
The Effect of Treatment-Associated Mutations on HIV Replication and Transmission Cycles



Abstract
1. Introduction
2. Transmission Cycle
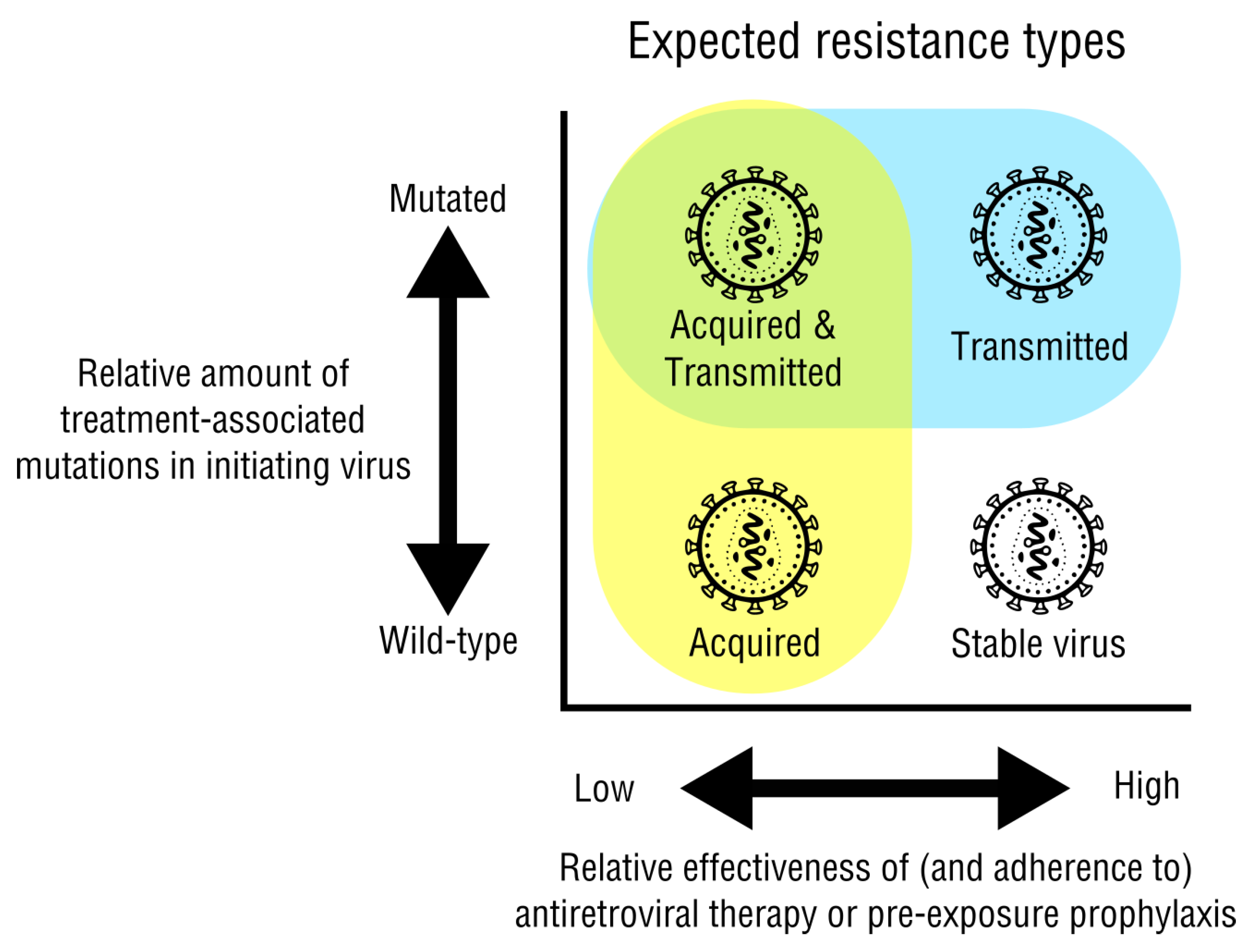
2.1. Acquired and Transmitted Drug Resistance
2.2. Drug Resistance Testing
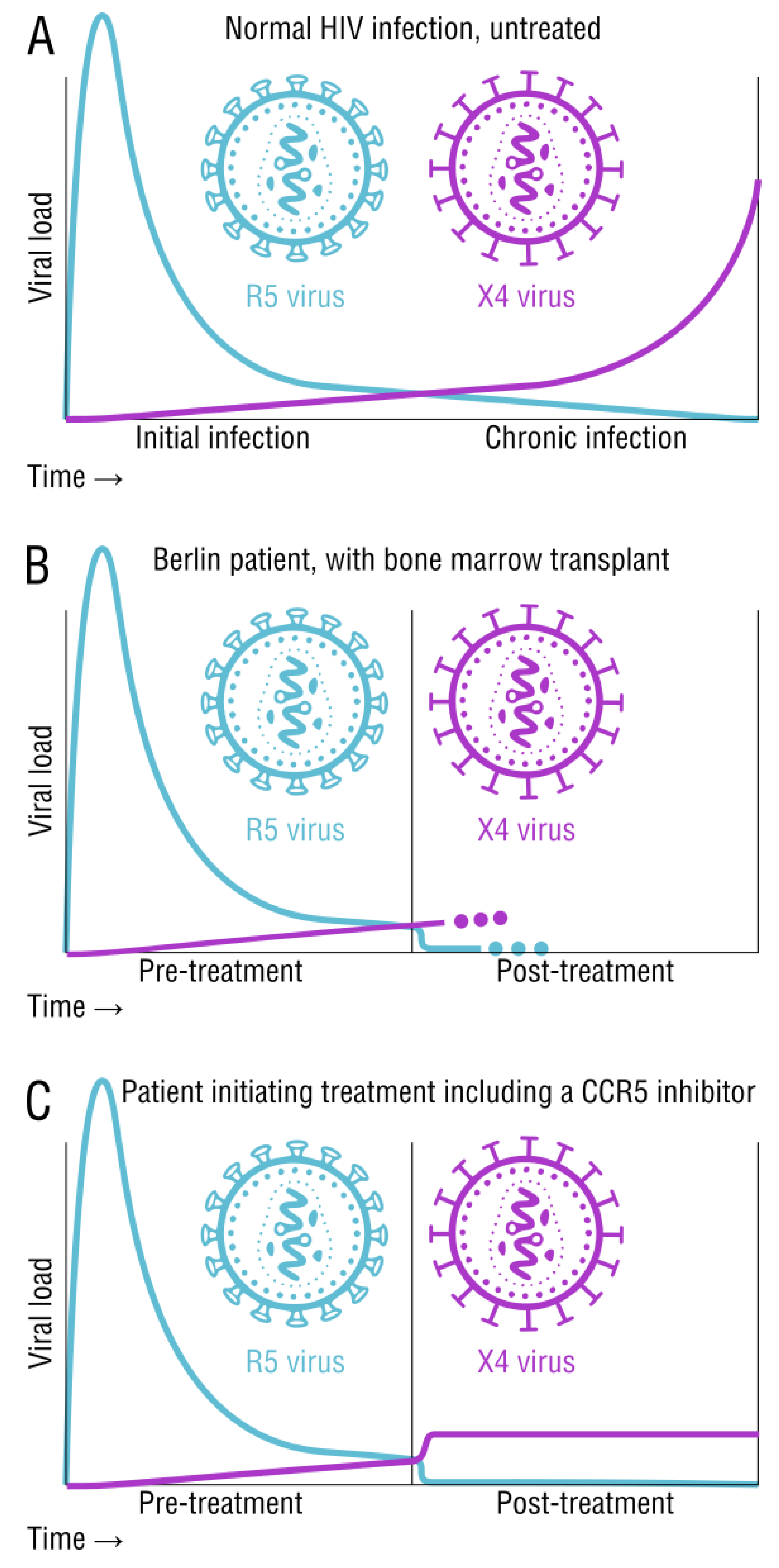
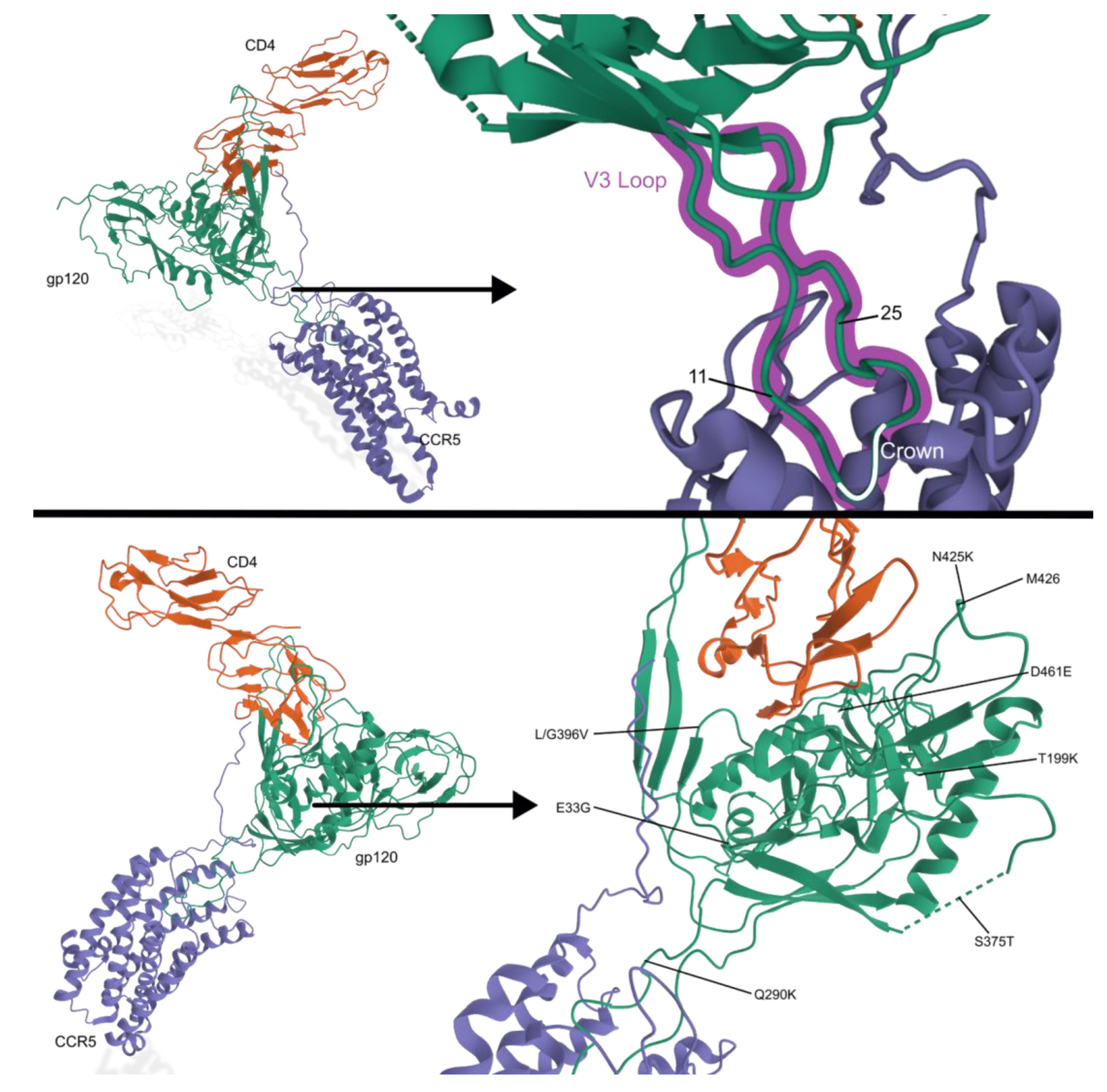
2.3. Tropism and Its Effects on Transmission and Disease Status
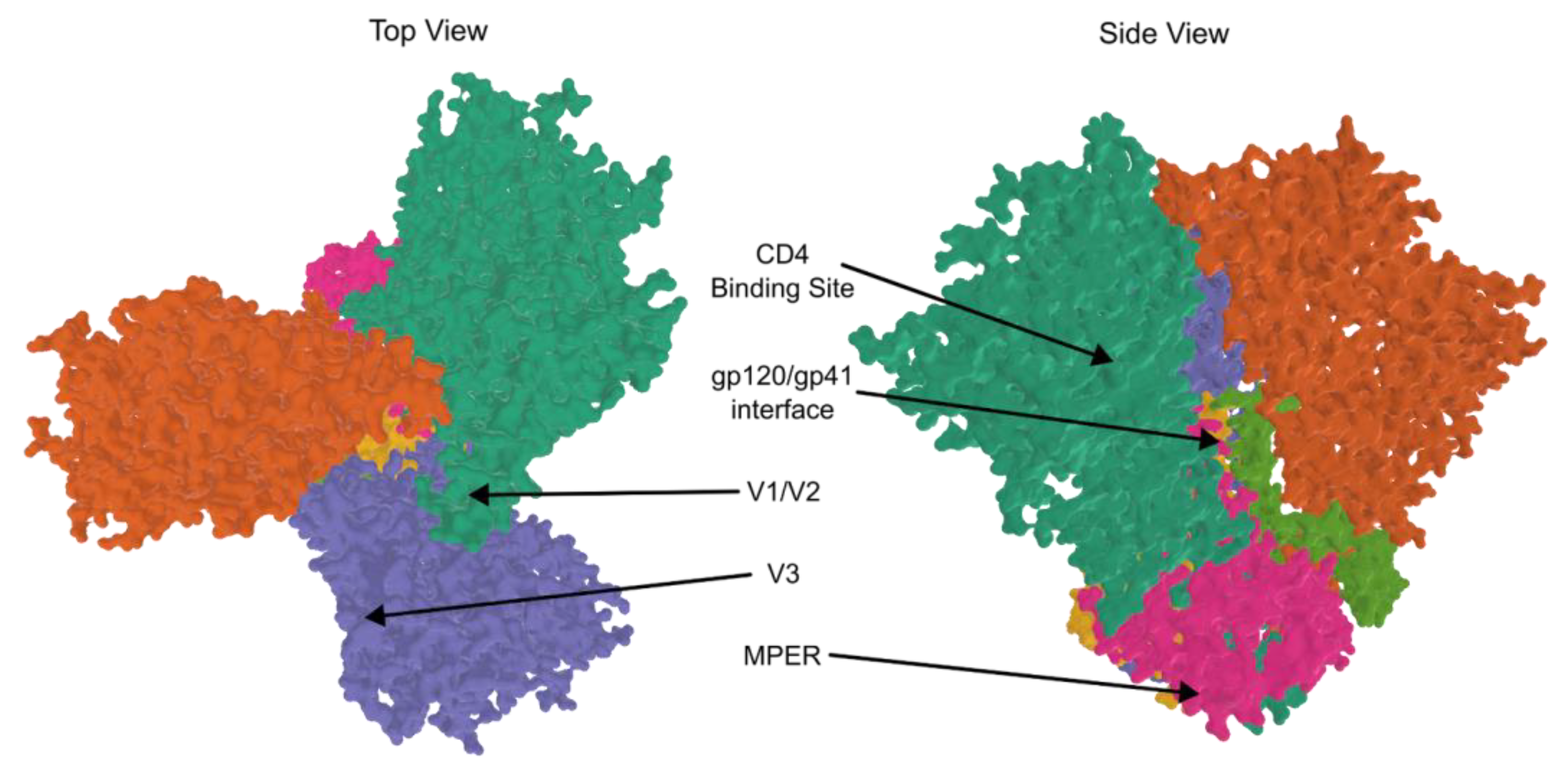
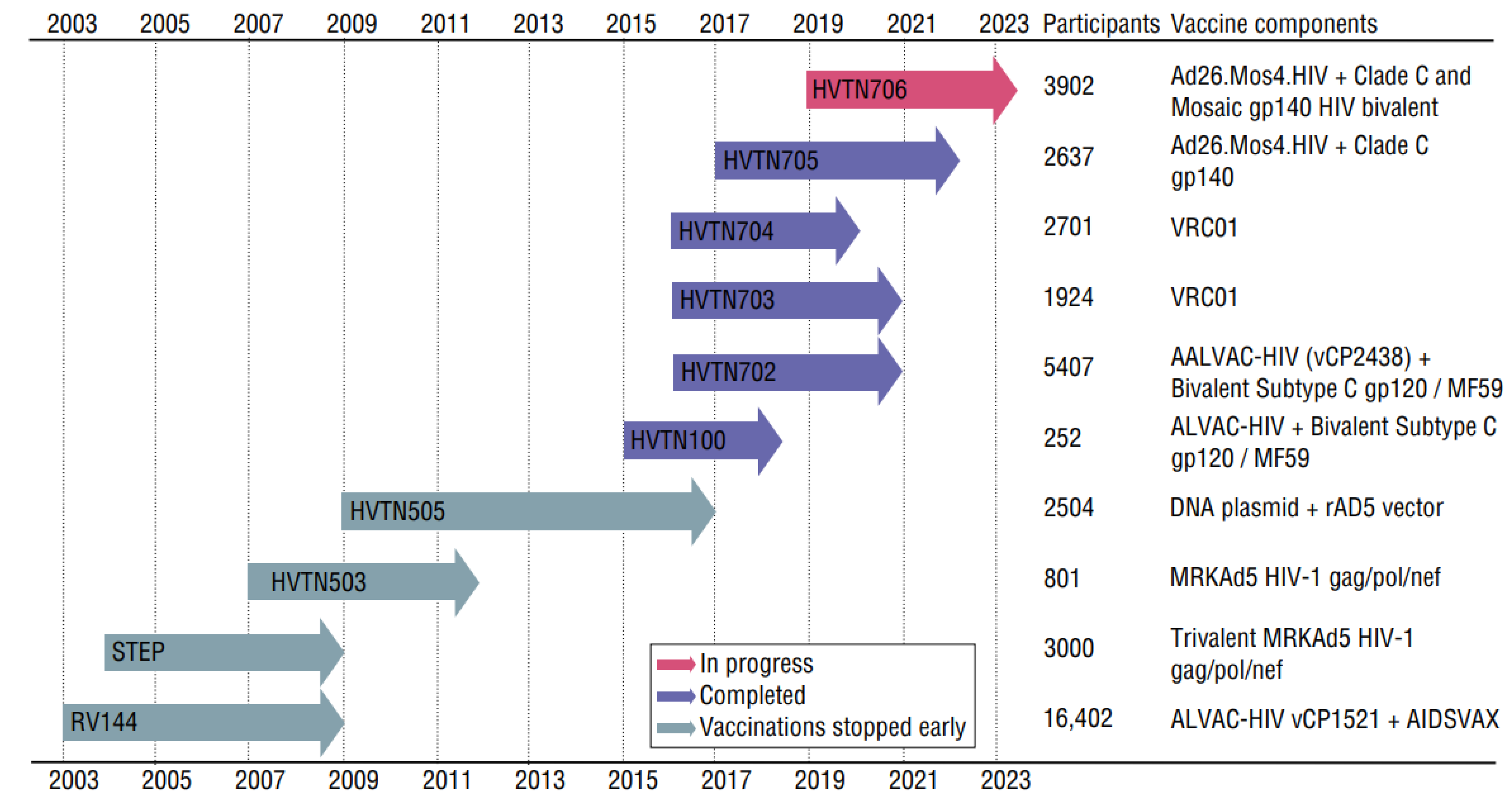
2.4. Vaccine and Prevention Strategies
2.4.1. Vaccine Trials and bNAbs
2.4.2. mRNA Vaccines
2.4.3. Preventative and Post-Exposure Treatments
2.4.4. CRISPR
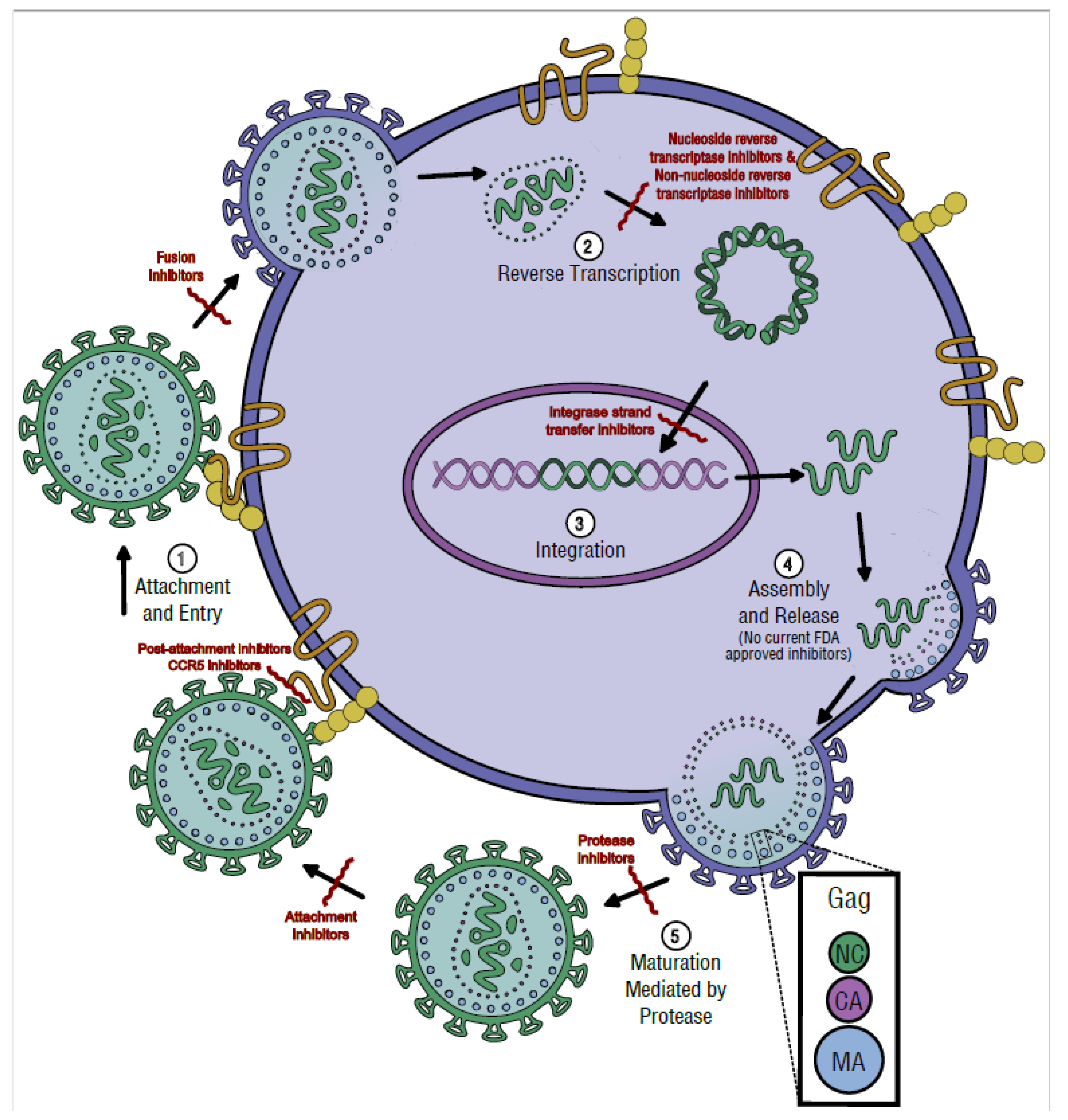
3. Replication Cycle
3.1. Attachment and Entry
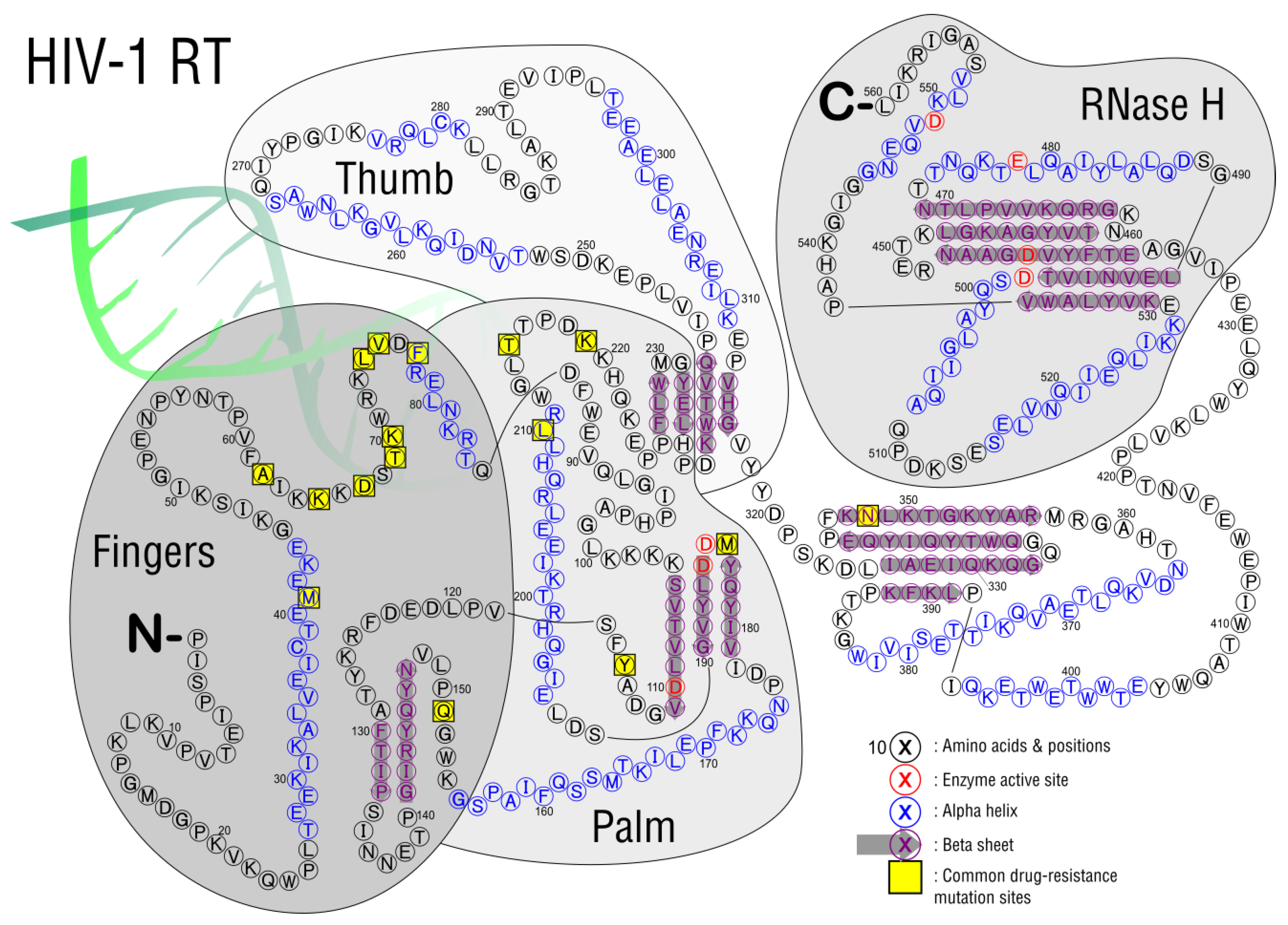
3.2. Reverse Transcription
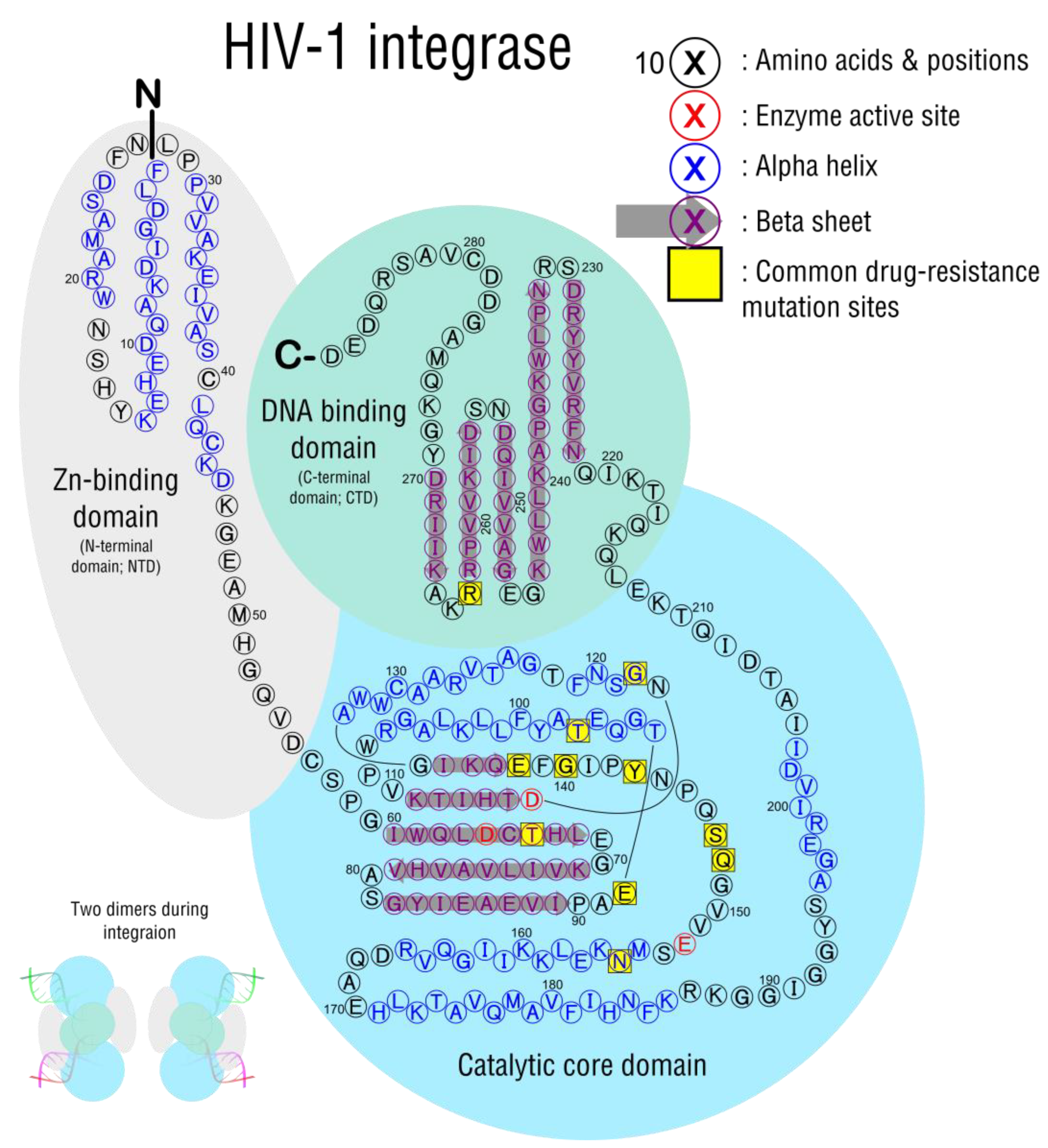
3.3. Integration
3.4. Assembly and Release
3.5. Maturation and Protease Activity
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Clutter, D.S.; Jordan, M.R.; Bertagnolio, S.; Shafer, R.W. HIV-1 Drug Resistance and Resistance Testing. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2016, 46, 292–307. [Google Scholar] [CrossRef]
- Gifford, R.J.; Liu, T.F.; Rhee, S.-Y.; Kiuchi, M.; Hue, S.; Pillay, D.; Shafer, R.W. The Calibrated Population Resistance Tool: Standardized Genotypic Estimation of Transmitted HIV-1 Drug Resistance. Bioinformatics 2009, 25, 1197–1198. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.F.; Shafer, R.W. Web Resources for HIV Type 1 Genotypic-Resistance Test Interpretation. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2006, 42, 1608–1618. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.-Y.; Gonzales, M.J.; Kantor, R.; Betts, B.J.; Ravela, J.; Shafer, R.W. Human Immunodeficiency Virus Reverse Transcriptase and Protease Sequence Database. Nucleic Acids Res. 2003, 31, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.-Y.; Kantor, R.; Katzenstein, D.A.; Camacho, R.; Morris, L.; Sirivichayakul, S.; Jorgensen, L.; Brigido, L.F.; Schapiro, J.M.; Shafer, R.W.; et al. HIV-1 Pol Mutation Frequency by Subtype and Treatment Experience: Extension of the HIVseq Program to Seven Non-B Subtypes. AIDS 2006, 20, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Shafer, R.W.; Jung, D.R.; Betts, B.J. Human Immunodeficiency Virus Type 1 Reverse Transcriptase and Protease Mutation Search Engine for Queries. Nat. Med. 2000, 6, 1290–1292. [Google Scholar] [CrossRef]
- Shafer, R.W. Rationale and Uses of a Public HIV Drug-Resistance Database. J. Infect. Dis. 2006, 194 (Suppl. S1), S51–S58. [Google Scholar] [CrossRef]
- Kawashima, Y.; Pfafferott, K.; Frater, J.; Matthews, P.; Payne, R.; Addo, M.; Gatanaga, H.; Fujiwara, M.; Hachiya, A.; Koizumi, H.; et al. Adaptation of HIV-1 to Human Leukocyte Antigen Class I. Nature 2009, 458, 641–645. [Google Scholar] [CrossRef]
- Klein, J.S.; Bjorkman, P.J. Few and Far between: How HIV May Be Evading Antibody Avidity. PLoS Pathog. 2010, 6, e1000908. [Google Scholar] [CrossRef]
- Gupta, R.K.; Hill, A.; Sawyer, A.W.; Cozzi-Lepri, A.; von Wyl, V.; Yerly, S.; Lima, V.D.; Günthard, H.F.; Gilks, C.; Pillay, D. Virological Monitoring and Resistance to First-Line Highly Active Antiretroviral Therapy in Adults Infected with HIV-1 Treated under WHO Guidelines: A Systematic Review and Meta-Analysis. Lancet Infect. Dis. 2009, 9, 409–417. [Google Scholar] [CrossRef]
- Atta, M.G.; De Seigneux, S.; Lucas, G.M. Clinical Pharmacology in HIV Therapy. Clin. J. Am. Soc. Nephrol. CJASN 2019, 14, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Le, T.; Chiarella, J.; Simen, B.B.; Hanczaruk, B.; Egholm, M.; Landry, M.L.; Dieckhaus, K.; Rosen, M.I.; Kozal, M.J. Low-Abundance HIV Drug-Resistant Viral Variants in Treatment-Experienced Persons Correlate with Historical Antiretroviral Use. PLoS ONE 2009, 4, e6079. [Google Scholar] [CrossRef] [PubMed]
- Simen, B.B.; Simons, J.F.; Hullsiek, K.H.; Novak, R.M.; Macarthur, R.D.; Baxter, J.D.; Huang, C.; Lubeski, C.; Turenchalk, G.S.; Braverman, M.S.; et al. Low-Abundance Drug-Resistant Viral Variants in Chronically HIV-Infected, Antiretroviral Treatment-Naive Patients Significantly Impact Treatment Outcomes. J. Infect. Dis. 2009, 199, 693–701. [Google Scholar] [CrossRef]
- Svarovskaia, E.S.; Margot, N.A.; Bae, A.S.; Waters, J.M.; Goodman, D.; Zhong, L.; Borroto-Esoda, K.; Miller, M.D. Low-Level K65R Mutation in HIV-1 Reverse Transcriptase of Treatment-Experienced Patients Exposed to Abacavir or Didanosine. J. Acquir. Immune Defic. Syndr. 1999 2007, 46, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Castro, H.; Pillay, D.; Cane, P.; Asboe, D.; Cambiano, V.; Phillips, A.; Dunn, D.T. UK Collaborative Group on HIV Drug Resistance Persistence of HIV-1 Transmitted Drug Resistance Mutations. J. Infect. Dis. 2013, 208, 1459–1463. [Google Scholar] [CrossRef]
- Guo, C.; Wu, Y.; Zhang, Y.; Liu, X.; Li, A.; Gao, M.; Zhang, T.; Wu, H.; Chen, G.; Huang, X. Transmitted Drug Resistance in Antiretroviral Therapy-Naive Persons With Acute/Early/Primary HIV Infection: A Systematic Review and Meta-Analysis. Front. Pharmacol. 2021, 12, 718763. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.-Y.; Blanco, J.L.; Jordan, M.R.; Taylor, J.; Lemey, P.; Varghese, V.; Hamers, R.L.; Bertagnolio, S.; Rinke de Wit, T.F.; Aghokeng, A.F.; et al. Geographic and Temporal Trends in the Molecular Epidemiology and Genetic Mechanisms of Transmitted HIV-1 Drug Resistance: An Individual-Patient- and Sequence-Level Meta-Analysis. PLoS Med. 2015, 12, e1001810. [Google Scholar] [CrossRef]
- Ávila-Ríos, S.; Parkin, N.; Swanstrom, R.; Paredes, R.; Shafer, R.; Ji, H.; Kantor, R. Next-Generation Sequencing for HIV Drug Resistance Testing: Laboratory, Clinical, and Implementation Considerations. Viruses 2020, 12, 617. [Google Scholar] [CrossRef]
