
Genomic Epidemiology of the SARS-CoV-2 Epidemic in Cyprus from November 2020 to October 2021: The Passage of Waves of Alpha and Delta Variants of Concern

 , , , ,
, , , ,  , , add
Show full author list
, , add
Show full author list
Abstract
1. Introduction
2. Materials and Methods
2.1. Sequences Used in the Study
2.2. Sample Collection, RNA Extraction and SARS-CoV-2 Real-Time RT–PCR
2.3. Next-Generation Sequencing (NGS)
2.3.1. NGS by Eurofins Genomics Sequencing Europe
2.3.2. NGS by NIPD Genetics and S.C.I.N.A. Bioanalysis Sciomedical Centre Ltd.
2.4. Bioinformatic Analysis
2.4.1. Lineage Classification
2.4.2. Mutation Calling
2.4.3. Dataset Compilation
2.4.4. Time-Scaled Phylogeographic Inference
2.4.5. Time-Scaled Phylogenetic Inference
2.5. Calculations and Figure Information
3. Results
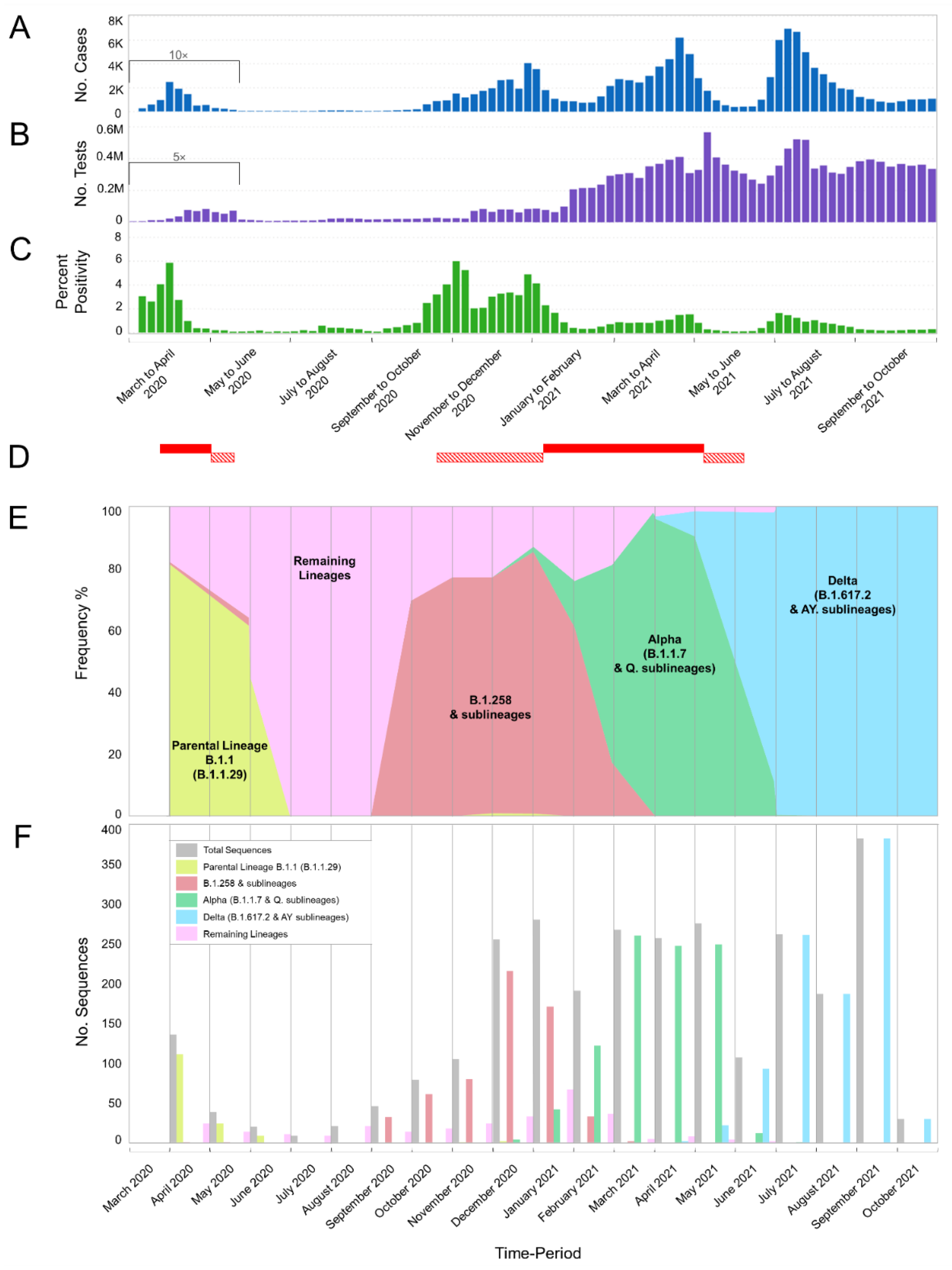
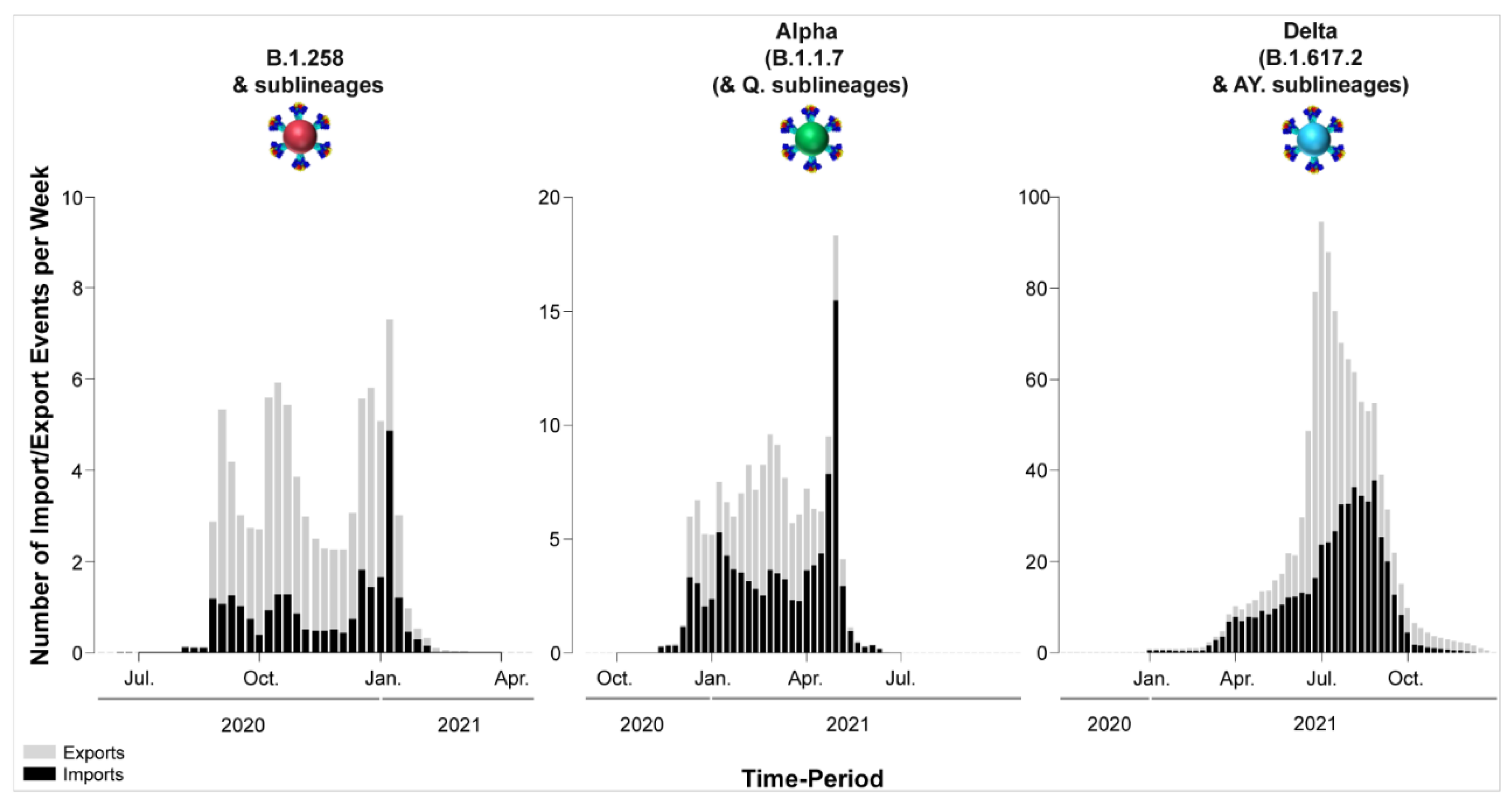
3.1. The Appearance of Lineages and the Waves of SARS-CoV-2 Infection in Cyprus

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time Period | Nov 2020–Jan 2021 | Feb–Apr 2021 | May–July 2021 | Aug–Oct 2021 | Total |
|---|---|---|---|---|---|
| Lineage | Νumber of Sequences per Lineage (%) | Νumber of Sequences per Lineage (%) | Νumber of Sequences per Lineage (%) | Νumber of Sequences per Lineage (%) | Νumber of Sequences per Lineage (%) |
| AD.2 | 1 (0.25) | - | - | - | 1 (0.04) |
| AY.1 | - | - | - | 1 (0.17) | 1 (0.04) |
| AY.4 | - | - | 5 (0.78) | 71 (11.85) | 76 (3.23) |
| AY.4.2 | - | - | - | 2 (0.33) | 2 (0.09) |
| AY.4.3 | - | - | - | 2 (0.33) | 2 (0.09) |
| AY.4.4 | - | - | - | 5 (0.83) | 5 (0.21) |
| AY.4.5 | - | - | - | 1 (0.17) | 1 (0.04) |
| AY.5 | - | - | - | 1 (0.17) | 1 (0.04) |
| AY.6 | - | - | - | 2 (0.33) | 2 (0.09) |
| AY.7 | - | - | 2 (0.31) | - | 2 (0.09) |
| AY.7.2 | - | - | - | 2 (0.33) | 2 (0.09) |
| AY.9 | - | - | 1 (0.16) | 8 (1.34) | 9 (0.38) |
| AY.9.2 | - | - | 1 (0.16) | 5 (0.83) | 6 (0.26) |
| AY.13 | - | - | - | 1 (0.17) | 1 (0.04) |
| AY.23 | - | - | 1 (0.16) | - | 1 (0.04) |
