

An Adenoviral Vector as a Versatile Tool for Delivery and Expression of miRNAs

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Cell Culture
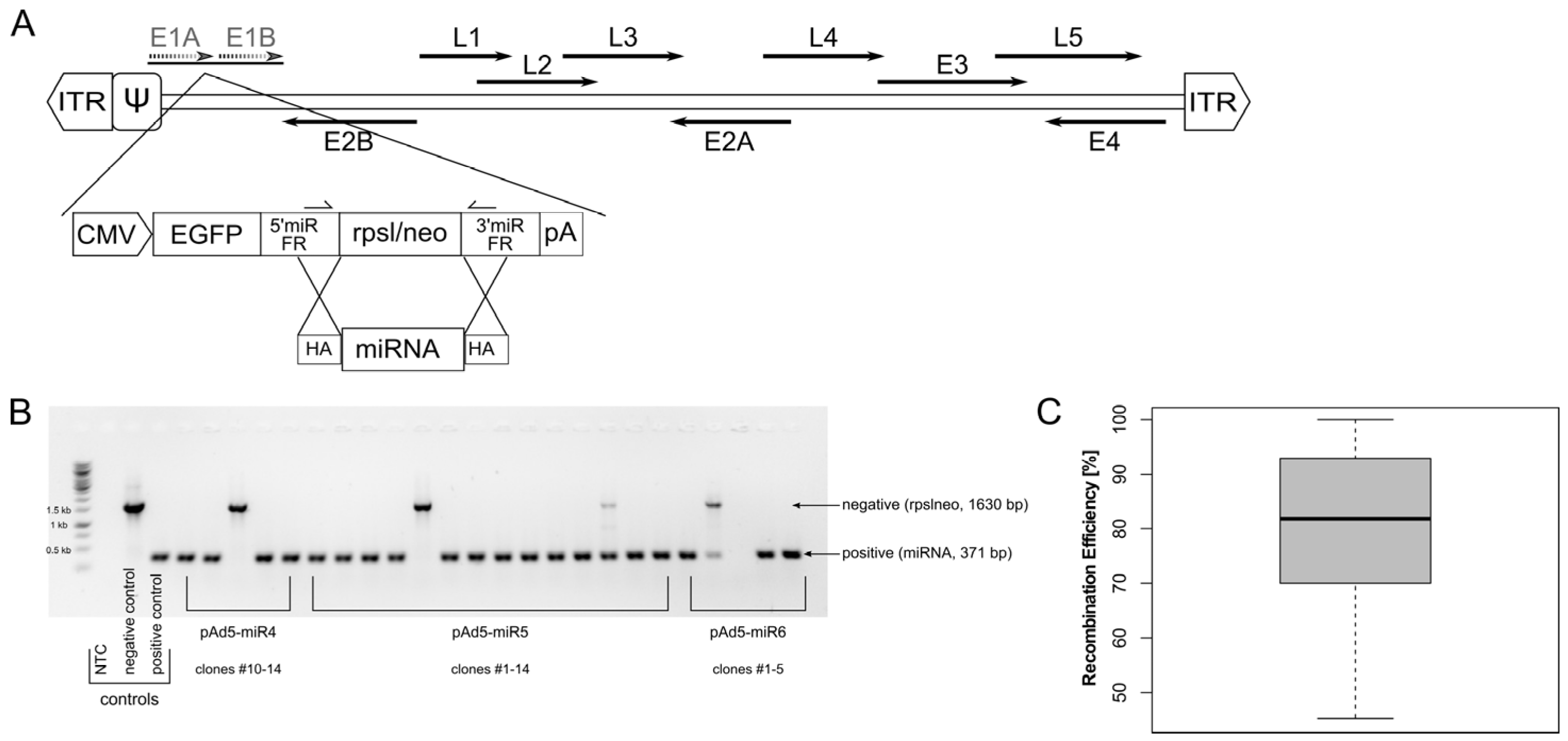
2.2. Vector Construction
2.3. Adenovirus Vector Purification
2.4. PEGylation of Vector Capsids
2.5. Polymerase Chain Reaction (PCR)
2.6. Transduction Assays
2.7. Isolation of Total RNA and cDNA Synthesis
2.8. Isolation of Total Genomic DNA
2.9. Real-Time Quantitative PCR Analysis
2.10. Statistical Analysis
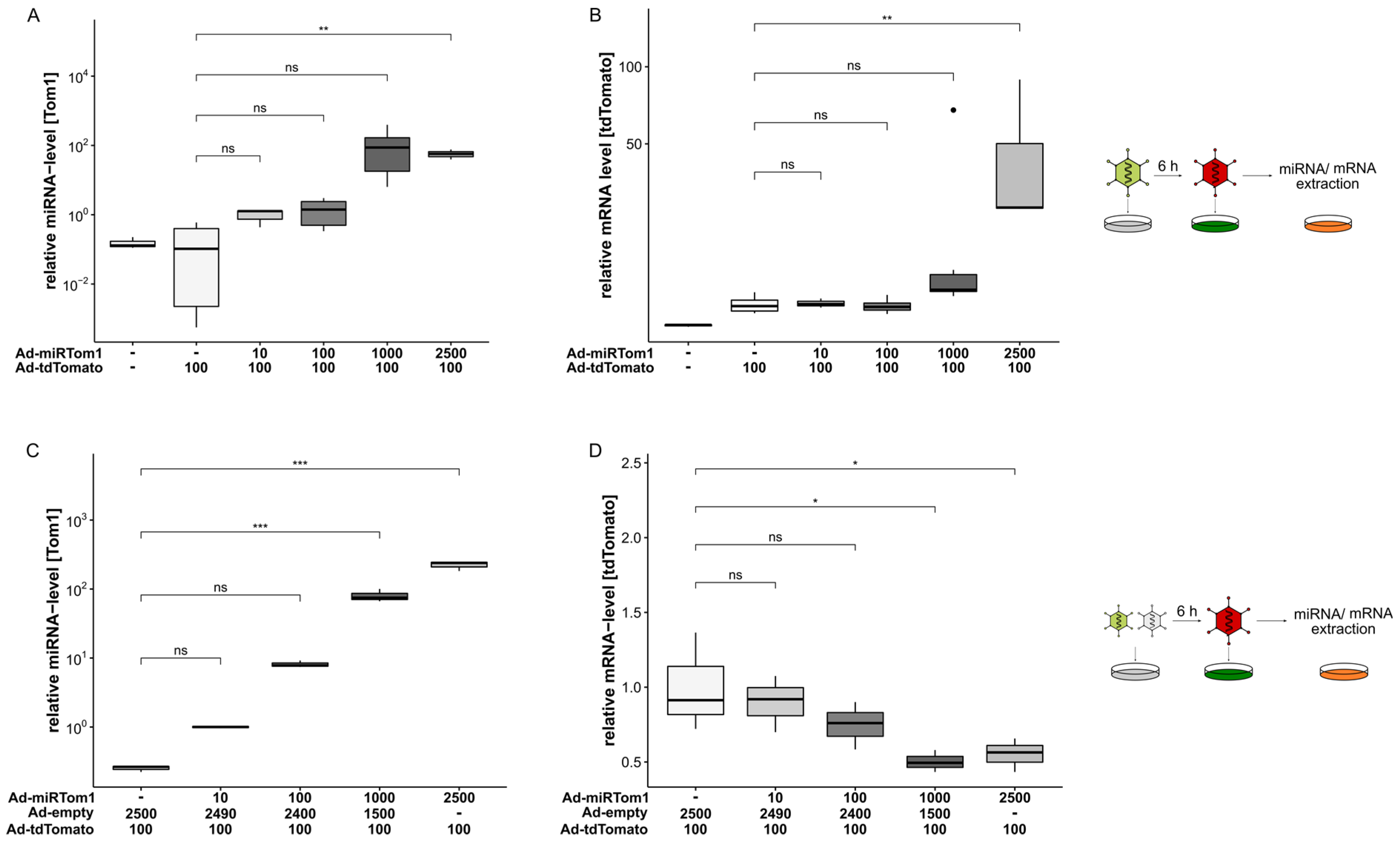
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wightman, B.; Ha, I.; Ruvkun, G. Posttranscriptional Regulation of the Heterochronic Gene Lin-14 by Lin-4 Mediates Temporal Pattern Formation in C. Elegans. Cell 1993, 75, 855–862. [Google Scholar] [CrossRef]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans Heterochronic Gene Lin-4 Encodes Small RNAs with Antisense Complementarity to Lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Plawgo, K.; Raczynska, K.D. Context-Dependent Regulation of Gene Expression by Non-Canonical Small RNAs. Noncoding RNA 2022, 8, 29. [Google Scholar] [CrossRef]
- Verdel, A.; Jia, S.; Gerber, S.; Sugiyama, T.; Gygi, S.; Grewal, S.I.S.; Moazed, D. RNAi-Mediated Targeting of Heterochromatin by the RITS Complex. Science 2004, 303, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Motamedi, M.R.; Verdel, A.; Colmenares, S.U.; Gerber, S.A.; Gygi, S.P.; Moazed, D. Two RNAi Complexes, RITS and RDRC, Physically Interact and Localize to Noncoding Centromeric RNAs. Cell 2004, 119, 789–802. [Google Scholar] [CrossRef]
- Hamilton, A.; Voinnet, O.; Chappell, L.; Baulcombe, D. Two Classes of Short Interfering RNA in RNA Silencing. EMBO J. 2002, 21, 4671–4679. [Google Scholar] [CrossRef]
- Zilberman, D.; Cao, X.; Jacobsen, S.E. ARGONAUTE4 Control of Locus-Specific SiRNA Accumulation and DNA and Histone Methylation. Science 2003, 299, 716–719. [Google Scholar] [CrossRef]
- Sijen, T.; Plasterk, R.H.A. Transposon Silencing in the Caenorhabditis Elegans Germ Line by Natural RNAi. Nature 2003, 426, 310–314. [Google Scholar] [CrossRef]
- Grishok, A.; Sinskey, J.L.; Sharp, P.A. Transcriptional Silencing of a Transgene by RNAi in the Soma of C. Elegans. Genes Dev. 2005, 19, 683–696. [Google Scholar] [CrossRef]
- Pal-Bhadra, M.; Leibovitch, B.A.; Gandhi, S.G.; Chikka, M.R.; Rao, M.; Bhadra, U.; Birchler, J.A.; Elgin, S.C.R. Heterochromatic Silencing and HP1 Localization in Drosophila Are Dependent on the RNAi Machinery. Science 2004, 303, 669–672. [Google Scholar] [CrossRef] [Green Version]