- Lee, E.R.; Parkin, N.; Jennings, C.; Brumme, C.J.; Enns, E.; Casadellà, M.; Howison, M.; Coetzer, M.; Avila-Rios, S.; Capina, R.; et al. Performance Comparison of next Generation Sequencing Analysis Pipelines for HIV-1 Drug Resistance Testing. Sci. Rep. 2020, 10, 1634. [Google Scholar] [CrossRef]
- King, M.S.; Rode, R.; Cohen-Codar, I.; Calvez, V.; Marcelin, A.-G.; Hanna, G.J.; Kempf, D.J. Predictive Genotypic Algorithm for Virologic Response to Lopinavir-Ritonavir in Protease Inhibitor-Experienced Patients. Antimicrob. Agents Chemother. 2007, 51, 3067–3074. [Google Scholar] [CrossRef][Green Version]
- Miller, M.D.; Margot, N.; Lu, B.; Zhong, L.; Chen, S.-S.; Cheng, A.; Wulfsohn, M. Genotypic and Phenotypic Predictors of the Magnitude of Response to Tenofovir Disoproxil Fumarate Treatment in Antiretroviral-Experienced Patients. J. Infect. Dis. 2004, 189, 837–846. [Google Scholar] [CrossRef] [PubMed]
- Little, S.J.; Holte, S.; Routy, J.-P.; Daar, E.S.; Markowitz, M.; Collier, A.C.; Koup, R.A.; Mellors, J.W.; Connick, E.; Conway, B.; et al. Antiretroviral-Drug Resistance among Patients Recently Infected with HIV. N. Engl. J. Med. 2002, 347, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Bronze, M.; Steegen, K.; Wallis, C.L.; De Wolf, H.; Papathanasopoulos, M.A.; Van Houtte, M.; Stevens, W.S.; de Wit, T.R.; Stuyver, L.J. ART-A Consortium HIV-1 Phenotypic Reverse Transcriptase Inhibitor Drug Resistance Test Interpretation Is Not Dependent on the Subtype of the Virus Backbone. PLoS ONE 2012, 7, e34708. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, C.; Hardy, I.; Lalonde, R.; Trottier, B.; Tsarevsky, I.; Vézina, L.-P.; Roger, M.; Wainberg, M.; Baril, J.-G. HIV-1 Tropism Testing and Clinical Management of CCR5 Antagonists: Quebec Review and Recommendations. Can. J. Infect. Dis. Med. Microbiol. J. Can. Mal. Infect. Microbiol. Med. 2013, 24, 202–208. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rosen, O.; Sharon, M.; Quadt-Akabayov, S.R.; Anglister, J. Molecular Switch for Alternative Conformations of the HIV-1 V3 Region: Implications for Phenotype Conversion. Proc. Natl. Acad. Sci. USA 2006, 103, 13950–13955. [Google Scholar] [CrossRef]
- Joseph, S.B.; Swanstrom, R. The Evolution of HIV-1 Entry Phenotypes as a Guide to Changing Target Cells. J. Leukoc. Biol. 2018, 103, 421–431. [Google Scholar] [CrossRef]
- Chandramouli, B.; Chillemi, G.; Abbate, I.; Capobianchi, M.R.; Rozera, G.; Desideri, A. Importance of V3 Loop Flexibility and Net Charge in the Context of Co-Receptor Recognition. A Molecular Dynamics Study on HIV Gp120. J. Biomol. Struct. Dyn. 2012, 29, 879–891. [Google Scholar] [CrossRef]
- Shen, H.-S.; Yin, J.; Leng, F.; Teng, R.-F.; Xu, C.; Xia, X.-Y.; Pan, X.-M. HIV Coreceptor Tropism Determination and Mutational Pattern Identification. Sci. Rep. 2016, 6, 21280. [Google Scholar] [CrossRef]
- Arif, M.S.; Hunter, J.; Léda, A.R.; Zukurov, J.P.L.; Samer, S.; Camargo, M.; Galinskas, J.; Kallás, E.G.; Komninakis, S.V.; Janini, L.M.; et al. Pace of Coreceptor Tropism Switch in HIV-1-Infected Individuals after Recent Infection. J. Virol. 2017, 91, e00793-17. [Google Scholar] [CrossRef]
- Regoes, R.R.; Bonhoeffer, S. The HIV Coreceptor Switch: A Population Dynamical Perspective. Trends Microbiol. 2005, 13, 269–277. [Google Scholar] [CrossRef]
- Connell, B.J.; Hermans, L.E.; Wensing, A.M.J.; Schellens, I.; Schipper, P.J.; van Ham, P.M.; de Jong, D.T.C.M.; Otto, S.; Mathe, T.; Moraba, R.; et al. Immune Activation Correlates with and Predicts CXCR4 Co-Receptor Tropism Switch in HIV-1 Infection. Sci. Rep. 2020, 10, 15866. [Google Scholar] [CrossRef] [PubMed]
- Cayota, A.; Vuillier, F.; Scott-Algara, D.; Dighiero, G. Preferential Replication of HIV-1 in Memory CD4+ Subpopulation. Lancet Lond. Engl. 1990, 336, 941. [Google Scholar] [CrossRef] [PubMed]
- Riley, J.L.; Levine, B.L.; Craighead, N.; Francomano, T.; Kim, D.; Carroll, R.G.; June, C.H. Naive and Memory CD4 T Cells Differ in Their Susceptibilities to Human Immunodeficiency Virus Type 1 Infection Following CD28 Costimulation: Implicatip6s for Transmission and Pathogenesis. J. Virol. 1998, 72, 8273–8280. [Google Scholar] [CrossRef] [PubMed]
- Cornelissen, M.; Mulder-Kampinga, G.; Veenstra, J.; Zorgdrager, F.; Kuiken, C.; Hartman, S.; Dekker, J.; van der Hoek, L.; Sol, C.; Coutinho, R. Syncytium-Inducing (SI) Phenotype Suppression at Seroconversion after Intramuscular Inoculation of a Non-Syncytium-Inducing/SI Phenotypically Mixed Human Immunodeficiency Virus Population. J. Virol. 1995, 69, 1810–1818. [Google Scholar] [CrossRef] [PubMed]
- Groenink, M.; Moore, J.P.; Broersen, S.; Schuitemaker, H. Equal Levels of Gp120 Retention and Neutralization Resistance of Phenotypically Distinct Primary Human Immunodeficiency Virus Type 1 Variants upon Soluble CD4 Treatment. J. Virol. 1995, 69, 523–527. [Google Scholar] [CrossRef]
- Liu, R.; Paxton, W.A.; Choe, S.; Ceradini, D.; Martin, S.R.; Horuk, R.; MacDonald, M.E.; Stuhlmann, H.; Koup, R.A.; Landau, N.R. Homozygous Defect in HIV-1 Coreceptor Accounts for Resistance of Some Multiply-Exposed Individuals to HIV-1 Infection. Cell 1996, 86, 367–377. [Google Scholar] [CrossRef]
- Michael, N.L.; Louie, L.G.; Rohrbaugh, A.L.; Schultz, K.A.; Dayhoff, D.E.; Wang, C.E.; Sheppard, H.W. The Role of CCR5 and CCR2 Polymorphisms in HIV-1 Transmission and Disease Progression. Nat. Med. 1997, 3, 1160–1162. [Google Scholar] [CrossRef]
- Brown, T.R. I Am the Berlin Patient: A Personal Reflection. AIDS Res. Hum. Retroviruses 2015, 31, 2–3. [Google Scholar] [CrossRef]
- McGovern, R.A.; Symons, J.; Poon, A.F.Y.; Harrigan, P.R.; van Lelyveld, S.F.L.; Hoepelman, A.I.M.; van Ham, P.M.; Dong, W.; Wensing, A.M.J.; Nijhuis, M. Maraviroc Treatment in Non-R5-HIV-1-Infected Patients Results in the Selection of Extreme CXCR4-Using Variants with Limited Effect on the Total Viral Setpoint. J. Antimicrob. Chemother. 2013, 68, 2007–2014. [Google Scholar] [CrossRef]