| AY.25.1 | - | - | - | 2 (0.33) | 2 (0.09) |
| AY.34.1 | - | - | - | 2 (0.33) | 2 (0.09) |
| AY.36 | - | - | - | 1 (0.17) | 1 (0.04) |
| AY.42 | - | - | - | 1 (0.17) | 1 (0.04) |
| AY.43 | - | - | 1 (0.16) | 46 (7.68) | 47 (2.00) |
| AY.44 | - | - | - | 2 (0.33) | 2 (0.09) |
| AY.46 | - | - | 1 (0.16) | - | 1 (0.04) |
| AY.46.6 | - | - | - | 3 (0.50) | 3 (0.13) |
| AY.60 | - | - | 106 (16.46) | 62 (10.35) | 168 (7.14) |
| AY.92 | - | - | - | 1 (0.17) | 1 (0.04) |
| AY.98 | - | - | 2 (0.31) | 1 (0.17) | 3 (0.13) |
| AY.98.1 | - | - | - | 9 (1.50) | 9 (0.38) |
| AY.103 | - | - | - | 1 (0.17) | 1 (0.04) |
| AY.116 | - | - | - | 1 (0.17) | 1 (0.04) |
| AY.120 | - | - | - | 3 (0.50) | 3 (0.13) |
| AY.122 | - | - | 246 (38.20) | 305 (50.92) | 551 (23.43) |
| AY.125 | - | - | - | 3 (0.50) | 3 (0.13) |
| AY.126 | - | - | - | 5 (0.83) | 5 (0.21) |
| AY.128 | - | - | - | 5 (0.83) | 5 (0.21) |
| B.1 | 5 (1.27) | - | - | - | 5 (0.21) |
| B.1.1.7 | 36 (9.14) | 627 (87.69) | 261 (40.53) | - | 924 (39.29) |
| B.1.1.25 | 8 (2.03) | 2 (0.28) | - | - | 10 (0.43) |
| B.1.1.219 | 1 (0.25) | - | - | - | 1 (0.04) |
| B.1.1.312 | 3 (0.76) | - | - | - | 3 (0.13) |
| B.1.1.317 | 1 (0.25) | 1 (0.14) | - | - | 2 (0.09) |
| B.1.1.487 | 2 (0.51) | - | - | - | 2 (0.09) |
| B.1.1.523 | - | 1 (0.14) | 2 (0.31) | - | 3 (0.13) |
| B.1.36.31 | 1 (0.25) | - | - | - | 1 (0.04) |
| B.1.160 | 14 (3.55) | 1 (0.14) | - | - | 15 (0.64) |
| B.1.177 | 41 (10.41) | 32 (4.48) | - | - | 73 (3.10) |
| B.1.177.15 | 1 (0.25) | - | - | - | 1 (0.04) |
| B.1.177.21 | 2 (0.51) | 4 (0.56) | - | - | 6 (0.26) |
| B.1.177.41 | 1 (0.25) | - | - | - | 1 (0.04) |
| B.1.177.82 | 7 (1.78) | - | - | - | 7 (0.30) |
| B.1.218 | 1 (0.25) | - | - | - | 1 (0.04) |
| B.1.258 | 263 (66.75) | 34 (4.76) | - | - | 297 (12.63) |
| B.1.258.17 | 3 (0.76) | - | - | - | 3 (0.13) |
| B.1.258.22 | 2 (0.51) | 1 (0.14) | - | - | 3 (0.13) |
| B.1.351 | - | - | 1 (0.16) | - | 1 (0.04) |
| B.1.398 | - | 1 (0.14) | - | - | 1 (0.04) |
| B.1.525 | - | 7 (0.98) | - | - | 7 (0.30) |