- Weinberg, M.S.; Villeneuve, L.M.; Ehsani, A.; Amarzguioui, M.; Aagaard, L.; Chen, Z.-X.; Riggs, A.D.; Rossi, J.J.; Morris, K.V. The Antisense Strand of Small Interfering RNAs Directs Histone Methylation and Transcriptional Gene Silencing in Human Cells. RNA 2006, 12, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Saetrom, P.; Snøve, O.; Rossi, J.J. MicroRNA-Directed Transcriptional Gene Silencing in Mammalian Cells. Proc. Natl. Acad. Sci. USA 2008, 105, 16230–16235. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Lee, Y.; Yeom, K.-H.; Kim, Y.-K.; Jin, H.; Kim, V.N. The Drosha-DGCR8 Complex in Primary MicroRNA Processing. Genes Dev. 2004, 18, 3016–3027. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Ahn, C.; Han, J.; Choi, H.; Kim, J.; Yim, J.; Lee, J.; Provost, P.; Rådmark, O.; Kim, S.; et al. The Nuclear RNase III Drosha Initiates MicroRNA Processing. Nature 2003, 425, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Grishok, A.; Pasquinelli, A.E.; Conte, D.; Li, N.; Parrish, S.; Ha, I.; Baillie, D.L.; Fire, A.; Ruvkun, G.; Mello, C.C. Genes and Mechanisms Related to RNA Interference Regulate Expression of the Small Temporal RNAs That Control C. Elegans Developmental Timing. Cell 2001, 106, 23–34. [Google Scholar] [CrossRef]
- Hutvágner, G.; McLachlan, J.; Pasquinelli, A.E.; Bálint, E.; Tuschl, T.; Zamore, P.D. A Cellular Function for the RNA-Interference Enzyme Dicer in the Maturation of the Let-7 Small Temporal RNA. Science 2001, 293, 834–838. [Google Scholar] [CrossRef]
- Zhang, H.; Kolb, F.A.; Jaskiewicz, L.; Westhof, E.; Filipowicz, W. Single Processing Center Models for Human Dicer and Bacterial RNase III. Cell 2004, 118, 57–68. [Google Scholar] [CrossRef]
- Schwarz, D.S.; Hutvágner, G.; Du, T.; Xu, Z.; Aronin, N.; Zamore, P.D. Asymmetry in the Assembly of the RNAi Enzyme Complex. Cell 2003, 115, 199–208. [Google Scholar] [CrossRef]
- Martinez, J.; Patkaniowska, A.; Urlaub, H.; Lührmann, R.; Tuschl, T. Single-Stranded Antisense SiRNAs Guide Target RNA Cleavage in RNAi. Cell 2002, 110, 563–574. [Google Scholar] [CrossRef]
- Hutvágner, G.; Zamore, P.D. A MicroRNA in a Multiple-Turnover RNAi Enzyme Complex. Science 2002, 297, 2056–2060. [Google Scholar] [CrossRef] [Green Version]
- Meister, G.; Landthaler, M.; Patkaniowska, A.; Dorsett, Y.; Teng, G.; Tuschl, T. Human Argonaute2 Mediates RNA Cleavage Targeted by MiRNAs and SiRNAs. Mol. Cell 2004, 15, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.; Jin, H.; Suh, D.-S.; Luo, Y.; Ha, H.-J.; Kim, T.H.; Hahn, Y.; Hyun, S.; Lee, K.; Bae, J. An Alternative MiRISC Targets a Cancer-Associated Coding Sequence Mutation in FOXL2. EMBO J. 2020, 39, e104719. [Google Scholar] [CrossRef] [PubMed]