- Tsibris, A.M.N.; Korber, B.; Arnaout, R.; Russ, C.; Lo, C.-C.; Leitner, T.; Gaschen, B.; Theiler, J.; Paredes, R.; Su, Z.; et al. Quantitative Deep Sequencing Reveals Dynamic HIV-1 Escape and Large Population Shifts during CCR5 Antagonist Therapy in Vivo. PLoS ONE 2009, 4, e5683. [Google Scholar] [CrossRef]
- Westby, M.; Lewis, M.; Whitcomb, J.; Youle, M.; Pozniak, A.L.; James, I.T.; Jenkins, T.M.; Perros, M.; van der Ryst, E. Emergence of CXCR4-Using Human Immunodeficiency Virus Type 1 (HIV-1) Variants in a Minority of HIV-1-Infected Patients Following Treatment with the CCR5 Antagonist Maraviroc Is from a Pretreatment CXCR4-Using Virus Reservoir. J. Virol. 2006, 80, 4909–4920. [Google Scholar] [CrossRef] [PubMed]
- Nankya, I.L.; Tebit, D.M.; Abraha, A.; Kyeyune, F.; Gibson, R.; Jegede, O.; Nickel, G.; Arts, E.J. Defining the Fitness of HIV-1 Isolates with Dual/Mixed Co-Receptor Usage. AIDS Res. Ther. 2015, 12, 34. [Google Scholar] [CrossRef]
- Gorman, J.; Chuang, G.-Y.; Lai, Y.-T.; Shen, C.-H.; Boyington, J.C.; Druz, A.; Geng, H.; Louder, M.K.; McKee, K.; Rawi, R.; et al. Structure of Super-Potent Antibody CAP256-VRC26.25 in Complex with HIV-1 Envelope Reveals a Combined Mode of Trimer-Apex Recognition. Cell Rep. 2020, 31, 107488. [Google Scholar] [CrossRef] [PubMed]
- Rolland, M.; Edlefsen, P.T.; Larsen, B.B.; Tovanabutra, S.; Sanders-Buell, E.; Hertz, T.; deCamp, A.C.; Carrico, C.; Menis, S.; Magaret, C.A.; et al. Increased HIV-1 Vaccine Efficacy against Viruses with Genetic Signatures in Env V2. Nature 2012, 490, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.; Henrick, K.; Nakamura, H. Announcing the Worldwide Protein Data Bank. Nat. Struct. Biol. 2003, 10, 980. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Sehnal, D.; Bittrich, S.; Deshpande, M.; Svobodová, R.; Berka, K.; Bazgier, V.; Velankar, S.; Burley, S.K.; Koča, J.; Rose, A.S. Mol* Viewer: Modern Web App for 3D Visualization and Analysis of Large Biomolecular Structures. Nucleic Acids Res. 2021, 49, W431–W437. [Google Scholar] [CrossRef]
- Ovchinnikov, V.; Louveau, J.E.; Barton, J.P.; Karplus, M.; Chakraborty, A.K. Role of Framework Mutations and Antibody Flexibility in the Evolution of Broadly Neutralizing Antibodies. eLife 2018, 7, e33038. [Google Scholar] [CrossRef]
- Liu, Y.; Cao, W.; Sun, M.; Li, T. Broadly Neutralizing Antibodies for HIV-1: Efficacies, Challenges and Opportunities. Emerg. Microbes Infect. 2020, 9, 194–206. [Google Scholar] [CrossRef]
- Edupuganti, S.; Mgodi, N.; Karuna, S.T.; Andrew, P.; Rudnicki, E.; Kochar, N.; deCamp, A.; De La Grecca, R.; Anderson, M.; Karg, C.; et al. Feasibility and Successful Enrollment in a Proof-of-Concept HIV Prevention Trial of VRC01, a Broadly Neutralizing HIV-1 Monoclonal Antibody. J. Acquir. Immune Defic. Syndr. 2021, 87, 671–679. [Google Scholar] [CrossRef]
- Rerks-Ngarm, S.; Pitisuttithum, P.; Nitayaphan, S.; Kaewkungwal, J.; Chiu, J.; Paris, R.; Premsri, N.; Namwat, C.; de Souza, M.; Adams, E.; et al. Vaccination with ALVAC and AIDSVAX to Prevent HIV-1 Infection in Thailand. N. Engl. J. Med. 2009, 361, 2209–2220. [Google Scholar] [CrossRef] [PubMed]
- Buchbinder, S.P.; Mehrotra, D.V.; Duerr, A.; Fitzgerald, D.W.; Mogg, R.; Li, D.; Gilbert, P.B.; Lama, J.R.; Marmor, M.; Del Rio, C.; et al. Efficacy Assessment of a Cell-Mediated Immunity HIV-1 Vaccine (the Step Study): A Double-Blind, Randomised, Placebo-Controlled, Test-of-Concept Trial. Lancet 2008, 372, 1881–1893. [Google Scholar] [CrossRef] [PubMed]
- Gray, G.; Buchbinder, S.; Duerr, A. Overview of STEP and Phambili Trial Results: Two Phase IIb Test-of-Concept Studies Investigating the Efficacy of MRK Adenovirus Type 5 Gag/Pol/Nef Subtype B HIV Vaccine. Curr. Opin. HIV AIDS 2010, 5, 357–361. [Google Scholar] [CrossRef] [PubMed]
- Gray, G.E.; Allen, M.; Moodie, Z.; Churchyard, G.; Bekker, L.-G.; Nchabeleng, M.; Mlisana, K.; Metch, B.; de Bruyn, G.; Latka, M.H.; et al. Safety and Efficacy of the HVTN 503/Phambili Study of a Clade-B-Based HIV-1 Vaccine in South Africa: A Double-Blind, Randomised, Placebo-Controlled Test-of-Concept Phase 2b Study. Lancet Infect. Dis. 2011, 11, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Hammer, S.M.; Sobieszczyk, M.E.; Janes, H.; Karuna, S.T.; Mulligan, M.J.; Grove, D.; Koblin, B.A.; Buchbinder, S.P.; Keefer, M.C.; Tomaras, G.D.; et al. Efficacy Trial of a DNA/RAd5 HIV-1 Preventive Vaccine. N. Engl. J. Med. 2013, 369, 2083–2092. [Google Scholar] [CrossRef] [PubMed]
- Laher, F.; Moodie, Z.; Cohen, K.W.; Grunenberg, N.; Bekker, L.-G.; Allen, M.; Frahm, N.; Yates, N.L.; Morris, L.; Malahleha, M.; et al. Safety and Immune Responses after a 12-Month Booster in Healthy HIV-Uninfected Adults in HVTN 100 in South Africa: A Randomized Double-Blind Placebo-Controlled Trial of ALVAC-HIV (VCP2438) and Bivalent Subtype C Gp120/MF59 Vaccines. PLoS Med. 2020, 17, e1003038. [Google Scholar] [CrossRef]
- Gray, G.E.; Bekker, L.-G.; Laher, F.; Malahleha, M.; Allen, M.; Moodie, Z.; Grunenberg, N.; Huang, Y.; Grove, D.; Prigmore, B.; et al. Vaccine Efficacy of ALVAC-HIV and Bivalent Subtype C Gp120-MF59 in Adults. N. Engl. J. Med. 2021, 384, 1089–1100. [Google Scholar] [CrossRef]
- Lee, J.H.; Crotty, S. HIV Vaccinology: 2021 Update. Semin. Immunol. 2021, 51, 101470. [Google Scholar] [CrossRef]
- Mdluli, T.; Jian, N.; Slike, B.; Paquin-Proulx, D.; Donofrio, G.; Alrubayyi, A.; Gift, S.; Grande, R.; Bryson, M.; Lee, A.; et al. RV144 HIV-1 Vaccination Impacts Post-Infection Antibody Responses. PLoS Pathog. 2020, 16, e1009101. [Google Scholar] [CrossRef]
- Easterhoff, D.; Pollara, J.; Luo, K.; Janus, B.; Gohain, N.; Williams, L.D.; Tay, M.Z.; Monroe, A.; Peachman, K.; Choe, M.; et al. HIV Vaccine Delayed Boosting Increases Env Variable Region 2-Specific Antibody Effector Functions. JCI Insight 2020, 5, 131437. [Google Scholar] [CrossRef]