| B.1.617.2 | - | 2 (0.28) | 10 (1.55) | 45 (7.51) | 57 (2.42) |
| C.36.3 | - | - | 3 (0.47) | - | 3 (0.13) |
| D.5 | 1 (0.25) | - | - | - | 1 (0.04) |
| Q.6 | - | 1 (0.14) | - | - | 1 (0.04) |
| Q.8 | - | 1 (0.14) | 1 (0.16) | - | 2 (0.09) |
| Total | 394 | 715 | 644 | 599 | 2352 |


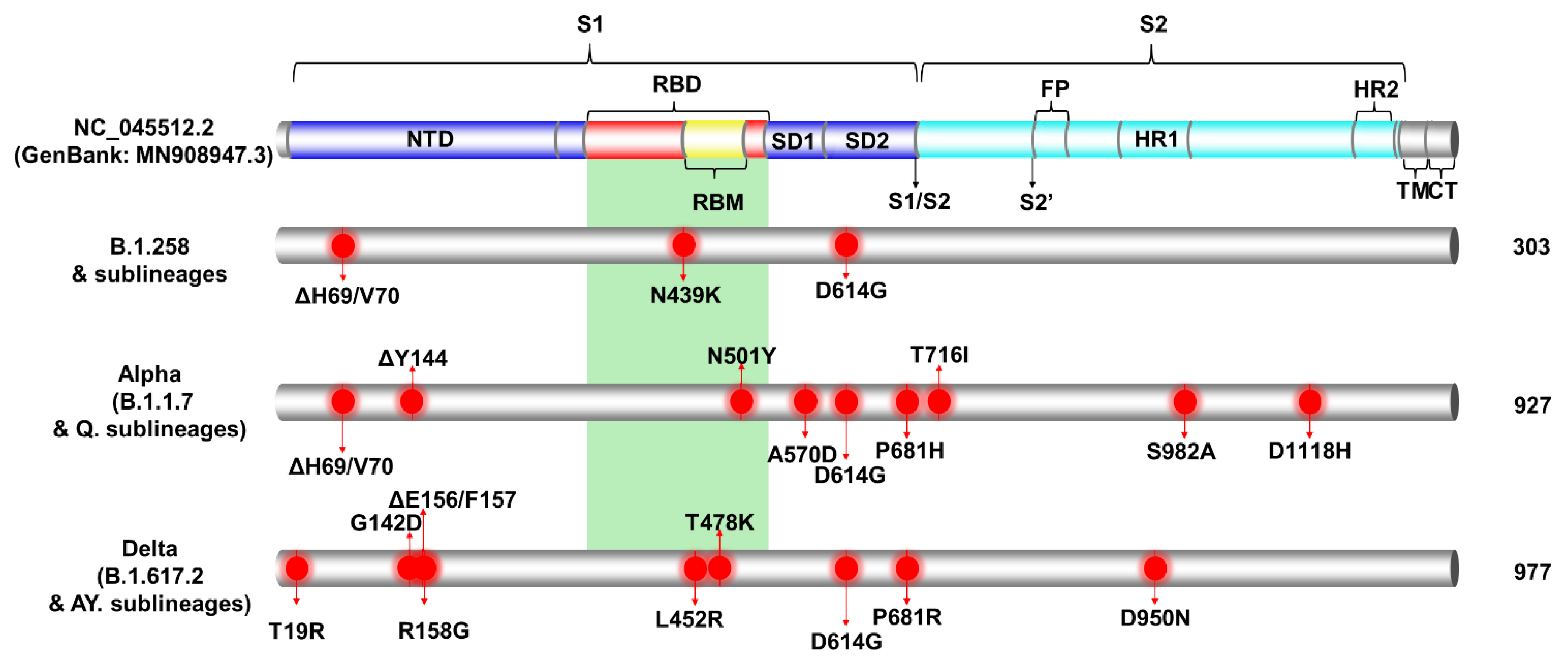
3.2. Spike Protein Mutations of the Most Prevalent Lineages/Variants in Cyprus


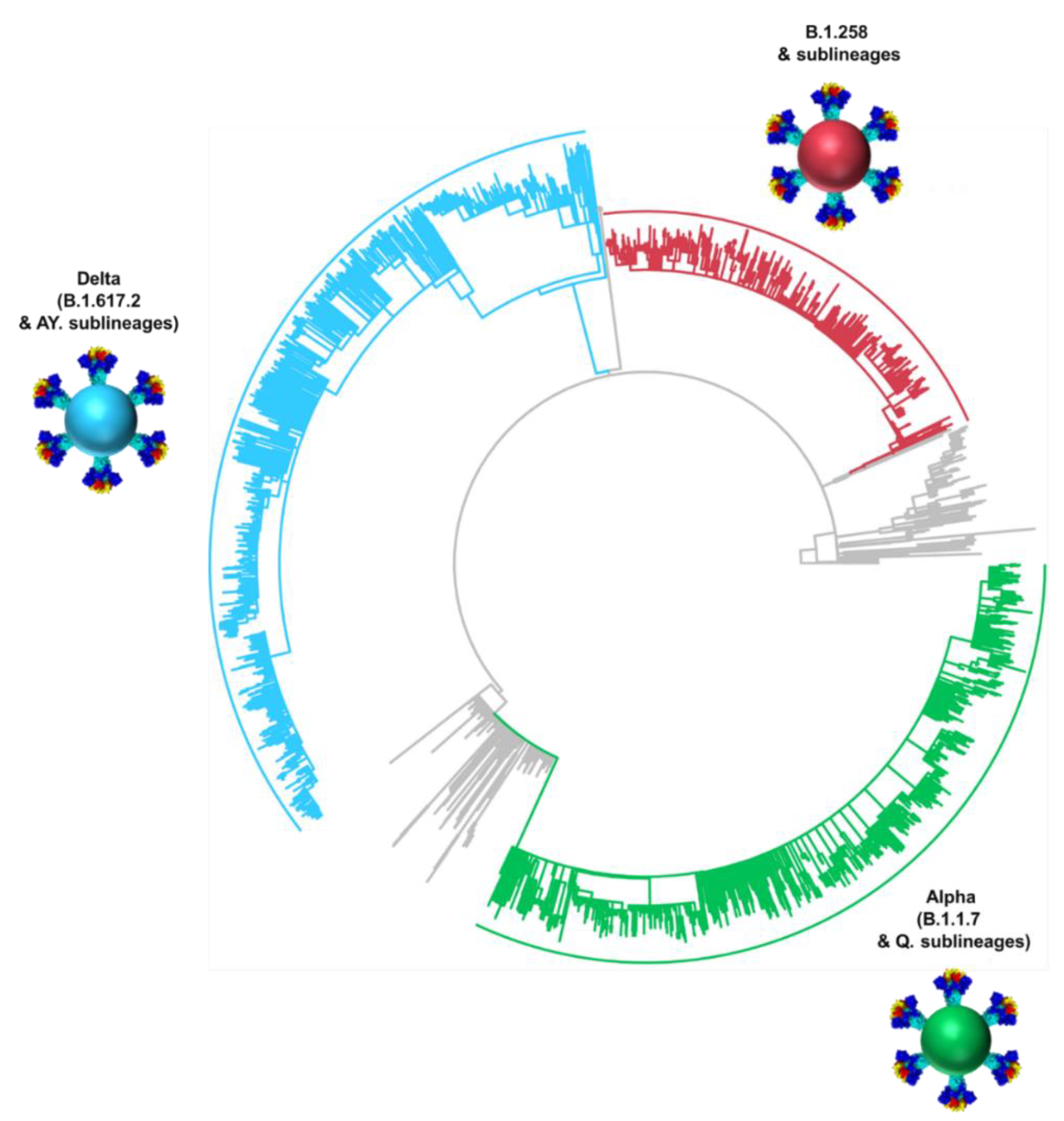
3.3. Phylogeny of Cypriot SARS-CoV-2 Sequences
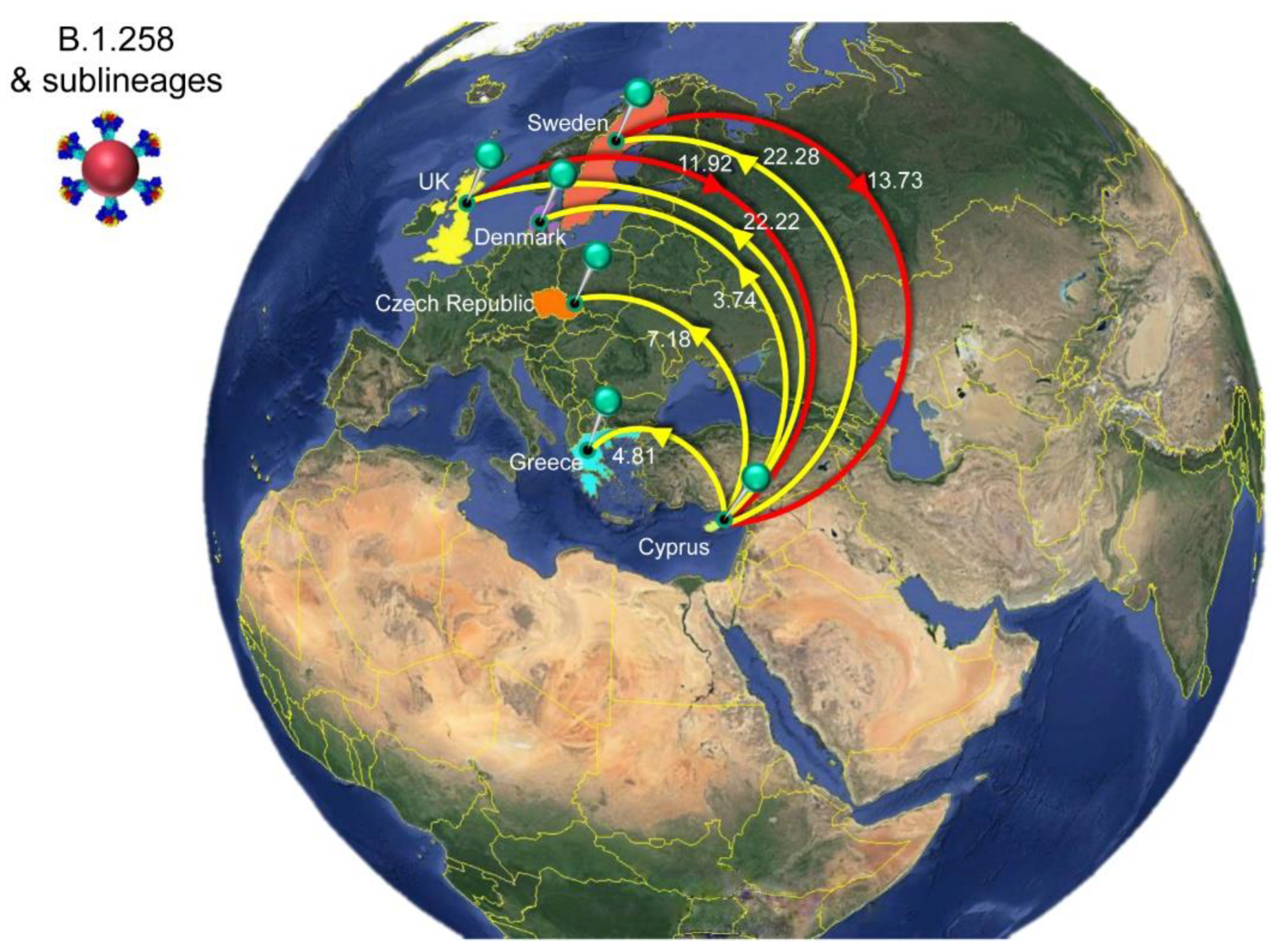
3.4. Timed Migration Histories




| Lineage/Variant a | From b | To c | Average d | Lower e | Upper f |
|---|---|---|---|---|---|
| B.1.258 & sublineages | All g | Cyprus | 25.65 | 18 | 46 |
| Sweden | Cyprus | 13.73 | 8 | 18 | |
| United Kingdom | Cyprus | 11.92 | 6 | 35 | |
| Cyprus | All | 60.23 | 39 | 68 | |
| Cyprus | Sweden | 22.28 | 18 | 26 | |
| Cyprus | United Kingdom | 22.22 | 11 | 27 | |
| Cyprus | Czech Republic | 7.18 | 0 | 10 | |
| Cyprus | Greece | 4.81 | 4 | 6 | |
| Cyprus | Denmark | 3.74 | 0 | 6 | |
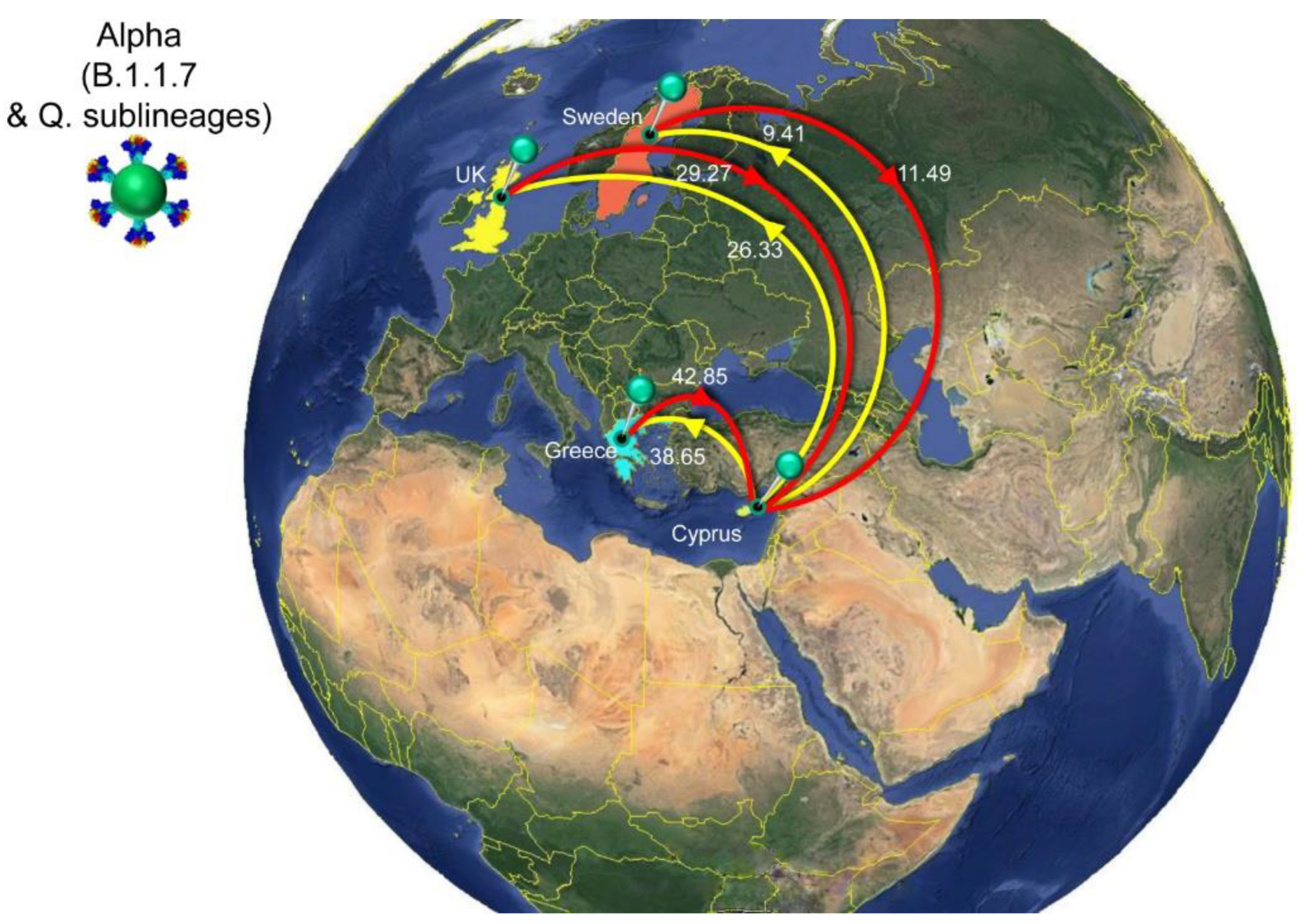
| Alpha (B.1.1.7 & Q. sublineages) | All | Cyprus | 94.09 | 87 | 100 |
| Greece | Cyprus | 42.85 | 37 | 48 | |
| United Kingdom | Cyprus | 29.27 | 22 | 36 | |
| Sweden | Cyprus | 11.49 | 5 | 16 | |
| Germany | Cyprus | 3.46 | 0 | 9 | |
| Bulgaria | Cyprus | 3.37 | 3 | 5 | |
| North America | Cyprus | 2.07 | 0 | 6 | |
| Africa | Cyprus | 1.12 | 0 | 3 | |
| Israel | Cyprus | 0.46 | 0 | 3 | |
| Cyprus | All | 74.40 | 67 | 81 | |
| Cyprus | Greece | 38.65 | 34 | 43 | |
| Cyprus | United Kingdom | 26.33 | 22 | 31 | |
| Cyprus | Sweden | 9.41 | 7 | 13 | |
| Delta (B.1.617.2 & AY. sublineages) | All | Cyprus | 521.05 | 487 | 557 |
| Switzerland | Cyprus | 93.18 | 69 | 118 | |
| Russia | Cyprus | 89.50 | 66 | 109 | |
| United Kingdom | Cyprus | 88.12 | 68 | 103 | |
| Germany | Cyprus | 86.66 | 58 | 117 | |
| Denmark | Cyprus | 57.71 | 42 | 72 | |
| Southern Asia | Cyprus | 38.20 | 21 | 52 | |
| Greece | Cyprus | 13.04 | 8 | 18 | |
| Italy | Cyprus | 11.96 | 5 | 21 | |