- Jo, M.H.; Shin, S.; Jung, S.-R.; Kim, E.; Song, J.-J.; Hohng, S. Human Argonaute 2 Has Diverse Reaction Pathways on Target RNAs. Mol. Cell 2015, 59, 117–124. [Google Scholar] [CrossRef] [PubMed]
- De, N.; Young, L.; Lau, P.-W.; Meisner, N.-C.; Morrissey, D.V.; MacRae, I.J. Highly Complementary Target RNAs Promote Release of Guide RNAs from Human Argonaute2. Mol. Cell 2013, 50, 344–355. [Google Scholar] [CrossRef]
- Frédérick, P.-M.; Simard, M.J. Regulation and Different Functions of the Animal MicroRNA-Induced Silencing Complex. WIREs RNA 2022, 13, e1701. [Google Scholar] [CrossRef]
- Li, X.; Wang, X.; Cheng, Z.; Zhu, Q. AGO2 and Its Partners: A Silencing Complex, a Chromatin Modulator, and New Features. Crit. Rev. Biochem. Mol. Biol. 2020, 55, 33–53. [Google Scholar] [CrossRef]
- Broughton, J.P.; Lovci, M.T.; Huang, J.L.; Yeo, G.W.; Pasquinelli, A.E. Pairing beyond the Seed Supports MicroRNA Targeting Specificity. Mol. Cell 2016, 64, 320–333. [Google Scholar] [CrossRef]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. (Lausanne) 2018, 9, 402. [Google Scholar] [CrossRef]
- Lauretti, E.; Dincer, O.; Praticò, D. Regional and Temporal MiRNAs Expression Profile in a Transgenic Mouse Model of Tauopathy: Implication for Its Pathogenesis. Mol. Psychiatry 2020, 26, 7020–7028. [Google Scholar] [CrossRef]
- Ludwig, N.; Leidinger, P.; Becker, K.; Backes, C.; Fehlmann, T.; Pallasch, C.; Rheinheimer, S.; Meder, B.; Stähler, C.; Meese, E.; et al. Distribution of MiRNA Expression across Human Tissues. Nucleic Acids Res. 2016, 44, 3865–3877. [Google Scholar] [CrossRef]
- Mitchell, P.S.; Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Peterson, A.; Noteboom, J.; O’Briant, K.C.; Allen, A.; et al. Circulating MicroRNAs as Stable Blood-Based Markers for Cancer Detection. Proc. Natl. Acad. Sci. USA 2008, 105, 10513–10518. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.A.; Baxter, D.H.; Zhang, S.; Huang, D.Y.; Huang, K.H.; Lee, M.J.; Galas, D.J.; Wang, K. The MicroRNA Spectrum in 12 Body Fluids. Clin. Chem. 2010, 56, 1733–1741. [Google Scholar] [CrossRef] [PubMed]
- Park, N.J.; Zhou, H.; Elashoff, D.; Henson, B.S.; Kastratovic, D.A.; Abemayor, E.; Wong, D.T. Salivary MicroRNA: Discovery, Characterization, and Clinical Utility for Oral Cancer Detection. Clin. Cancer Res. 2009, 15, 5473–5477. [Google Scholar] [CrossRef]
- Gallo, A.; Tandon, M.; Alevizos, I.; Illei, G.G. The Majority of MicroRNAs Detectable in Serum and Saliva Is Concentrated in Exosomes. PLoS ONE 2012, 7, e30679. [Google Scholar] [CrossRef]