- Sekaly, R.-P. The Failed HIV Merck Vaccine Study: A Step Back or a Launching Point for Future Vaccine Development? J. Exp. Med. 2008, 205, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Altfeld, M.; Goulder, P.J. The STEP Study Provides a Hint That Vaccine Induction of the Right CD8+ T Cell Responses Can Facilitate Immune Control of HIV. J. Infect. Dis. 2011, 203, 753–755. [Google Scholar] [CrossRef] [PubMed]
- Ko, E.-J.; Kang, S.-M. Immunology and Efficacy of MF59-Adjuvanted Vaccines. Hum. Vaccines Immunother. 2018, 14, 3041–3045. [Google Scholar] [CrossRef] [PubMed]
- Bloom, K.; van den Berg, F.; Arbuthnot, P. Self-Amplifying RNA Vaccines for Infectious Diseases. Gene Ther. 2021, 28, 117–129. [Google Scholar] [CrossRef]
- Mu, Z.; Haynes, B.F.; Cain, D.W. HIV MRNA Vaccines-Progress and Future Paths. Vaccines 2021, 9, 134. [Google Scholar] [CrossRef]
- Bogers, W.M.; Oostermeijer, H.; Mooij, P.; Koopman, G.; Verschoor, E.J.; Davis, D.; Ulmer, J.B.; Brito, L.A.; Cu, Y.; Banerjee, K.; et al. Potent Immune Responses in Rhesus Macaques Induced by Nonviral Delivery of a Self-Amplifying RNA Vaccine Expressing HIV Type 1 Envelope with a Cationic Nanoemulsion. J. Infect. Dis. 2015, 211, 947–955. [Google Scholar] [CrossRef]
- Barbier, A.J.; Jiang, A.Y.; Zhang, P.; Wooster, R.; Anderson, D.G. The Clinical Progress of MRNA Vaccines and Immunotherapies. Nat. Biotechnol. 2022, 40, 840–854. [Google Scholar] [CrossRef]
- Fogel, J.M.; Sivay, M.V.; Cummings, V.; Wilson, E.A.; Hart, S.; Gamble, T.; Laeyendecker, O.; Fernandez, R.E.; Del Rio, C.; Batey, D.S.; et al. HIV Drug Resistance in a Cohort of HIV-Infected MSM in the United States. AIDS 2020, 34, 91–101. [Google Scholar] [CrossRef]
- Tanner, M.R.; Miele, P.; Carter, W.; Valentine, S.S.; Dunville, R.; Kapogiannis, B.G.; Smith, D.K. Preexposure Prophylaxis for Prevention of HIV Acquisition Among Adolescents: Clinical Considerations, 2020. MMWR Recomm. Rep. Morb. Mortal. Wkly. Rep. Recomm. Rep. 2020, 69, 1–12. [Google Scholar] [CrossRef]
- Camp, C.; Saberi, P. Facilitators and Barriers of 2-1-1 HIV Pre-Exposure Prophylaxis. PLoS ONE 2021, 16, e0251917. [Google Scholar] [CrossRef]
- Sewell, W.C.; Powell, V.E.; Mayer, K.H.; Ochoa, A.; Krakower, D.S.; Marcus, J.L. Nondaily Use of HIV Preexposure Prophylaxis in a Large Online Survey of Primarily Men Who Have Sex With Men in the United States. J. Acquir. Immune Defic. Syndr. 1999 2020, 84, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Beymer, M.R.; Holloway, I.W.; Pulsipher, C.; Landovitz, R.J. Current and Future PrEP Medications and Modalities: On-Demand, Injectables, and Topicals. Curr. HIV/AIDS Rep. 2019, 16, 349–358. [Google Scholar] [CrossRef]
- Rizzardini, G.; Overton, E.T.; Orkin, C.; Swindells, S.; Arasteh, K.; Górgolas Hernández-Mora, M.; Pokrovsky, V.; Girard, P.-M.; Oka, S.; Andrade-Villanueva, J.F.; et al. Long-Acting Injectable Cabotegravir + Rilpivirine for HIV Maintenance Therapy: Week 48 Pooled Analysis of Phase 3 ATLAS and FLAIR Trials. J. Acquir. Immune Defic. Syndr. 2020, 85, 498–506. [Google Scholar] [CrossRef] [PubMed]
- Roland, M.E.; Neilands, T.B.; Krone, M.R.; Katz, M.H.; Franses, K.; Grant, R.M.; Busch, M.P.; Hecht, F.M.; Shacklett, B.L.; Kahn, J.O.; et al. Seroconversion Following Nonoccupational Postexposure Prophylaxis against HIV. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2005, 41, 1507–1513. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Q.; Guo, D.; Chen, S. Application of CRISPR/Cas9-Based Gene Editing in HIV-1/AIDS Therapy. Front. Cell Infect. Microbiol. 2019, 9, 69. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, W.; Cui, Y.C.; Pan, Q.; Zhu, W.; Gendron, P.; Guo, F.; Cen, S.; Witcher, M.; Liang, C. HIV-1 Employs Multiple Mechanisms To Resist Cas9/Single Guide RNA Targeting the Viral Primer Binding Site. J. Virol. 2018, 92, e01135-18. [Google Scholar] [CrossRef]
- Yoder, K.E.; Bundschuh, R. Host Double Strand Break Repair Generates HIV-1 Strains Resistant to CRISPR/Cas9. Sci. Rep. 2016, 6, 29530. [Google Scholar] [CrossRef]
- Yin, L.; Zhao, F.; Sun, H.; Wang, Z.; Huang, Y.; Zhu, W.; Xu, F.; Mei, S.; Liu, X.; Zhang, D.; et al. CRISPR-Cas13a Inhibits HIV-1 Infection. Mol. Ther. Nucleic Acids 2020, 21, 147–155. [Google Scholar] [CrossRef]
- Ambrose, Z.; Aiken, C. HIV-1 Uncoating: Connection to Nuclear Entry and Regulation by Host Proteins. Virology 2014, 454–455, 371–379. [Google Scholar] [CrossRef]
- Chen, B. Molecular Mechanism of HIV-1 Entry. Trends Microbiol. 2019, 27, 878–891. [Google Scholar] [CrossRef]
- Wilen, C.B.; Tilton, J.C.; Doms, R.W. HIV: Cell Binding and Entry. Cold Spring Harb. Perspect. Med. 2012, 2, a006866. [Google Scholar] [CrossRef] [PubMed]
- Dingens, A.S.; Arenz, D.; Overbaugh, J.; Bloom, J.D. Massively Parallel Profiling of HIV-1 Resistance to the Fusion Inhibitor Enfuvirtide. Viruses 2019, 11, 439. [Google Scholar] [CrossRef] [PubMed]
- Pu, J.; Wang, Q.; Xu, W.; Lu, L.; Jiang, S. Development of Protein- and Peptide-Based HIV Entry Inhibitors Targeting Gp120 or Gp41. Viruses 2019, 11, 705. [Google Scholar] [CrossRef] [PubMed]
- Woollard, S.M.; Kanmogne, G.D. Maraviroc: A Review of Its Use in HIV Infection and Beyond. Drug Des. Devel. Ther. 2015, 9, 5447–5468. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.G.; Guo, J.; Wu, Y. Chemokine Receptor CCR5 Antagonist Maraviroc: Medicinal Chemistry and Clinical Applications. Curr. Top. Med. Chem. 2014, 14, 1504–1514. [Google Scholar] [CrossRef]
- Lai, Y.-T. Small Molecule HIV-1 Attachment Inhibitors: Discovery, Mode of Action and Structural Basis of Inhibition. Viruses 2021, 13, 843. [Google Scholar] [CrossRef]
- Yuan, C.; Wang, J.-Y.; Zhao, H.-J.; Li, Y.; Li, D.; Ling, H.; Zhuang, M. Mutations of Glu560 within HIV-1 Envelope Glycoprotein N-Terminal Heptad Repeat Region Contribute to Resistance to Peptide Inhibitors of Virus Entry. Retrovirology 2019, 16, 36. [Google Scholar] [CrossRef]