| Belgium | Cyprus | 9.02 | 0 | 16 | |
| Israel | Cyprus | 7.81 | 4 | 11 | |
| Sweden | Cyprus | 6.91 | 0 | 16 | |
| Northern America | Cyprus | 4.22 | 0 | 13 | |
| Romania | Cyprus | 3.66 | 0 | 9 | |
| Finland | Cyprus | 3.25 | 0 | 14 | |
| France | Cyprus | 2.75 | 0 | 11 | |
| Bulgaria | Cyprus | 2.55 | 0 | 5 | |
| Lithuania | Cyprus | 1.82 | 0 | 4 | |
| South-Eastern Asia | Cyprus | 0.68 | 0 | 4 | |
| Cyprus | All | 576.49 | 524 | 623 | |
| Cyprus | Denmark | 141.53 | 118 | 166 | |
| Cyprus | Sweden | 91.58 | 78 | 108 | |
| Cyprus | United Kingdom | 87.96 | 69 | 107 | |
| Cyprus | Germany | 54.36 | 30 | 78 | |
| Cyprus | Switzerland | 48.35 | 26 | 70 | |
| Cyprus | Greece | 40.71 | 32 | 49 | |
| Cyprus | Italy | 20.12 | 11 | 29 | |
| Cyprus | Netherlands | 13.57 | 6 | 21 | |
| Cyprus | Eastern Asia | 13.39 | 8 | 18 | |
| Cyprus | Israel | 12.49 | 6 | 18 | |
| Cyprus | Slovakia | 11.30 | 6 | 16 | |
| Cyprus | Bulgaria | 9.54 | 5 | 14 | |
| Cyprus | Western Europe | 8.97 | 5 | 12 | |
| Cyprus | Finland | 7.27 | 0 | 16 | |
| Cyprus | Croatia | 5.47 | 0 | 11 | |
| Cyprus | France | 5.05 | 0 | 16 | |
| Cyprus | Spain | 2.42 | 0 | 8 | |
| Cyprus | Western Asia | 1.77 | 0 | 4 | |
| Cyprus | Lithuania | 0.62 | 0 | 5 |

4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Rabi, F.A.; Al Zoubi, M.S.; Al-Nasser, A.D.; Kasasbeh, G.A.; Salameh, D.M. SARS-CoV-2 and coronavirus disease 2019: What we know so far. Pathogens 2020, 9, 231. [Google Scholar] [CrossRef] [PubMed]
- Demoliner, M.; Gularte, J.S.; Girardi, V.; Almeida, P.R.; de Weber, M.N.; Eisen, A.K.A.; Fleck, J.D.; Spilki, F.R. SARS-CoV-2 and COVID-19: A perspective from environmental virology. Genet. Mol. Biol. 2021, 44, e20200228. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, H.; Mathieu, E.; Rodés-Guirao, L.; Appel, C.; Giattino, C.; Ortiz-Ospina, E.; Hasell, J.; Macdonald, B.; Beltekian, D.; Roser, M.; et al. Coronavirus Pandemic (COVID-19). Available online: https://ourworldindata.org/coronavirus (accessed on 19 May 2022).