- van Rooij, E.; Sutherland, L.B.; Liu, N.; Williams, A.H.; McAnally, J.; Gerard, R.D.; Richardson, J.A.; Olson, E.N. A Signature Pattern of Stress-Responsive MicroRNAs That Can Evoke Cardiac Hypertrophy and Heart Failure. Proc. Natl. Acad. Sci. USA 2006, 103, 18255–18260. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Peng, W.; Wang, Z.; Zhang, L.; Liu, K. Identification of Biomarkers Associated with Metabolic Cardiovascular Disease Using MRNA-SNP-MiRNA Regulatory Network Analysis. BMC Cardiovasc. Disord. 2021, 21, 351. [Google Scholar] [CrossRef]
- Bazrgar, M.; Khodabakhsh, P.; Prudencio, M.; Mohagheghi, F.; Ahmadiani, A. The Role of MicroRNA-34 Family in Alzheimer’s Disease: A Potential Molecular Link between Neurodegeneration and Metabolic Disorders. Pharmacol. Res. 2021, 172, 105805. [Google Scholar] [CrossRef]
- Wu, T.; Lei, Y.; Jin, S.; Zhao, Q.; Cheng, W.; Xi, Y.; Wang, L.; Wang, Z.; Niu, X.; Chen, G. MiRNA-467b Inhibits Th17 Differentiation by Targeting EIF4E in Experimental Autoimmune Encephalomyelitis. Mol. Immunol. 2021, 133, 23–33. [Google Scholar] [CrossRef]
- Sproviero, D.; Gagliardi, S.; Zucca, S.; Arigoni, M.; Giannini, M.; Garofalo, M.; Olivero, M.; Dell’Orco, M.; Pansarasa, O.; Bernuzzi, S.; et al. Different MiRNA Profiles in Plasma Derived Small and Large Extracellular Vesicles from Patients with Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 2737. [Google Scholar] [CrossRef]
- Schaefer, A.; O’Carroll, D.; Tan, C.L.; Hillman, D.; Sugimori, M.; Llinas, R.; Greengard, P. Cerebellar Neurodegeneration in the Absence of MicroRNAs. J. Exp. Med. 2007, 204, 1553–1558. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Inoue, K.; Ishii, J.; Vanti, W.B.; Voronov, S.V.; Murchison, E.; Hannon, G.; Abeliovich, A. A MicroRNA Feedback Circuit in Midbrain Dopamine Neurons. Science 2007, 317, 1220–1224. [Google Scholar] [CrossRef] [PubMed]
- Sonkoly, E.; Wei, T.; Janson, P.C.J.; Sääf, A.; Lundeberg, L.; Tengvall-Linder, M.; Norstedt, G.; Alenius, H.; Homey, B.; Scheynius, A.; et al. MicroRNAs: Novel Regulators Involved in the Pathogenesis of Psoriasis? PLoS ONE 2007, 2, e610. [Google Scholar] [CrossRef] [PubMed]
- Sonkoly, E.; Ståhle, M.; Pivarcsi, A. MicroRNAs and Immunity: Novel Players in the Regulation of Normal Immune Function and Inflammation. Semin Cancer Biol. 2008, 18, 131–140. [Google Scholar] [CrossRef]
- Tang, W.; Lv, Q.; Huang, X.; Li, Y.; Zou, J.; Zheng, J.; Sun, L.; Bao, Y.; Chen, H.; Li, T.; et al. MiR-143 Targets IGF-1R to Suppress Autoimmunity in Thyroid-Associated Ophthalmopathy. J. Inflamm. Res. 2022, 15, 1543–1554. [Google Scholar] [CrossRef] [PubMed]
- Xia, P.; Pasquali, L.; Gao, C.; Srivastava, A.; Khera, N.; Freisenhausen, J.C.; Luo, L.; Rosén, E.; van Lierop, A.; Homey, B.; et al. MiR-378a Regulates Keratinocyte Responsiveness to Interleukin-17A in Psoriasis. Br. J. Dermatol. 2022, 187, 211–222. [Google Scholar] [CrossRef]