- Aquaro, S.; D’Arrigo, R.; Svicher, V.; Perri, G.D.; Caputo, S.L.; Visco-Comandini, U.; Santoro, M.; Bertoli, A.; Mazzotta, F.; Bonora, S.; et al. Specific Mutations in HIV-1 Gp41 Are Associated with Immunological Success in HIV-1-Infected Patients Receiving Enfuvirtide Treatment. J. Antimicrob. Chemother. 2006, 58, 714–722. [Google Scholar] [CrossRef]
- Rose, R.; Gartland, M.; Li, Z.; Zhou, N.; Cockett, M.; Beloor, J.; Lataillade, M.; Ackerman, P.; Krystal, M. Clinical Evidence for a Lack of Cross-Resistance between Temsavir and Ibalizumab or Maraviroc. AIDS Lond. Engl. 2022, 36, 11–18. [Google Scholar] [CrossRef]
- Ratcliff, A.N.; Shi, W.; Arts, E.J. HIV-1 Resistance to Maraviroc Conferred by a CD4 Binding Site Mutation in the Envelope Glycoprotein Gp120. J. Virol. 2013, 87, 923–934. [Google Scholar] [CrossRef]
- Yuan, Y.; Yokoyama, M.; Maeda, Y.; Terasawa, H.; Harada, S.; Sato, H.; Yusa, K. Structure and Dynamics of the Gp120 V3 Loop That Confers Noncompetitive Resistance in R5 HIV-1(JR-FL) to Maraviroc. PLoS ONE 2013, 8, e65115. [Google Scholar] [CrossRef]
- Alessandri-Gradt, E.; Charpentier, C.; Leoz, M.; Mourez, T.; Descamps, D.; Plantier, J.-C. Impact of Natural Polymorphisms of HIV-1 Non-Group M on Genotypic Susceptibility to the Attachment Inhibitor Fostemsavir. J. Antimicrob. Chemother. 2018, 73, 2716–2720. [Google Scholar] [CrossRef] [PubMed]
- Bouba, Y.; Berno, G.; Fabeni, L.; Carioti, L.; Salpini, R.; Aquaro, S.; Svicher, V.; Perno, C.F.; Ceccherini-Silberstein, F.; Santoro, M.M. Identification of Gp120 Polymorphisms in HIV-1 B Subtype Potentially Associated with Resistance to Fostemsavir. J. Antimicrob. Chemother. 2020, 75, 1778–1786. [Google Scholar] [CrossRef] [PubMed]
- Lepore, L.; Fabrizio, C.; Bavaro, D.F.; Milano, E.; Volpe, A.; Lagioia, A.; Angarano, G.; Saracino, A.; Monno, L. Gp120 Substitutions at Positions Associated with Resistance to Fostemsavir in Treatment-Naive HIV-1-Positive Individuals. J. Antimicrob. Chemother. 2020, 75, 1580–1587. [Google Scholar] [CrossRef]
- Pace, C.S.; Fordyce, M.W.; Franco, D.; Kao, C.-Y.; Seaman, M.S.; Ho, D.D. Anti-CD4 Monoclonal Antibody Ibalizumab Exhibits Breadth and Potency against HIV-1, with Natural Resistance Mediated by the Loss of a V5 Glycan in Envelope. J. Acquir. Immune Defic. Syndr. 2013, 62, 1–9. [Google Scholar] [CrossRef]
- Shaik, M.M.; Peng, H.; Lu, J.; Rits-Volloch, S.; Xu, C.; Liao, M.; Chen, B. Structural Basis of Coreceptor Recognition by HIV-1 Envelope Spike. Nature 2019, 565, 318–323. [Google Scholar] [CrossRef]
- Le Grice, S.F.; Naas, T.; Wohlgensinger, B.; Schatz, O. Subunit-Selective Mutagenesis Indicates Minimal Polymerase Activity in Heterodimer-Associated P51 HIV-1 Reverse Transcriptase. EMBO J. 1991, 10, 3905–3911. [Google Scholar] [CrossRef]
- Nowotny, M.; Gaidamakov, S.A.; Crouch, R.J.; Yang, W. Crystal Structures of RNase H Bound to an RNA/DNA Hybrid: Substrate Specificity and Metal-Dependent Catalysis. Cell 2005, 121, 1005–1016. [Google Scholar] [CrossRef]
- Basu, V.P.; Song, M.; Gao, L.; Rigby, S.T.; Hanson, M.N.; Bambara, R.A. Strand Transfer Events during HIV-1 Reverse Transcription. Virus Res. 2008, 134, 19–38. [Google Scholar] [CrossRef]
- Arhel, N. Revisiting HIV-1 Uncoating. Retrovirology 2010, 7, 96. [Google Scholar] [CrossRef]
- Dyda, F.; Hickman, A.B.; Jenkins, T.M.; Engelman, A.; Craigie, R.; Davies, D.R. Crystal Structure of the Catalytic Domain of HIV-1 Integrase: Similarity to Other Polynucleotidyl Transferases. Science 1994, 266, 1981–1986. [Google Scholar] [CrossRef] [PubMed]
- Chiu, T.K.; Davies, D.R. Structure and Function of HIV-1 Integrase. Curr. Top. Med. Chem. 2004, 4, 965–977. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.; Zheng, R.; Caffrey, M.; Craigie, R.; Clore, G.M.; Gronenborn, A.M. Solution Structure of the N-Terminal Zinc Binding Domain of HIV-1 Integrase. Nat. Struct. Biol. 1997, 4, 567–577. [Google Scholar] [CrossRef]
- Lodi, P.J.; Ernst, J.A.; Kuszewski, J.; Hickman, A.B.; Engelman, A.; Craigie, R.; Clore, G.M.; Gronenborn, A.M. Solution Structure of the DNA Binding Domain of HIV-1 Integrase. Biochemistry 1995, 34, 9826–9833. [Google Scholar] [CrossRef] [PubMed]
- Hare, S.; Gupta, S.S.; Valkov, E.; Engelman, A.; Cherepanov, P. Retroviral Intasome Assembly and Inhibition of DNA Strand Transfer. Nature 2010, 464, 232–236. [Google Scholar] [CrossRef]
- Christ, F.; Voet, A.; Marchand, A.; Nicolet, S.; Desimmie, B.A.; Marchand, D.; Bardiot, D.; Van der Veken, N.J.; Van Remoortel, B.; Strelkov, S.V.; et al. Rational Design of Small-Molecule Inhibitors of the LEDGF/P75-Integrase Interaction and HIV Replication. Nat. Chem. Biol. 2010, 6, 442–448. [Google Scholar] [CrossRef]
- De Rijck, J.; Vandekerckhove, L.; Christ, F.; Debyser, Z. Lentiviral Nuclear Import: A Complex Interplay between Virus and Host. BioEssays News Rev. Mol. Cell Dev. Biol. 2007, 29, 441–451. [Google Scholar] [CrossRef]
- Stremlau, M.; Perron, M.; Lee, M.; Li, Y.; Song, B.; Javanbakht, H.; Diaz-Griffero, F.; Anderson, D.J.; Sundquist, W.I.; Sodroski, J. Specific Recognition and Accelerated Uncoating of Retroviral Capsids by the TRIM5alpha Restriction Factor. Proc. Natl. Acad. Sci. USA 2006, 103, 5514–5519. [Google Scholar] [CrossRef]
- Van Maele, B.; Debyser, Z. HIV-1 Integration: An Interplay between HIV-1 Integrase, Cellular and Viral Proteins. AIDS Rev. 2005, 7, 26–43. [Google Scholar]
- Haffar, O.K.; Popov, S.; Dubrovsky, L.; Agostini, I.; Tang, H.; Pushkarsky, T.; Nadler, S.G.; Bukrinsky, M. Two Nuclear Localization Signals in the HIV-1 Matrix Protein Regulate Nuclear Import of the HIV-1 Pre-Integration Complex. J. Mol. Biol. 2000, 299, 359–368. [Google Scholar] [CrossRef]
- Craigie, R.; Bushman, F.D. HIV DNA Integration. Cold Spring Harb. Perspect. Med. 2012, 2, a006890. [Google Scholar] [CrossRef] [PubMed]
- Hazuda, D.J. HIV Integrase as a Target for Antiretroviral Therapy. Curr. Opin. HIV AIDS 2012, 7, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Johnson, A.A.; Marchand, C. Integrase Inhibitors to Treat HIV/AIDS. Nat. Rev. Drug Discov. 2005, 4, 236–248. [Google Scholar] [CrossRef] [PubMed]
