Curr. Opin. Virol. 2019, 34, 39–49. [Google Scholar] [CrossRef]
- Yang, Y.-L.; Yu, J.-Q.; Huang, Y.-W. Swine enteric alphacoronavirus (swine acute diarrhea syndrome coronavirus): An update three years after its discovery. Virus Res. 2020, 285, 198024. [Google Scholar] [CrossRef]
- Vlasova, A.N.; Saif, L.J. Bovine coronavirus and the associated diseases. Front. Vet. Sci. 2021, 8, 643220. [Google Scholar] [CrossRef]
- Samad, A.; Naveed, A.; Alam, A.N.; Atique, R.; Muazzam, A.; Anwar, B.; Saeed, H.A.; Zahra, M.; Rana, T.; Hossain, M.J. Brief Overview on Rabies: A Fatal and Preventable Virus. Indones. Health J. 2024, 3, 162–170. [Google Scholar] [CrossRef]
- Nadin-Davis, S.A.; Huang, W.; Armstrong, J.; Casey, G.A.; Bahloul, C.; Tordo, N.; Wandeler, A.I. Antigenic and genetic divergence of rabies viruses from bat species indigenous to Canada. Virus Res. 2001, 74, 139–156. [Google Scholar] [CrossRef]
- Lung, O.; Nadin-Davis, S.; Fisher, M.; Erickson, A.; Knowles, M.K.; Furukawa-Stoffer, T.; Ambagala, A. Microarray for identification of the chiropteran host species of rabies virus in Canada. Microarrays 2013, 2, 153–169. [Google Scholar] [CrossRef] [PubMed]
- Xiu, L.; Binder, R.A.; Alarja, N.A.; Kochek, K.; Coleman, K.K.; Than, S.T.; Bailey, E.S.; Bui, V.N.; Toh, T.-H.; Erdman, D.D. A RT-PCR assay for the detection of coronaviruses from four genera. J. Clin. Virol. 2020, 128, 104391. [Google Scholar] [CrossRef] [PubMed]
- Shahhosseini, N.; Lühken, R.; Jöst, H.; Jansen, S.; Börstler, J.; Rieger, T.; Krüger, A.; Yadouleton, A.; de Mendonça Campos, R.; Cirne-Santos, C.C. Detection and characterization of a novel rhabdovirus in Aedes cantans mosquitoes and evidence for a mosquito-associated new genus in the family Rhabdoviridae. Infect. Genet. Evol. 2017, 55, 260–268. [Google Scholar] [CrossRef] [PubMed]
- Shahhosseini, N.; Chinikar, S.; Nowotny, N.; Fooks, A.R.; Schmidt-Chanasit, J. Genetic analysis of imported dengue virus strains by Iranian travelers. Asian Pac. J. Trop. Dis. 2016, 6, 850–853. [Google Scholar] [CrossRef]
- Schaeffer, R.; Temeeyasen, G.; Hause, B.M. Alphacoronaviruses are common in bats in the upper midwestern United States. Viruses 2022, 14, 184. [Google Scholar] [CrossRef]
- Thakor, J.C.; Dinesh, M.; Manikandan, R.; Bindu, S.; Sahoo, M.; Sahoo, D.; Dhawan, M.; Pandey, M.K.; Tiwari, R.; Emran, T.B. Swine coronaviruses (SCoVs) and their emerging threats to swine population, inter-species transmission, exploring the susceptibility of pigs for SARS-CoV-2 and zoonotic concerns. Vet. Q. 2022, 42, 125–147. [Google Scholar] [CrossRef]
- Zhou, L.; Li, Q.N.; Su, J.N.; Chen, G.H.; Wu, Z.X.; Luo, Y.; Wu, R.T.; Sun, Y.; Lan, T.; Ma, J.Y. The re-emerging of SADS-CoV infection in pig herds in Southern China. Transbound. Emerg. Dis. 2019, 66, 2180–2183. [Google Scholar] [CrossRef]
- Liu, C.; Huang, W.; He, X.; Feng, Z.; Chen, Q. Research Advances on Swine Acute Diarrhea Syndrome Coronavirus. Animals 2024, 14, 448. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Zhu, W.; Yang, J.; Lu, S.; Jin, D.; Wu, S.; Pu, J.; Luo, X.-l.; Liu, L.; Li, Z.; Xu, J. Discovery and evolution of a divergent coronavirus in the plateau pika from China that extends the host range of alphacoronaviruses. Front. Microbiol. 2021, 12, 755599. [Google Scholar] [CrossRef]
- Menachery, V.D.; Yount, B.L.; Debbink, K.; Agnihothram, S.; Gralinski, L.E.; Plante, J.A.; Graham, R.L.; Scobey, T.; Ge, X.-Y.; Donaldson, E.F. A SARS-like cluster of circulating bat coronaviruses shows potential for human emergence. Nat. Med. 2015, 21, 1508–1513. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Aravena, M.; McKee, C.; Gamble, A.; Lunn, T.; Morris, A.; Snedden, C.E.; Yinda, C.K.; Port, J.R.; Buchholz, D.W.; Yeo, Y.Y. Ecology, evolution and spillover of coronaviruses from bats. Nat. Rev. Microbiol. 2022, 20, 299–314. [Google Scholar] [CrossRef] [PubMed]