- CSSE JHU, C. for S.S. and E. (CSSE) at J.H.U. (JHU) COVID-19 Dashboard. Available online: https://gisanddata.maps.arcgis.com/apps/dashboards/bda7594740fd40299423467b48e9ecf6 (accessed on 16 June 2022).
- Dong, E.; Du, H.; Gardner, L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 2020, 20, 533–534. [Google Scholar] [CrossRef]
- KIOS Research and Innovation Center of Excellence (KIOS CoE). H εξάπλωση της COVID-19 στη Κύπρο (The spread of COVID-19 in Cyprus). Available online: https://covid19.ucy.ac.cy/ (accessed on 30 May 2022).
- Pachetti, M.; Marini, B.; Benedetti, F.; Giudici, F.; Mauro, E.; Storici, P.; Masciovecchio, C.; Angeletti, S.; Ciccozzi, M.; Gallo, R.C.; et al. Emerging SARS-CoV-2 mutation hot spots include a novel RNA-dependent-RNA polymerase variant. J. Transl. Med. 2020, 18, 179. [Google Scholar] [CrossRef]
- Wang, R.; Hozumi, Y.; Yin, C.; Wei, G.W. Decoding SARS-CoV-2 Transmission and Evolution and Ramifications for COVID-19 Diagnosis, Vaccine, and Medicine. J. Chem. Inf. Model. 2020, 60, 5853–5865. [Google Scholar] [CrossRef]
- Ghadimi-Moghadam, A.; Haghani, M.; Bevelacqua, J.J.; Jafarzadeh, A.; Kaveh-Ahangar, A.; Mortazavi, S.M.J.; Ghadimi-Moghadam, A.; Mortazavi, S.A.R. COVID-19 tragic pandemic: Concerns over unintentional “directed accelerated evolution” of novel coronavirus (SARS-CoV-2) and introducing a modified treatment method for ards. J. Biomed. Phys. Eng. 2020, 10, 241–246. [Google Scholar] [CrossRef]
- Tao, K.; Tzou, P.L.; Nouhin, J.; Gupta, R.K.; de Oliveira, T.; Kosakovsky Pond, S.L.; Fera, D.; Shafer, R.W. The biological and clinical significance of emerging SARS-CoV-2 variants. Nat. Rev. Genet. 2021, 22, 757–773. [Google Scholar] [CrossRef]
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef]
- Cosar, B.; Karagulleoglu, Z.Y.; Unal, S.; Ince, A.T.; Uncuoglu, D.B.; Tuncer, G.; Kilinc, B.R.; Ozkan, Y.E.; Ozkoc, H.C.; Demir, I.N.; et al. SARS-CoV-2 Mutations and their Viral Variants. Cytokine Growth Factor Rev. 2022, 63, 10–22. [Google Scholar] [CrossRef]
- Telenti, A.; Hodcroft, E.B.; Robertson, D.L. The Evolution and Biology of SARS-CoV-2 Variants. Cold Spring Harb. Perspect. Med. 2022, 12, a041390. [Google Scholar] [CrossRef] [PubMed]
- Markov, P.V.; Katzourakis, A.; Stilianakis, N.I. Antigenic evolution will lead to new SARS-CoV-2 variants with unpredictable severity. Nat. Rev. Microbiol. 2022, 20, 251–252. [Google Scholar] [CrossRef] [PubMed]
- The World Health Organization (WHO). Tracking SARS-CoV-2 Variants. Available online: https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/ (accessed on 20 June 2022).
- Choi, J.Y.; Smith, D.M. SARS-CoV-2 variants of concern. Yonsei Med. J. 2021, 62, 961–968. [Google Scholar] [CrossRef] [PubMed]
- Gómez, C.E.; Perdiguero, B.; Esteban, M. Emerging SARS-CoV-2 variants and impact in global vaccination programs against SARS-CoV-2/COVID-19. Vaccines 2021, 9, 243. [Google Scholar] [CrossRef]
- Ali, M.; Nas, F.; Mu’azu, L.; Abdallah, M. SARS-CoV-2 Variants of Concern (VOC): A Review. Clin. Res. Immunol. 2021, 4, 13–18. [Google Scholar] [CrossRef]
- Giovanetti, M.; Fonseca, V.; Wilkinson, E.; Tegally, H.; San, E.J.; Althaus, C.L.; Xavier, J.; Nanev Slavov, S.; Viala, V.L.; Ranieri Jerônimo Lima, A.; et al. Replacement of the Gamma by the Delta variant in Brazil: Impact of lineage displacement on the ongoing pandemic. Virus Evol. 2022, 8, veac024. [Google Scholar] [CrossRef]
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Detection of a SARS-CoV-2 variant of concern in South Africa. Nature 2021, 592, 438–443. [Google Scholar] [CrossRef]
- Grubaugh, N.D.; Cobey, S. Of variants and vaccines. Cell 2021, 184, 6222–6223. [Google Scholar] [CrossRef]
- Chen, C.; Nadeau, S.; Yared, M.; Voinov, P.; Xie, N.; Roemer, C.; Stadler, T. CoV-Spectrum: Analysis of globally shared SARS-CoV-2 data to identify and characterize new variants. Bioinformatics 2022, 38, 1735–1737. [Google Scholar] [CrossRef]
- Salehi-Vaziri, M.; Fazlalipour, M.; Seyed Khorrami, S.M.; Azadmanesh, K.; Pouriayevali, M.H.; Jalali, T.; Shoja, Z.; Maleki, A. The ins and outs of SARS-CoV-2 variants of concern (VOCs). Arch. Virol. 2022, 167, 327–344. [Google Scholar] [CrossRef]
- Lino, A.; Cardoso, M.A.; Martins-Lopes, P.; Gonçalves, H.M.R. Omicron—The new SARS-CoV-2 challenge? Rev. Med. Virol. 2022, 32, e2358. [Google Scholar] [CrossRef] [PubMed]
- Viana, R.; Moyo, S.; Amoako, D.G.; Tegally, H.; Scheepers, C.; Althaus, C.L.; Anyaneji, U.J.; Bester, P.A.; Boni, M.F.; Chand, M.; et al. Rapid epidemic expansion of the SARS-CoV-2 Omicron variant in southern Africa. Nature 2022, 603, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.L. What Makes a “Wave” of Disease? An Epidemiologist Explains. Available online: https://theconversation.com/what-makes-a-wave-of-disease-an-epidemiologist-explains-141573 (accessed on 25 July 2022).
- Akande, O.W.; Elimian, K.O.; Igumbor, E.; Dunkwu, L.; Kaduru, C.; Olopha, O.O.; Ohanu, D.O.; Nwozor, L.; Agogo, E.; Aruna, O.; et al. Epidemiological comparison of the first and second waves of the COVID-19 pandemic in Nigeria, February 2020–April 2021. BMJ Glob. Health 2021, 6, e007076. [Google Scholar] [CrossRef] [PubMed]
- Chrysostomou, A.C.; Vrancken, B.; Koumbaris, G.; Themistokleous, G.; Aristokleous, A.; Masia, C.; Eleftheriou, C.; Iοannou, C.; Stylianou, D.C.; Ioannides, M.; et al. A Comprehensive Molecular Epidemiological Analysis of SARS-CoV-2 Infection in Cyprus from April 2020 to January 2021: Evidence of a Highly Polyphyletic and Evolving Epidemic. Viruses 2021, 13, 1098. [Google Scholar] [CrossRef] [PubMed]
- O’Toole, Á.; Scher, E.; Underwood, A.; Jackson, B.; Hill, V.; McCrone, J.T.; Colquhoun, R.; Ruis, C.; Abu-Dahab, K.; Taylor, B.; et al. Assignment of epidemiological lineages in an emerging pandemic using the pangolin tool. Virus Evol. 2021, 7, veab064. [Google Scholar] [CrossRef]
- Quick, J. Artic-ncov2019 Primer Schemes. Available online: https://github.com/artic-network/artic-ncov2019/blob/master/primer_schemes/nCoV-2019/V3/nCoV-2019.tsv (accessed on 13 February 2022).