- Fransen, S.; Gupta, S.; Danovich, R.; Hazuda, D.; Miller, M.; Witmer, M.; Petropoulos, C.J.; Huang, W. Loss of Raltegravir Susceptibility by Human Immunodeficiency Virus Type 1 Is Conferred via Multiple Nonoverlapping Genetic Pathways. J. Virol. 2009, 83, 11440–11446. [Google Scholar] [CrossRef] [PubMed]
- Goethals, O.; Clayton, R.; Van Ginderen, M.; Vereycken, I.; Wagemans, E.; Geluykens, P.; Dockx, K.; Strijbos, R.; Smits, V.; Vos, A.; et al. Resistance Mutations in Human Immunodeficiency Virus Type 1 Integrase Selected with Elvitegravir Confer Reduced Susceptibility to a Wide Range of Integrase Inhibitors. J. Virol. 2008, 82, 10366–10374. [Google Scholar] [CrossRef]
- Smith, S.J.; Zhao, X.Z.; Burke, T.R.; Hughes, S.H. Efficacies of Cabotegravir and Bictegravir against Drug-Resistant HIV-1 Integrase Mutants. Retrovirology 2018, 15, 37. [Google Scholar] [CrossRef]
- Yoshinaga, T.; Seki, T.; Miki, S.; Miyamoto, T.; Suyama-Kagitani, A.; Kawauchi-Miki, S.; Kobayashi, M.; Sato, A.; Stewart, E.; Underwood, M.; et al. Novel Secondary Mutations C56S and G149A Confer Resistance to HIV-1 Integrase Strand Transfer Inhibitors. Antiviral Res. 2018, 152, 1–9. [Google Scholar] [CrossRef]
- Cook, N.J.; Li, W.; Berta, D.; Badaoui, M.; Ballandras-Colas, A.; Nans, A.; Kotecha, A.; Rosta, E.; Engelman, A.N.; Cherepanov, P. Structural Basis of Second-Generation HIV Integrase Inhibitor Action and Viral Resistance. Science 2020, 367, 806–810. [Google Scholar] [CrossRef]
- Smith, S.J.; Zhao, X.Z.; Passos, D.O.; Lyumkis, D.; Burke, T.R.; Hughes, S.H. HIV-1 Integrase Inhibitors That Are Active against Drug-Resistant Integrase Mutants. Antimicrob. Agents Chemother. 2020, 64, e00611-20. [Google Scholar] [CrossRef]
- Krishnan, L.; Li, X.; Naraharisetty, H.L.; Hare, S.; Cherepanov, P.; Engelman, A. Structure-Based Modeling of the Functional HIV-1 Intasome and Its Inhibition. Proc. Natl. Acad. Sci. USA 2010, 107, 15910–15915. [Google Scholar] [CrossRef]
- Espeseth, A.S.; Felock, P.; Wolfe, A.; Witmer, M.; Grobler, J.; Anthony, N.; Egbertson, M.; Melamed, J.Y.; Young, S.; Hamill, T.; et al. HIV-1 Integrase Inhibitors That Compete with the Target DNA Substrate Define a Unique Strand Transfer Conformation for Integrase. Proc. Natl. Acad. Sci. USA 2000, 97, 11244–11249. [Google Scholar] [CrossRef]
- Malet, I.; Subra, F.; Charpentier, C.; Collin, G.; Descamps, D.; Calvez, V.; Marcelin, A.-G.; Delelis, O. Mutations Located Outside the Integrase Gene Can Confer Resistance to HIV-1 Integrase Strand Transfer Inhibitors. mBio 2017, 8, e00922-17. [Google Scholar] [CrossRef]
- Wijting, I.E.A.; Lungu, C.; Rijnders, B.J.A.; van der Ende, M.E.; Pham, H.T.; Mesplede, T.; Pas, S.D.; Voermans, J.J.C.; Schuurman, R.; van de Vijver, D.A.M.C.; et al. HIV-1 Resistance Dynamics in Patients With Virologic Failure to Dolutegravir Maintenance Monotherapy. J. Infect. Dis. 2018, 218, 688–697. [Google Scholar] [CrossRef]
- Freed, E.O. HIV-1 Assembly, Release and Maturation. Nat. Rev. Microbiol. 2015, 13, 484–496. [Google Scholar] [CrossRef]
- Kleinpeter, A.B.; Freed, E.O. HIV-1 Maturation: Lessons Learned from Inhibitors. Viruses 2020, 12, 940. [Google Scholar] [CrossRef]
- Waki, K.; Durell, S.R.; Soheilian, F.; Nagashima, K.; Butler, S.L.; Freed, E.O. Structural and Functional Insights into the HIV-1 Maturation Inhibitor Binding Pocket. PLoS Pathog. 2012, 8, e1002997. [Google Scholar] [CrossRef]
- Castro-Gonzalez, S.; Shi, Y.; Colomer-Lluch, M.; Song, Y.; Mowery, K.; Almodovar, S.; Bansal, A.; Kirchhoff, F.; Sparrer, K.; Liang, C.; et al. HIV-1 Nef Counteracts Autophagy Restriction by Enhancing the Association between BECN1 and Its Inhibitor BCL2 in a PRKN-Dependent Manner. Autophagy 2021, 17, 553–577. [Google Scholar] [CrossRef]
- Frankel, A.D.; Young, J.A. HIV-1: Fifteen Proteins and an RNA. Annu. Rev. Biochem. 1998, 67, 1–25. [Google Scholar] [CrossRef]
- Welker, R.; Kottler, H.; Kalbitzer, H.R.; Kräusslich, H.G. Human Immunodeficiency Virus Type 1 Nef Protein Is Incorporated into Virus Particles and Specifically Cleaved by the Viral Proteinase. Virology 1996, 219, 228–236. [Google Scholar] [CrossRef]
- Alvarez, E.; Castelló, A.; Menéndez-Arias, L.; Carrasco, L. HIV Protease Cleaves Poly(A)-Binding Protein. Biochem. J. 2006, 396, 219–226. [Google Scholar] [CrossRef]
- Tabler, C.O.; Wegman, S.J.; Chen, J.; Shroff, H.; Alhusaini, N.; Tilton, J.C. The HIV-1 Viral Protease Is Activated during Assembly and Budding Prior to Particle Release. J. Virol. 2022, 96, e0219821. [Google Scholar] [CrossRef] [PubMed]
- Prabu-Jeyabalan, M.; Nalivaika, E.; Schiffer, C.A. Substrate Shape Determines Specificity of Recognition for HIV-1 Protease: Analysis of Crystal Structures of Six Substrate Complexes. Structure 2002, 10, 369–381. [Google Scholar] [CrossRef]
- Mimoto, T.; Hattori, N.; Takaku, H.; Kisanuki, S.; Fukazawa, T.; Terashima, K.; Kato, R.; Nojima, S.; Misawa, S.; Ueno, T.; et al. Structure-Activity Relationship of Orally Potent Tripeptide-Based HIV Protease Inhibitors Containing Hydroxymethylcarbonyl Isostere. Chem. Pharm. Bull. 2000, 48, 1310–1326. [Google Scholar] [CrossRef][Green Version]
- Rutenber, E.; Fauman, E.B.; Keenan, R.J.; Fong, S.; Furth, P.S.; Ortiz de Montellano, P.R.; Meng, E.; Kuntz, I.D.; DeCamp, D.L.; Salto, R. Structure of a Non-Peptide Inhibitor Complexed with HIV-1 Protease. Developing a Cycle of Structure-Based Drug Design. J. Biol. Chem. 1993, 268, 15343–15346. [Google Scholar] [CrossRef]
- Lu, S.; Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; Hurwitz, D.I.; Marchler, G.H.; Song, J.S.; et al. CDD/SPARCLE: The Conserved Domain Database in 2020. Nucleic Acids Res. 2020, 48, D265–D268. [Google Scholar] [CrossRef] [PubMed]