- Ma, L.; Teruya-Feldstein, J.; Weinberg, R.A. Tumour Invasion and Metastasis Initiated by MicroRNA-10b in Breast Cancer. Nature 2007, 449, 682–688. [Google Scholar] [CrossRef]
- He, L.; Thomson, J.M.; Hemann, M.T.; Hernando-Monge, E.; Mu, D.; Goodson, S.; Powers, S.; Cordon-Cardo, C.; Lowe, S.W.; Hannon, G.J.; et al. A MicroRNA Polycistron as a Potential Human Oncogene. Nature 2005, 435, 828–833. [Google Scholar] [CrossRef]
- Pickering, M.T.; Stadler, B.M.; Kowalik, T.F. MiR-17 and MiR-20a Temper an E2F1-Induced G1 Checkpoint to Regulate Cell Cycle Progression. Oncogene 2009, 28, 140–145. [Google Scholar] [CrossRef]
- Yan, L.-X.; Huang, X.-F.; Shao, Q.; Huang, M.-Y.; Deng, L.; Wu, Q.-L.; Zeng, Y.-X.; Shao, J.-Y. MicroRNA MiR-21 Overexpression in Human Breast Cancer Is Associated with Advanced Clinical Stage, Lymph Node Metastasis and Patient Poor Prognosis. RNA 2008, 14, 2348–2360. [Google Scholar] [CrossRef]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent Deletions and Down-Regulation of Micro- RNA Genes MiR15 and MiR16 at 13q14 in Chronic Lymphocytic Leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar] [CrossRef] [Green Version]
- Bhattarai, S.; Sugita, B.M.; Bortoletto, S.M.; Fonseca, A.S.; Cavalli, L.R.; Aneja, R. QNBC Is Associated with High Genomic Instability Characterized by Copy Number Alterations and MiRNA Deregulation. Int. J. Mol. Sci. 2021, 22, 11548. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.; Cristóbal, I.; Rubio, J.; Caramés, C.; Luque, M.; Sanz-Alvarez, M.; Morales-Gallego, M.; Madoz-Gúrpide, J.; Rojo, F.; García-Foncillas, J. MicroRNA-199b Deregulation Shows Oncogenic Properties and Promising Clinical Value as Circulating Marker in Locally Advanced Rectal Cancer Patients. Int. J. Mol. Sci. 2022, 23, 2203. [Google Scholar] [CrossRef] [PubMed]
- van der Ree, M.H.; van der Meer, A.J.; de Bruijne, J.; Maan, R.; van Vliet, A.; Welzel, T.M.; Zeuzem, S.; Lawitz, E.J.; Rodriguez-Torres, M.; Kupcova, V.; et al. Long-Term Safety and Efficacy of MicroRNA-Targeted Therapy in Chronic Hepatitis C Patients. Antivir. Res. 2014, 111, 53–59. [Google Scholar] [CrossRef]
- Kristen, A.V.; Ajroud-Driss, S.; Conceição, I.; Gorevic, P.; Kyriakides, T.; Obici, L. Patisiran, an RNAi Therapeutic for the Treatment of Hereditary Transthyretin-Mediated Amyloidosis. Neurodegener Dis. Manag. 2019, 9, 5–23. [Google Scholar] [CrossRef] [PubMed]
- Coelho, T.; Adams, D.; Conceição, I.; Waddington-Cruz, M.; Schmidt, H.H.; Buades, J.; Campistol, J.; Berk, J.L.; Polydefkis, M.; Wang, J.J.; et al. A Phase II, Open-Label, Extension Study of Long-Term Patisiran Treatment in Patients with Hereditary Transthyretin-Mediated (HATTR) Amyloidosis. Orphanet J. Rare Dis. 2020, 15, 179. [Google Scholar] [CrossRef]