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Chen, S.; Zhou, Y.; Chen, Y.; Huang, T.; Liao, W.; Xu, Y.; Li, Z.; Gu, J. Gencore: An efficient tool to generate consensus reads for error suppressing and duplicate removing of NGS data. BMC Bioinform. 2019, 20, 606. [Google Scholar] [CrossRef]
- Töpfer, A. ConsensusFixer. Available online: https://github.com/cbg-ethz/consensusfixer (accessed on 8 July 2022).
- Aksamentov, I.; Roemer, C.; Hodcroft, E.; Neher, R. Nextclade: Clade assignment, mutation calling and quality control for viral genomes. J. Open Source Softw. 2021, 6, 3773. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Larsson, A. AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Khare, S.; Gurry, C.; Freitas, L.; Schultz, M.B.; Bach, G.; Diallo, A.; Akite, N.; Ho, J.; Lee, R.T.C.; Yeo, W. GISAID’s Role in Pandemic Response. China CDC Wkly. 2021, 3, 1049. [Google Scholar] [CrossRef]
- GISAID. GISAID Initiative. Available online: https://www.gisaid.org/ (accessed on 24 February 2021).
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- To, T.-H.; Jung, M.; Lycett, S.; Gascuel, O. Fast Dating Using Least-Squares Criteria and Algorithms. Syst. Biol. 2016, 65, 82–97. [Google Scholar] [CrossRef]
- Worobey, M.; Pekar, J.; Larsen, B.B.; Nelson, M.I.; Hill, V.; Joy, J.B.; Rambaut, A.; Suchard, M.A.; Wertheim, J.O.; Lemey, P. The emergence of SARS-CoV-2 in Europe and North America. Science 2020, 370, 564–570. [Google Scholar] [CrossRef]
- Lemey, P.; Rambaut, A.; Bedford, T.; Faria, N.; Bielejec, F.; Baele, G.; Russell, C.A.; Smith, D.J.; Pybus, O.G.; Brockmann, D.; et al. Unifying Viral Genetics and Human Transportation Data to Predict the Global Transmission Dynamics of Human Influenza H3N2. PLoS Pathog. 2014, 10, e1003932. [Google Scholar] [CrossRef]
- Lemey, P.; Rambaut, A.; Drummond, A.J.; Suchard, M.A. Bayesian phylogeography finds its roots. PLoS Comput. Biol. 2009, 5, e1000520. [Google Scholar] [CrossRef]
- Edwards, C.J.; Suchard, M.A.; Lemey, P.; Welch, J.J.; Barnes, I.; Fulton, T.L.; Barnett, R.; O’Connell, T.C.; Coxon, P.; Monaghan, N.; et al. Ancient hybridization and an irish origin for the modern polar bear matriline. Curr. Biol. 2011, 21, 1251–1258. [Google Scholar] [CrossRef]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [PubMed]
- Minin, V.N.; Suchard, M.A. Fast, accurate and simulation-free stochastic mapping. Philos. Trans. R. Soc. B Biol. Sci. 2008, 363, 3985–3995. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Kishino, H.; Yano, T. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 1985, 22, 160–174. [Google Scholar] [CrossRef] [PubMed]
- Uzzell, T.; Corbin, K.W. Fitting Discrete Probability Distributions to Evolutionary Events. Science 1971, 172, 1089–1096. [Google Scholar] [CrossRef] [PubMed]
- Gill, M.S.; Lemey, P.; Faria, N.R.; Rambaut, A.; Shapiro, B.; Suchard, M.A. Improving bayesian population dynamics inference: A coalescent-based model for multiple loci. Mol. Biol. Evol. 2013, 30, 713–724. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention (CDC). Calculating SARS-CoV-2 Laboratory Test Percent Positivity: CDC Methods and Considerations for Comparisons and Interpretation. Available online: https://www.cdc.gov/coronavirus/2019-ncov/lab/resources/calculating-percent-positivity.html (accessed on 15 June 2022).
- Press and Information Office Aνακοινωθέντα (Press Releases)-Aνακοίνωση του Υπουργείου Υγείας για νέα περιστατικά της νόσου COVID-19 (Announcement of the Ministry of Health of New COVID-19 Incidents. Available online: https://www.pio.gov.cy/ανακοινωθέντα/?keyword=Aνακοίνωση+του+Υπουργείου+Υγείας+για+νέα+περιστατικά+της+νόσου+COVID-19&startdate=&enddate=&category=&submitbtn=Aναζήτηση (accessed on 15 June 2022).
- Bateman, A.; Martin, M.J.; O’Donovan, C.; Magrane, M.; Alpi, E.; Antunes, R.; Bely, B.; Bingley, M.; Bonilla, C.; Britto, R.; et al. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef]
- Liu, L.; Wang, P.; Nair, M.S.; Yu, J.; Rapp, M.; Wang, Q.; Luo, Y.; Chan, J.F.W.; Sahi, V.; Figueroa, A.; et al. Potent neutralizing antibodies against multiple epitopes on SARS-CoV-2 spike. Nature 2020, 584, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Ma, J.; Acevedo, A. High-Potency Polypeptide-based Interference for Coronavirus Spike Glycoproteins. BioRxiv 2021. [Google Scholar] [CrossRef]
- Khelashvili, G.; Plante, A.; Doktorova, M.; Weinstein, H. Ca(2+)-dependent mechanism of membrane insertion and destabilization by the SARS-CoV-2 fusion peptide. Biophys. J. 2021, 120, 1105–1119. [Google Scholar] [CrossRef]
- Wang, P.; Nair, M.S.; Liu, L.; Iketani, S.; Luo, Y.; Guo, Y.; Wang, M.; Yu, J.; Zhang, B.; Kwong, P.D.; et al. Antibody Resistance of SARS-CoV-2 Variants, B.1.351 and B.1.1.7. Nature 2021, 593, 130–135. [Google Scholar] [CrossRef]
- Kim, S.; Lee, J.H.; Lee, S.; Shim, S.; Nguyen, T.T.; Hwang, J.; Kim, H.; Choi, Y.O.; Hong, J.; Bae, S.; et al. The progression of sars coronavirus 2 (SARS-CoV-2): Mutation in the receptor binding domain of spike gene. Immune Netw. 2020, 20, e41. [Google Scholar] [CrossRef] [PubMed]
- Mittal, A.; Manjunath, K.; Ranjan, R.K.; Kaushik, S.; Kumar, S.; Verma, V. COVID-19 pandemic: Insights into structure, function, and hACE2 receptor recognition by SARS-CoV-2. PLoS Pathog. 2020, 16, e1008762. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yang, C.; Xu, X.F.; Xu, W.; Liu, S.W. Structural and functional properties of SARS-CoV-2 spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Xia, X. Domains and Functions of Spike Protein in SARS-CoV-2 in the Context of Vaccine Design. Viruses 2021, 13, 109. [Google Scholar] [CrossRef]
- Gobeil, S.M.C.; Janowska, K.; McDowell, S.; Mansouri, K.; Parks, R.; Manne, K.; Stalls, V.; Kopp, M.F.; Henderson, R.; Edwards, R.J.; et al. D614G Mutation Alters SARS-CoV-2 Spike Conformation and Enhances Protease Cleavage at the S1/S2 Junction. Cell Rep. 2021, 34, 108630. [Google Scholar] [CrossRef]
- Sasaki, M.; Uemura, K.; Sato, A.; Toba, S.; Sanaki, T.; Maenaka, K.; Hall, W.W.; Orba, Y.; Sawa, H. SARS-CoV-2 variants with mutations at the S1/ S2 cleavage site are generated in vitro during propagation in TMPRSS2-deficient cells. PLoS Pathog. 2021, 17, e1009233. [Google Scholar] [CrossRef]
- Kapoor, K.; Chen, T.; Tajkhorshid, E. Posttranslational modifications optimize the ability of SARS-CoV-2 spike for effective interaction with host cell receptors. Proc. Natl. Acad. Sci. USA 2022, 119, e2119761119. [Google Scholar] [CrossRef]