- Ofotokun, I.; Na, L.H.; Landovitz, R.J.; Ribaudo, H.J.; McComsey, G.A.; Godfrey, C.; Aweeka, F.; Cohn, S.E.; Sagar, M.; Kuritzkes, D.R.; et al. Comparison of the Metabolic Effects of Ritonavir-Boosted Darunavir or Atazanavir versus Raltegravir, and the Impact of Ritonavir Plasma Exposure: ACTG 5257. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2015, 60, 1842–1851. [Google Scholar] [CrossRef] [PubMed]
- Ragland, D.A.; Whitfield, T.W.; Lee, S.-K.; Swanstrom, R.; Zeldovich, K.B.; Kurt-Yilmaz, N.; Schiffer, C.A. Elucidating the Interdependence of Drug Resistance from Combinations of Mutations. J. Chem. Theory Comput. 2017, 13, 5671–5682. [Google Scholar] [CrossRef]
- Aoki, M.; Das, D.; Hayashi, H.; Aoki-Ogata, H.; Takamatsu, Y.; Ghosh, A.K.; Mitsuya, H. Mechanism of Darunavir (DRV)’s High Genetic Barrier to HIV-1 Resistance: A Key V32I Substitution in Protease Rarely Occurs, but Once It Occurs, It Predisposes HIV-1 To Develop DRV Resistance. mBio 2018, 9, e02425-17. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, Z.; Brunzelle, J.S.; Kovari, I.A.; Dewdney, T.G.; Reiter, S.J.; Kovari, L.C. The Higher Barrier of Darunavir and Tipranavir Resistance for HIV-1 Protease. Biochem. Biophys. Res. Commun. 2011, 412, 737–742. [Google Scholar] [CrossRef]
- Wlodawer, A. Rational Approach to AIDS Drug Design through Structural Biology. Annu. Rev. Med. 2002, 53, 595–614. [Google Scholar] [CrossRef]
- Paulsen, D.; Liao, Q.; Fusco, G.; St Clair, M.; Shaefer, M.; Ross, L. Genotypic and Phenotypic Cross-Resistance Patterns to Lopinavir and Amprenavir in Protease Inhibitor-Experienced Patients with HIV Viremia. AIDS Res. Hum. Retrovir. 2002, 18, 1011–1019. [Google Scholar] [CrossRef]
- Colonno, R.; Rose, R.; McLaren, C.; Thiry, A.; Parkin, N.; Friborg, J. Identification of I50L as the Signature Atazanavir (ATV)-Resistance Mutation in Treatment-Naive HIV-1-Infected Patients Receiving ATV-Containing Regimens. J. Infect. Dis. 2004, 189, 1802–1810. [Google Scholar] [CrossRef]
- Yanchunas, J.; Langley, D.R.; Tao, L.; Rose, R.E.; Friborg, J.; Colonno, R.J.; Doyle, M.L. Molecular Basis for Increased Susceptibility of Isolates with Atazanavir Resistance-Conferring Substitution I50L to Other Protease Inhibitors. Antimicrob. Agents Chemother. 2005, 49, 3825–3832. [Google Scholar] [CrossRef]
- Van Duyne, R.; Kuo, L.S.; Pham, P.; Fujii, K.; Freed, E.O. Mutations in the HIV-1 Envelope Glycoprotein Can Broadly Rescue Blocks at Multiple Steps in the Virus Replication Cycle. Proc. Natl. Acad. Sci. USA 2019, 116, 9040–9049. [Google Scholar] [CrossRef]
- Graci, J.D.; Cameron, C.E. Therapeutically Targeting RNA Viruses via Lethal Mutagenesis. Future Virol. 2008, 3, 553–566. [Google Scholar] [CrossRef]
- Hadj Hassine, I.; Ben M’hadheb, M.; Menéndez-Arias, L. Lethal Mutagenesis of RNA Viruses and Approved Drugs with Antiviral Mutagenic Activity. Viruses 2022, 14, 841. [Google Scholar] [CrossRef]
- Bai, R.; Lv, S.; Wu, H.; Dai, L. Insights into the HIV-1 Latent Reservoir and Strategies to Cure HIV-1 Infection. Dis. Markers 2022, 2022, 6952286. [Google Scholar] [CrossRef]
- Chen, J.; Zhou, T.; Zhang, Y.; Luo, S.; Chen, H.; Chen, D.; Li, C.; Li, W. The Reservoir of Latent HIV. Front. Cell Infect. Microbiol. 2022, 12, 945956. [Google Scholar] [CrossRef]
- Siliciano, J.D.; Kajdas, J.; Finzi, D.; Quinn, T.C.; Chadwick, K.; Margolick, J.B.; Kovacs, C.; Gange, S.J.; Siliciano, R.F. Long-Term Follow-up Studies Confirm the Stability of the Latent Reservoir for HIV-1 in Resting CD4+ T Cells. Nat. Med. 2003, 9, 727–728. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Class | Name | Trade Name | Use | FDA Approval |
|---|---|---|---|---|
| Fusion Inhibitor | Enfuvirtide (T20) | Fuzeon | Patients who are treatment experienced and who currently have virological failure. | 13 March 2003 |
| Attachment Inhibitor | Fostemsavir (FTR) | Rukobia | Patients who are treatment experienced, have multi-drug resistant HIV, and who currently have virological failure. | 2 July 2020 |
| Post-Attachment Inhibitors | Ibalizumab (IBA) | Trogarzo | Patients who are treatment experienced, have multi-drug resistant HIV, and who currently have virological failure. | 6 March 2018 |
| CCR5 Antagonist | Maraviroc (MVC) | Selzentry | Patients who have CCR5-tropic infection, given in combination with other medicines. | 6 August 2007 |
| Name | Significant Mutations | Effect of Mutations |
|---|---|---|
| Enfuvirtide | N126K, E137K, S138A | Reduced viral susceptibility and increased viral fusion |
| R46K/M on gp41 | Potential resistance | |
| L44M on gp41 | Major resistance (1.8 fold) | |
| V38A/E/K/M on gp41 | Increase in CD4 cell count without significant viremia | |
| Q40H/K/P/T | CD4 loss without significant viremia | |
| L45Q/M | CD4 loss without significant viremia | |
| E560K/D/G | Escape peptide inhibitors | |
| Fostemsavir | S375H/I/M/N/T in gp120 | Reduced viral susceptibility and may reduce binding of the drug |
| M426L/P in gp120 | Reduced viral susceptibility and may reduce binding of the drug | |
| M434I/K in gp120 | Reduced viral susceptibility | |
| M475I in gp120 | Reduced viral susceptibility | |
| L116P | Reduced viral susceptibility | |
| Ibalizumab | N460Q | Disrupts PNGS in V5 loop |
| N464Q | Disrupts PNGS in V5 loop | |
| Maraviroc | N425K | May impact CD4 interactions; primary resistance mutation |
| E33G, R117Q, Q290K, L/G396V, D461E | May work to stabilize N425K mutation | |
| T199K in C2 region | Enhances viral fitness | |
| I318V in V3 | Necessary for extensive replication | |
| F312W, T314A, E317D in V3 | Necessary for noncompetitive resistance | |
| I304V in V3 | Enhances viral fitness |
| Drug Class | Name | Trade Name | Use | FDA Approval |
|---|---|---|---|---|
| NRTI | Abacavir (ABC) | Ziagen | As part of initial regimen, given in combination with other medicines. Cannot have HLA-B*5701 allele. | 17 December 1998 |
| NRTI | Emtricitabine (FTC) | Emtriva | As part of initial regimen, given in combination with other medicines. | 2 July 2003 |