- Idris, A.; Davis, A.; Supramaniam, A.; Acharya, D.; Kelly, G.; Tayyar, Y.; West, N.; Zhang, P.; McMillan, C.L.D.; Soemardy, C.; et al. A SARS-CoV-2 Targeted SiRNA-Nanoparticle Therapy for COVID-19. Mol. Ther. 2021, 29, 2219–2226. [Google Scholar] [CrossRef]
- Fu, Y.; Chen, J.; Huang, Z. Recent Progress in MicroRNA-Based Delivery Systems for the Treatment of Human Disease. ExRNA 2019, 1, 24. [Google Scholar] [CrossRef]
- Tuttolomondo, M.; Casella, C.; Hansen, P.L.; Polo, E.; Herda, L.M.; Dawson, K.A.; Ditzel, H.J.; Mollenhauer, J. Human DMBT1-Derived Cell-Penetrating Peptides for Intracellular SiRNA Delivery. Mol. Ther. Nucleic Acids 2017, 8, 264–276. [Google Scholar] [CrossRef]
- Alemany, R.; Curiel, D. CAR-Binding Ablation Does Not Change Biodistribution and Toxicity of Adenoviral Vectors. Gene Ther. 2001, 8, 1347–1353. [Google Scholar] [CrossRef]
- Doronin, K.; Flatt, J.W.; Di Paolo, N.C.; Khare, R.; Kalyuzhniy, O.; Acchione, M.; Sumida, J.P.; Ohto, U.; Shimizu, T.; Akashi-Takamura, S.; et al. Coagulation Factor X Activates Innate Immunity to Human Species C Adenovirus. Science 2012, 338, 795–798. [Google Scholar] [CrossRef] [Green Version]
- Kratzer, R.F.; Kreppel, F. Production, Purification, and Titration of First-Generation Adenovirus Vectors. Methods Mol. Biol. (Clifton N.J.) 2017, 1654, 377–388. [Google Scholar] [CrossRef]
- Graham, F.L.; Smiley, J.; Russell, W.C.; Nairn, R. Characteristics of a Human Cell Line Transformed by DNA from Human Adenovirus Type 5. J. Gen. Virol. 1977, 36, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Kreppel, F.; Gackowski, J.; Schmidt, E.; Kochanek, S. Combined Genetic and Chemical Capsid Modifications Enable Flexible and Efficient De- and Retargeting of Adenovirus Vectors. Mol. Ther. 2005, 12, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Maizel, J.V.; White, D.O.; Scharff, M.D. The Polypeptides of Adenovirus. I. Evidence for Multiple Protein Components in the Virion and a Comparison of Types 2, 7A, and 12. Virology 1968, 36, 115–125. [Google Scholar] [CrossRef]
- Jönsson, F.; Hagedorn, C.; Kreppel, F. Combined Genetic and Chemical Capsid Modifications of Adenovirus-Based Gene Transfer Vectors for Shielding and Targeting. J. Vis. Exp. 2018, 140, e58480. [Google Scholar] [CrossRef]
- Muyrers, J.P.; Zhang, Y.; Testa, G.; Stewart, A.F. Rapid Modification of Bacterial Artificial Chromosomes by ET-Recombination. Nucleic Acids Res. 1999, 27, 1555–1557. [Google Scholar] [CrossRef] [PubMed]