- De Marco, C.; Veneziano, C.; Massacci, A.; Pallocca, M.; Marascio, N.; Quirino, A.; Barreca, G.S.; Giancotti, A.; Gallo, L.; Lamberti, A.G.; et al. Dynamics of Viral Infection and Evolution of SARS-CoV-2 Variants in the Calabria Area of Southern Italy. Front. Microbiol. 2022, 13, 934993. [Google Scholar] [CrossRef]
- de Hoffer, A.; Vatani, S.; Cot, C.; Cacciapaglia, G.; Chiusano, M.L.; Cimarelli, A.; Conventi, F.; Giannini, A.; Hohenegger, S.; Sannino, F. Variant-driven early warning via unsupervised machine learning analysis of spike protein mutations for COVID-19. Sci. Rep. 2022, 12, 9275. [Google Scholar] [CrossRef]
- Mathias, M.; do Nascimento, G.M.; Nooruzzaman, M.; Yuan, F.; Chen, C.; Caserta, L.C.; Miller, A.D.; Whittaker, G.R.; Fang, Y.; Diel, D.G. The Omicron Variant BA.1.1 Presents a Lower Pathogenicity than B.1 D614G and Delta Variants in a Feline Model of SARS-CoV-2 Infection. J. Virol. 2022, 96, e0096122. [Google Scholar] [CrossRef]
- Meng, B.; Kemp, S.A.; Papa, G.; Datir, R.; Ferreira, I.A.T.M.; Marelli, S.; Harvey, W.T.; Lytras, S.; Mohamed, A.; Gallo, G.; et al. Recurrent emergence of SARS-CoV-2 spike deletion H69/V70 and its role in the Alpha variant B.1.1.7. Cell Rep. 2021, 35, 109292. [Google Scholar] [CrossRef] [PubMed]
- Brejová, B.; Boršová, K.; Hodorová, V.; Čabanová, V.; Reizigová, L.; Paul, E.D.; Čekan, P.; Klempa, B.; Nosek, J.; Vinař, T. A SARS-CoV-2 mutant from B.1.258 lineage with ∆H69/∆V70 deletion in the Spike protein circulating in Central Europe in the fall 2020. Virus Genes 2021, 57, 556–560. [Google Scholar] [CrossRef] [PubMed]
- Thakur, V.; Bhola, S.; Thakur, P.; Patel, S.K.S.; Kulshrestha, S.; Ratho, R.K.; Kumar, P. Waves and variants of SARS-CoV-2: Understanding the causes and effect of the COVID-19 catastrophe. Infection 2022, 50, 309–325. [Google Scholar] [CrossRef]
- Republic of Cyprus Ministry of Health. New Coronavirus Disease (COVID-19). Available online: https://www.pio.gov.cy/coronavirus/eng/categories/important-announcements (accessed on 16 November 2022).
- Wu, J.; Zhang, L.; Zhang, Y.; Wang, H.; Ding, R.; Nie, J.; Li, Q.; Liu, S.; Yu, Y.; Yang, X.; et al. The Antigenicity of Epidemic SARS-CoV-2 Variants in the United Kingdom. Front. Immunol. 2021, 12, 2205. [Google Scholar] [CrossRef]
- Tande, A.J.; Binnicker, M.J.; Ting, H.H.; Del Rio, C.; Jalil, L.; Brawner, M.; Carter, P.W.; Toomey, K.; Shah, N.D.; Berbari, E.F. SARS-CoV-2 Testing Before International Airline Travel, December 2020 to May 2021. Mayo Clin. Proc. 2021, 96, 2856–2860. [Google Scholar] [CrossRef] [PubMed]
- Kubik, S.; Arrigo, N.; Bonet, J.; Xu, Z. Mutational Hotspot in the SARS-CoV-2 Spike Protein N-Terminal Domain Conferring Immune Escape Potential. Viruses 2021, 13, 2114. [Google Scholar] [CrossRef] [PubMed]
- Klinakis, A.; Cournia, Z.; Rampias, T. N-terminal domain mutations of the spike protein are structurally implicated in epitope recognition in emerging SARS-CoV-2 strains. Comput. Struct. Biotechnol. J. 2021, 19, 5556–5567. [Google Scholar] [CrossRef]
- Awasthi, M.; Gulati, S.; Sarkar, D.P.; Tiwari, S.; Kateriya, S.; Ranjan, P.; Verma, S.K. The Sialoside-Binding Pocket of SARS-CoV-2 Spike Glycoprotein Structurally Resembles MERS-CoV. Viruses 2020, 12, 909. [Google Scholar] [CrossRef]
- Peng, Q.; Zhou, R.; Liu, N.; Wang, H.; Xu, H.; Zhao, M.; Yang, D.; Au, K.-K.; Huang, H.; Liu, L.; et al. Naturally occurring spike mutations influence the infectivity and immunogenicity of SARS-CoV-2. Cell. Mol. Immunol. 2022, 19, 1302–1310. [Google Scholar] [CrossRef]
- McMillen, T.; Jani, K.; Robilotti, E.V.; Kamboj, M.; Babady, N.E. The spike gene target failure (SGTF) genomic signature is highly accurate for the identification of Alpha and Omicron SARS-CoV-2 variants. Sci. Rep. 2022, 12, 18968. [Google Scholar] [CrossRef]
- Torbati, E.; Krause, K.L.; Ussher, J.E. The Immune Response to SARS-CoV-2 and Variants of Concern. Viruses 2021, 13, 1911. [Google Scholar] [CrossRef]
- Thomson, E.C.; Rosen, L.E.; Shepherd, J.G.; Spreafico, R.; da Silva Filipe, A.; Wojcechowskyj, J.A.; Davis, C.; Piccoli, L.; Pascall, D.J.; Dillen, J.; et al. Circulating SARS-CoV-2 spike N439K variants maintain fitness while evading antibody-mediated immunity. Cell 2021, 184, 1171–1187.e20. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, K.R.; Rennick, L.J.; Nambulli, S.; Robinson-McCarthy, L.R.; Bain, W.G.; Haidar, G.; Duprex, W.P. Recurrent deletions in the SARS-CoV-2 spike glycoprotein drive antibody escape. Science 2021, 371, 1139–1142. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Mangalaganesh, S.; Wilson, L.O.W.; Kuiper, M.J.; Drew, T.W.; Vasan, S.S. Tracking Co-Occurrence of N501Y, P681R, and Other Key Mutations in SARS-CoV-2 Spike for Surveillance. Zoonotic Dis. 2022, 2, 147–162. [Google Scholar] [CrossRef]
- McCallum, M.; Czudnochowski, N.; Rosen, L.E.; Zepeda, S.K.; Bowen, J.E.; Walls, A.C.; Hauser, K.; Joshi, A.; Stewart, C.; Dillen, J.R.; et al. Structural basis of SARS-CoV-2 Omicron immune evasion and receptor engagement. Science 2022, 375, 864–868. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.-J.; Yu, P.-Y.; Chang, Y.-C.; Liang, K.-H.; Tso, H.-C.; Ho, M.-R.; Chen, W.-Y.; Lin, H.-T.; Wu, H.-C.; Hsu, S.-T.D. Effect of SARS-CoV-2 B.1.1.7 mutations on spike protein structure and function. Nat. Struct. Mol. Biol. 2021, 28, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Eslami, S.; Glassy, M.C.; Ghafouri-Fard, S. A comprehensive overview of identified mutations in SARS CoV-2 spike glycoprotein among Iranian patients. Gene 2022, 813, 146113. [Google Scholar] [CrossRef] [PubMed]
- Lubinski, B.; Fernandes, M.H.V.; Frazier, L.; Tang, T.; Daniel, S.; Diel, D.G.; Jaimes, J.A.; Whittaker, G.R. Functional evaluation of the P681H mutation on the proteolytic activation of the SARS-CoV-2 variant B.1.1.7 (Alpha) spike. iScience 2022, 25, 103589. [Google Scholar] [CrossRef]
- Gobeil, S.M.-C.; Janowska, K.; McDowell, S.; Mansouri, K.; Parks, R.; Stalls, V.; Kopp, M.F.; Manne, K.; Li, D.; Wiehe, K.; et al. Effect of natural mutations of SARS-CoV-2 on spike structure, conformation, and antigenicity. Science 2022, 373, eabi6226. [Google Scholar] [CrossRef]
- Domingo, P.; de Benito, N. Alpha variant SARS-CoV-2 infection: How it all starts. eBioMedicine 2021, 74, 103703. [Google Scholar] [CrossRef]
- Cyprus Statistical Service. Arrivals of Tourists by Country of Usual Residence. 2021. Available online: https://www.cystat.gov.cy/en/KeyFiguresList?s=51&fbclid=IwAR0mThPdhjg-Uj64Q2kAW7ibreEZgx1i4PftJZ_orJimRXU1AOKpbUXExB0 (accessed on 18 November 2022).