- Decaro, N.; Lorusso, A. Novel human coronavirus (SARS-CoV-2): A lesson from animal coronaviruses. Vet. Microbiol. 2020, 244, 108693. [Google Scholar] [CrossRef] [PubMed]
- Do, H.-Q.; Nguyen, V.-G.; Chung, C.-U.; Jeon, Y.-S.; Shin, S.; Jang, K.-C.; Pham, L.B.H.; Kong, A.; Kim, C.-U.; Park, Y.-H. Genomic characterization of a novel alphacoronavirus isolated from bats, Korea, 2020. Viruses 2021, 13, 2041. [Google Scholar] [CrossRef] [PubMed]
- Letko, M.; Seifert, S.N.; Olival, K.J.; Plowright, R.K.; Munster, V.J. Bat-borne virus diversity, spillover and emergence. Nat. Rev. Microbiol. 2020, 18, 461–471. [Google Scholar] [CrossRef]
- Shahhosseini, N.J.G.R. Characterization of mutations modulating enhanced transmissibility of SARS-CoV-2 B. 1.617+(Delta) variant using In Silico tools. Gene Rep. 2022, 27, 101636. [Google Scholar] [CrossRef]
- Misra, V.; Dumonceaux, T.; Dubois, J.; Willis, C.; Nadin-Davis, S.; Severini, A.; Wandeler, A.; Lindsay, R.; Artsob, H. Detection of polyoma and corona viruses in bats of Canada. J. Gen. Virol. 2009, 90, 2015–2022. [Google Scholar] [CrossRef]
- Subudhi, S.; Rapin, N.; Bollinger, T.K.; Hill, J.E.; Donaldson, M.E.; Davy, C.M.; Warnecke, L.; Turner, J.M.; Kyle, C.J.; Willis, C.K. A persistently infecting coronavirus in hibernating Myotis lucifugus, the North American little brown bat. J. Gen. Virol. 2017, 98, 2297–2309. [Google Scholar] [CrossRef]
- Goldstein, S.A.; Brown, J.; Pedersen, B.S.; Quinlan, A.R.; Elde, N.C. Extensive recombination-driven coronavirus diversification expands the pool of potential pandemic pathogens. Genome Biol. Evol. 2022, 14, evac161. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bat Species | Date of Collection | City/Town of Collection | Province Location | Accession Number |
|---|---|---|---|---|
| Eptesicus fuscus | 04/08/2021 | Saskatoon | Saskatchewan | OR365502 |
| Eptesicus fuscus | 13/10/2021 | Moose Jaw | Saskatchewan | OR371600/PP621596 * |
| Eptesicus fuscus | 08/06/2021 | Regina | Saskatchewan | OR371597 |
| Eptesicus fuscus | 21/01/2022 | Stratford | Ontario | OR371949 |
| Eptesicus fuscus | 26/05/2022 | Summerland | British Columbia | OR371596 |
| Myotis lucifugus | 09/06/2021 | Sylvan lake | Alberta | OR365503 |
| Myotis californicus | 31/05/2022 | Pender Island | British Columbia | OR371599 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simon, A.Y.; Badmalia, M.D.; Paquette, S.-J.; Manalaysay, J.; Czekay, D.; Kandel, B.S.; Sultana, A.; Lung, O.; Babuadze, G.G.; Shahhosseini, N. Evolutionary Relationships of Unclassified Coronaviruses in Canadian Bat Species. Viruses 2024, 16, 1878. https://doi.org/10.3390/v16121878
Simon AY, Badmalia MD, Paquette S-J, Manalaysay J, Czekay D, Kandel BS, Sultana A, Lung O, Babuadze GG, Shahhosseini N. Evolutionary Relationships of Unclassified Coronaviruses in Canadian Bat Species. Viruses. 2024; 16(12):1878. https://doi.org/10.3390/v16121878
Chicago/Turabian StyleSimon, Ayo Yila, Maulik D. Badmalia, Sarah-Jo Paquette, Jessica Manalaysay, Dominic Czekay, Bishnu Sharma Kandel, Asma Sultana, Oliver Lung, George Giorgi Babuadze, and Nariman Shahhosseini. 2024. "Evolutionary Relationships of Unclassified Coronaviruses in Canadian Bat Species" Viruses 16, no. 12: 1878. https://doi.org/10.3390/v16121878
APA StyleSimon, A. Y., Badmalia, M. D., Paquette, S.-J., Manalaysay, J., Czekay, D., Kandel, B. S., Sultana, A., Lung, O., Babuadze, G. G., & Shahhosseini, N. (2024). Evolutionary Relationships of Unclassified Coronaviruses in Canadian Bat Species. Viruses, 16(12), 1878. https://doi.org/10.3390/v16121878