- Weil, P.P.; Reincke, S.; Hirsch, C.A.; Giachero, F.; Aydin, M.; Scholz, J.; Jönsson, F.; Hagedorn, C.; Nguyen, D.N.; Thymann, T.; et al. Uncovering the Gastrointestinal Passage, Intestinal Epithelial Cellular Uptake and Ago2 Loading of Milk MiRNAs in Neonates Using Xenobiotic Tracers. medRxiv 2022. [Google Scholar] [CrossRef]
- Kirby, I.; Davison, E.; Beavil, A.J.; Soh, C.P.C.; Wickham, T.J.; Roelvink, P.W.; Kovesdi, I.; Sutton, B.J.; Santis, G. Identification of Contact Residues and Definition of the CAR-Binding Site of Adenovirus Type 5 Fiber Protein. J. Virol. 2000, 74, 2804–2813. [Google Scholar] [CrossRef]
- Alba, R.; Bradshaw, A.C.; Parker, A.L.; Bhella, D.; Waddington, S.N.; Nicklin, S.A.; van Rooijen, N.; Custers, J.; Goudsmit, J.; Barouch, D.H.; et al. Identification of Coagulation Factor (F)X Binding Sites on the Adenovirus Serotype 5 Hexon: Effect of Mutagenesis on FX Interactions and Gene Transfer. Blood 2009, 114, 965–971. [Google Scholar] [CrossRef]
- Krutzke, L.; Prill, J.M.; Engler, T.; Schmidt, C.Q.; Xu, Z.; Byrnes, A.P.; Simmet, T.; Kreppel, F. Substitution of Blood Coagulation Factor X-Binding to Ad5 by Position-Specific PEGylation: Preventing Vector Clearance and Preserving Infectivity. J. Control Release 2016, 235, 379–392. [Google Scholar] [CrossRef] [Green Version]
- Prill, J.-M.; Subr, V.; Pasquarelli, N.; Engler, T.; Hoffmeister, A.; Kochanek, S.; Ulbrich, K.; Kreppel, F. Traceless Bioresponsive Shielding of Adenovirus Hexon with HPMA Copolymers Maintains Transduction Capacity in Vitro and in Vivo. PLoS ONE 2014, 9, e82716. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.M.; Bahal, R.; Rasmussen, T.P.; Manautou, J.E.; Zhong, X.-B. The Growth of SiRNA-Based Therapeutics: Updated Clinical Studies. Biochem. Pharmacol. 2021, 189, 114432. [Google Scholar] [CrossRef]
- Dasgupta, I.; Chatterjee, A. Recent Advances in MiRNA Delivery Systems. Methods Protoc. 2021, 4, 10. [Google Scholar] [CrossRef] [PubMed]
- Brachtlova, T.; van Ginkel, J.-W.; Luinenburg, M.J.; de Menezes, R.X.; Koppers-Lalic, D.; Pegtel, D.M.; Dong, W.; de Gruijl, T.D.; Beusechem, V.W. van Expression of Oncolytic Adenovirus-Encoded RNAi Molecules Is Most Effective in a Pri-MiRNA Precursor Format. Mol. Ther. -Oncolytics 2020, 19, 332–343. [Google Scholar] [CrossRef]
- Lagos-Quintana, M.; Rauhut, R.; Yalcin, A.; Meyer, J.; Lendeckel, W.; Tuschl, T. Identification of Tissue-Specific MicroRNAs from Mouse. Curr. Biol. 2002, 12, 735–739. [Google Scholar] [CrossRef]
- Leopold, P.L.; Ferris, B.; Grinberg, I.; Worgall, S.; Hackett, N.R.; Crystal, R.G. Fluorescent Virions: Dynamic Tracking of the Pathway of Adenoviral Gene Transfer Vectors in Living Cells. Hum. Gene Ther. 1998, 9, 367–378. [Google Scholar] [CrossRef]