- Callaway, E. The mutation that helps Delta spread like wildfire. Nature 2021, 596, 472–473. [Google Scholar] [CrossRef] [PubMed]
- von Wintersdorff, C.J.H.; Dingemans, J.; van Alphen, L.B.; Wolffs, P.F.G.; van der Veer, B.M.J.W.; Hoebe, C.J.P.A.; Savelkoul, P.H.M. Infections with the SARS-CoV-2 Delta variant exhibit fourfold increased viral loads in the upper airways compared to Alpha or non-variants of concern. Sci. Rep. 2022, 12, 13922. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Huang, J.; Li, C.; Zhao, Y.; Wang, D.; Huang, Z.; Yang, K. The role of seasonality in the spread of COVID-19 pandemic. Environ. Res. 2021, 195, 110874. [Google Scholar] [CrossRef] [PubMed]
- McCallum, M.; Walls, A.C.; Sprouse, K.R.; Bowen, J.E.; Rosen, L.E.; Dang, H.V.; De Marco, A.; Franko, N.; Tilles, S.W.; Logue, J.; et al. Molecular basis of immune evasion by the Delta and Kappa SARS-CoV-2 variants. Science 2021, 374, 1621–1626. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Sharma, K.; Singh, P.; Bhargava, A.; Negi, S.S.; Sharma, P.; Bhise, M.; Tripathi, M.K.; Jindal, A.; Nagarkar, N.M. Genomic characterization unravelling the causative role of SARS-CoV-2 Delta variant of lineage B.1.617.2 in 2nd wave of COVID-19 pandemic in Chhattisgarh, India. Microb. Pathog. 2022, 164, 105404. [Google Scholar] [CrossRef]
- Mishra, T.; Dalavi, R.; Joshi, G.; Kumar, A.; Pandey, P.; Shukla, S.; Mishra, R.K.; Chande, A. SARS-CoV-2 spike E156G/Δ157-158 mutations contribute to increased infectivity and immune escape. Life Sci. Alliance 2022, 5, e202201415. [Google Scholar] [CrossRef]
- Dhawan, M.; Sharma, A.; Priyanka; Thakur, N.; Rajkhowa, T.K.; Choudhary, O.P. Delta variant (B.1.617.2) of SARS-CoV-2: Mutations, impact, challenges and possible solutions. Hum. Vaccin. Immunother. 2022, 18, 2068883. [Google Scholar] [CrossRef]
- Bhattacharya, M.; Chatterjee, S.; Sharma, A.R.; Lee, S.S.; Chakraborty, C. Delta variant (B. 1.617. 2) of SARS-CoV-2: Current understanding of infection, transmission, immune escape, and mutational landscape. Folia Microbiol. 2022, 12, 1–2. [Google Scholar]
- Wilhelm, A.; Toptan, T.; Pallas, C.; Wolf, T.; Goetsch, U.; Gottschalk, R.; Vehreschild, M.J.G.T.; Ciesek, S.; Widera, M. Antibody-Mediated Neutralization of Authentic SARS-CoV-2 B.1.617 Variants Harboring L452R and T478K/E484Q. Viruses 2021, 13, 1693. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, J.; Johnson, B.A.; Xia, H.; Ku, Z.; Schindewolf, C.; Widen, S.G.; An, Z.; Weaver, S.C.; Menachery, V.D.; et al. Delta spike P681R mutation enhances SARS-CoV-2 fitness over Alpha variant. Cell Rep. 2022, 39, 110829. [Google Scholar] [CrossRef]
- Saito, A.; Irie, T.; Suzuki, R.; Maemura, T.; Nasser, H.; Uriu, K.; Kosugi, Y.; Shirakawa, K.; Sadamasu, K.; Kimura, I.; et al. Enhanced fusogenicity and pathogenicity of SARS-CoV-2 Delta P681R mutation. Nature 2022, 602, 300–306. [Google Scholar] [CrossRef] [PubMed]
- He, X.; He, C.; Hong, W.; Zhang, K.; Wei, X. The challenges of COVID-19 Delta variant: Prevention and vaccine development. MedComm 2021, 2, 846–854. [Google Scholar] [CrossRef] [PubMed]
- Focosi, D.; Maggi, F.; Mcconnell, S.; Casadevall, A. Spike mutations in SARS-CoV-2 AY sublineages of the Delta variant of concern: Implications for the future of the pandemic. Future Microbiol. 2022, 17, 219–221. [Google Scholar] [CrossRef]
- Saunders, N.; Planas, D.; Bolland, W.H.; Rodriguez, C.; Fourati, S.; Buchrieser, J.; Planchais, C.; Prot, M.; Staropoli, I.; Guivel-Benhassine, F.; et al. Fusogenicity and neutralization sensitivity of the SARS-CoV-2 Delta sublineage AY.4.2. eBioMedicine 2022, 77, 103934. [Google Scholar] [CrossRef] [PubMed]
- Arora, P.; Kempf, A.; Nehlmeier, I.; Graichen, L.; Winkler, M.S.; Lier, M.; Schulz, S.; Jäck, H.-M.; Pöhlmann, S.; Hoffmann, M. No evidence for increased cell entry or antibody evasion by Delta sublineage AY.4.2. Cell. Mol. Immunol. 2022, 19, 449–452. [Google Scholar] [CrossRef] [PubMed]
- Chugh, A.; Khurana, N.; Verma, K.; Sehgal, I.; Rolta, R.; Vats, P.; Phartyal, R.; Salaria, D.; Kaushik, N.; Choi, E.H.; et al. Changing Dynamics of SARS-CoV-2: A Global Challenge. Appl. Sci. 2022, 12, 5546. [Google Scholar] [CrossRef]
- Fan, Y.; Li, X.; Zhang, L.; Wan, S.; Zhang, L.; Zhou, F. SARS-CoV-2 Omicron variant: Recent progress and future perspectives. Signal Transduct. Target. Ther. 2022, 7, 141. [Google Scholar] [CrossRef]
- Darwin, C. On the Origin of Species, 1859; Routledge: London, UK, 2004. [Google Scholar]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chrysostomou, A.C.; Vrancken, B.; Haralambous, C.; Alexandrou, M.; Aristokleous, A.; Christodoulou, C.; Gregoriou, I.; Ioannides, M.; Kalakouta, O.; Karagiannis, C.; et al. Genomic Epidemiology of the SARS-CoV-2 Epidemic in Cyprus from November 2020 to October 2021: The Passage of Waves of Alpha and Delta Variants of Concern. Viruses 2023, 15, 108. https://doi.org/10.3390/v15010108
Chrysostomou AC, Vrancken B, Haralambous C, Alexandrou M, Aristokleous A, Christodoulou C, Gregoriou I, Ioannides M, Kalakouta O, Karagiannis C, et al. Genomic Epidemiology of the SARS-CoV-2 Epidemic in Cyprus from November 2020 to October 2021: The Passage of Waves of Alpha and Delta Variants of Concern. Viruses. 2023; 15(1):108. https://doi.org/10.3390/v15010108
Chicago/Turabian StyleChrysostomou, Andreas C., Bram Vrancken, Christos Haralambous, Maria Alexandrou, Antonia Aristokleous, Christina Christodoulou, Ioanna Gregoriou, Marios Ioannides, Olga Kalakouta, Christos Karagiannis, and et al. 2023. "Genomic Epidemiology of the SARS-CoV-2 Epidemic in Cyprus from November 2020 to October 2021: The Passage of Waves of Alpha and Delta Variants of Concern" Viruses 15, no. 1: 108. https://doi.org/10.3390/v15010108
APA StyleChrysostomou, A. C., Vrancken, B., Haralambous, C., Alexandrou, M., Aristokleous, A., Christodoulou, C., Gregoriou, I., Ioannides, M., Kalakouta, O., Karagiannis, C., Koumbaris, G., Loizides, C., Mendris, M., Papastergiou, P., Patsalis, P. C., Pieridou, D., Richter, J., Schmitt, M., Shammas, C., ... Kostrikis, L. G. (2023). Genomic Epidemiology of the SARS-CoV-2 Epidemic in Cyprus from November 2020 to October 2021: The Passage of Waves of Alpha and Delta Variants of Concern. Viruses, 15(1), 108. https://doi.org/10.3390/v15010108