| NRTI | Lamivudine (3TC) | Epivir | As part of initial regimen, given in combination with other medicines. | 17 November 1995 |
| NRTI | Tenofovir (TDF) | Viread | As part of initial regimen. | 26 October 2001 |
| NRTI | Zidovudine (AZT) | Retrovir | No longer used/rarely prescribed. | 19 March 1987 |
| NNRTI | Doravirine | Pifeltro | As part of initial regimen, given in combination with other medicines. | 30 August 2018 |
| NNRTI | Efavirenz (EFV) | Sustiva | As part of initial regimen, given in combination with other medicines. | 17 September 1998 |
| NNRTI | Etravirine (ETR) | Intelence | For treatment-experienced patients, given in combination with other medicines. | 18 January 2008 |
| NNRTI | Nevirapine (NVP | Viramune | Given in combination with other medicines. | 21 June 1996 |
| NNRTI | Rilpivirine | Edurant | For treatment-naive patients, given in combination with other medicines. | 20 May 2011 |
| Mutation | Drug Inhibition | Response/Resistance | Increase Susceptibility |
|---|---|---|---|
| M184V/I | 3TC, FTC | Reduces viral replication in vitro and in vivo | AZT, d4T, TDF |
| K65R | TDF, ABC, ddl, 3TC, FTC | AZT | |
| M41L and T215Y | AZT, d4T, ABC, ddL, TDF | High resistance: AZT, d4T Low resistance: ABC, ddI, TDF | |
| D67N | AZT, d4T | ||
| K70R | AZT, d4T, TDF | Intermediate Resistance: AZT Low resistance: d4T, TDF | |
| L210W, M41L, and T215Y | AZT, d4T, ABC, ddI, TDF | High resistance: AZT, d4T Intermediate Resistance: ABC, ddI, TDF | |
| T215Y/F | AZT, d4T, ABC, ddI, TDF | Intermediate resistance: AZT, d4T Low resistance: ABC, ddI, TDF | |
| K219Q/E | AZT, d4T | ||
| K70G/Q/E/T/N/S | D4T, ABC, TDF, 3TC/FTC, | Low resistance: 3TC/FTC | AZT |
| L74V/I and M184V | ABC, ddl | AZT, TDF | |
| Y115F | ABC, TDF | ||
| G151M with either A62V, V75I, F77L, and F116Y | 3TC/FTC, TDF | Intermediate resistance: 3TC/FTC, TDF | |
| G151M | AZT, d4T, ddI, ABC, 3TC/FTC, TDF | High resistance: AZT, d4T, ddI, ABC Low resistance: 3TC/FTC, TDF | |
| T69D/N/G | Ddl, d4T | ||
| T69D/N/G and TAMS | AZT | ||
| V75T/M/A/S | D4T, ddl | ||
| N348I | AZT, NVP, EFV | Reduces rate of RNA template degradation |
| Drug Class | Name | Trade Name | Use | FDA Approval |
|---|---|---|---|---|
| INSTI | Cabotegravir (CAB) | Vocabria | For patients who will miss planned cabenuva injection, in combination with rilpivirine, or as short-term pre-exposure prophylaxis. | 22 January 2021 |
| INSTI | Dolutegravir (DTG) | Tivicay | As part of initial regimen, given in combination with other medicines. | 12 August 2013 |
| INSTI | Raltegravir (RAL) | Isentress | As part of initial regimen; post-exposure prophylaxis. | 12 October 2007 |
| Significant Mutations | Drug Inhibitions | Response/Resistance |
|---|---|---|
| T66A/I/K | BIC, DTG, EVG, RAL | High: EVG Intermediate: RAL Low: DTG |
| E92Q/G/V | BIC, DTG, EVG, RAL | High: EVG Intermediate: RAL Low: DTG |
| G118R | BIC, DTG, EVG, RAL | Intermediate: RAL, EVG, DTG Low: BIC |
| E138KAT with Q148 | BIC, DTG, EVG, RAL | High: RAL, EVG Intermediate: DTG |
| G140SAC with Q148H/R/K | BIC, DTG, EVG, RAL | High: RAL Intermediate DTG |
| Y143C/R with T97A | RAL | High: RAL |
| S147G | EVG | Intermediate: EVG |
| Q148H/R/K/N with G140S/A or E138K | BIC, DTG, EVG, RAL | High: RAL, EVG Intermediate: DTG, BIC |
| N155H/S/T/D | BIC, DTG, EVG, RAL | High: EVG Intermediate: RAL |
| R263K | BIC, DTG, EVG, RAL | Intermediate: BIC, DTG, EVG |
| Drug Class | Name | Trade Name | Use | FDA Approval |
|---|---|---|---|---|
| PI | Atazanavir (ATV) | Reyataz | Given in combination with other medicines. | 20 June 2003 |
| PI | Darunavir (DRV) | Prezista | Given in combination with other medicines. | 23 June 2006 |
| PI | Fosamprenavir | Lexiva | Given in combination with other medicines. | 20 October 2003 |
| PI | Ritonavir (RTV) | Norvir | Given in combination with other medicines. | 1 March 1996 |
| PI | Tipranavir (TPV) | Aptivus | For treatment-experienced patients, given in combination with other medicines. | 22 June 2005 |
| Mutations | Drug Inhibitions | Response/Resistance | Increased Susceptibility |
|---|---|---|---|
| D30N | NFV | High: NFV | N/A |
| V32I with I47V/A | IDV, FPV, LPV, DRV, ATV, LPV, NFV, TPV | High: LPV Intermediate: DRV | N/A |
| L33F with other PI-resistance mutations | DRV, FPV, LPV, NFV, TPV | N/A | N/A |
| M46I/L/V alone or in combination with V32I, I47V, I84V, L90M, I54V, or V82A | IDV, NFV, FPV, ATV, LPV, TPV | N/A | N/A |
| I47V/A alone or with V32I | IDV, FPV, LPV, DRV, NFV, TPV | High: LPV, FPV Low/intermediate: IDV, DRV, NFV, TPV | SQV |
| G48V/M/A/S/T/Q | SQV, IDV, LPV, NFV, ATV | High: SQV Intermediate: ATV Low: NFV, IDV, LPV | N/A |
| I50V/L | FPV, LPV, DRV, ATV | High: ATV | TPV |
| I54V/T/A/L/M | IDV, LPV, FPV, DRV, ATV, NFV, SQV, TPV | N/A | N/A |
| L76V | IDV, LPV, DRV, FPV | N/A | ATV, SQV, TPV |
| V82A/T/S/F/L/M/C | IDV, LPV, ATV, NFV, SQV, FPV, TPV, DRV | N/A | N/A |
| I84V/A/C | IDV, LPV, DRV, SQV, TPV, FPV, NFV, ATV | High: IDV, LPV, DRV, SQV, TPV, FPV, NFV, ATV | N/A |
| N88DS | NFV, ATV, IDV, SQV | N/A | FPV |
| L90M | SQV, NFV, IDV, LPV, ATV, FPV | N/A | N/A |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Johnson, M.M.; Jones, C.E.; Clark, D.N. The Effect of Treatment-Associated Mutations on HIV Replication and Transmission Cycles. Viruses 2023, 15, 107. https://doi.org/10.3390/v15010107
Johnson MM, Jones CE, Clark DN. The Effect of Treatment-Associated Mutations on HIV Replication and Transmission Cycles. Viruses. 2023; 15(1):107. https://doi.org/10.3390/v15010107
Chicago/Turabian StyleJohnson, Madison M., Carson Everest Jones, and Daniel N. Clark. 2023. "The Effect of Treatment-Associated Mutations on HIV Replication and Transmission Cycles" Viruses 15, no. 1: 107. https://doi.org/10.3390/v15010107
APA StyleJohnson, M. M., Jones, C. E., & Clark, D. N. (2023). The Effect of Treatment-Associated Mutations on HIV Replication and Transmission Cycles. Viruses, 15(1), 107. https://doi.org/10.3390/v15010107