- Pied, N.; Wodrich, H. Imaging the Adenovirus Infection Cycle. FEBS Lett. 2019, 593, 3419–3448. [Google Scholar] [CrossRef]
- Li, X.; Zhao, X.; Fang, Y.; Jiang, X.; Duong, T.; Fan, C.; Huang, C.C.; Kain, S.R. Generation of Destabilized Green Fluorescent Protein as a Transcription Reporter. J. Biol. Chem. 1998, 273, 34970–34975. [Google Scholar] [CrossRef]
- Kauffman, K.J.; Oberli, M.A.; Dorkin, J.R.; Hurtado, J.E.; Kaczmarek, J.C.; Bhadani, S.; Wyckoff, J.; Langer, R.; Jaklenec, A.; Anderson, D.G. Rapid, Single-Cell Analysis and Discovery of Vectored MRNA Transfection In Vivo with a LoxP-Flanked TdTomato Reporter Mouse. Mol. Ther. -Nucleic Acids 2018, 10, 55–63. [Google Scholar] [CrossRef]
- Lu, S.; Cullen, B.R. Adenovirus VA1 Noncoding RNA Can Inhibit Small Interfering RNA and MicroRNA Biogenesis. J. Virol. 2004, 78, 12868–12876. [Google Scholar] [CrossRef] [Green Version]
- Bennasser, Y.; Chable-Bessia, C.; Triboulet, R.; Gibbings, D.; Gwizdek, C.; Dargemont, C.; Kremer, E.J.; Voinnet, O.; Benkirane, M. Competition for XPO5 Binding between Dicer MRNA, Pre-MiRNA and Viral RNA Regulates Human Dicer Levels. Nat. Struct. Mol. Biol. 2011, 18, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Kawabata, K.; Kouyama, E.; Ishii, K.J.; Katayama, K.; Suzuki, T.; Kurachi, S.; Sakurai, F.; Akira, S.; Mizuguchi, H. Induction of Type I Interferon by Adenovirus-Encoded Small RNAs. Proc. Natl. Acad. Sci. USA 2010, 107, 17286–17291. [Google Scholar] [CrossRef] [PubMed]
- Kamel, W.; Segerman, B.; Öberg, D.; Punga, T.; Akusjärvi, G. The Adenovirus VA RNA-Derived MiRNAs Are Not Essential for Lytic Virus Growth in Tissue Culture Cells. Nucleic Acids Res. 2013, 41, 4802–4812. [Google Scholar] [CrossRef] [PubMed]
- Pei, Z.; Shi, G.; Kondo, S.; Ito, M.; Maekawa, A.; Suzuki, M.; Saito, I.; Suzuki, T.; Kanegae, Y. Adenovirus Vectors Lacking Virus-Associated RNA Expression Enhance ShRNA Activity to Suppress Hepatitis C Virus Replication. Sci. Rep. 2013, 3, 3575. [Google Scholar] [CrossRef] [PubMed]
- Machitani, M.; Sakurai, F.; Katayama, K.; Tachibana, M.; Suzuki, T.; Matsui, H.; Yamaguchi, T.; Mizuguchi, H. Improving Adenovirus Vector-Mediated RNAi Efficiency by Lacking the Expression of Virus-Associated RNAs. Virus Res. 2013, 178, 357–363. [Google Scholar] [CrossRef]
- Elbashir, S.M.; Martinez, J.; Patkaniowska, A.; Lendeckel, W.; Tuschl, T. Functional Anatomy of SiRNAs for Mediating Efficient RNAi in Drosophila Melanogaster Embryo Lysate. EMBO J. 2001, 20, 6877–6888. [Google Scholar] [CrossRef]
- Baccarini, A.; Chauhan, H.; Gardner, T.J.; Jayaprakash, A.D.; Sachidanandam, R.; Brown, B.D. Kinetic Analysis Reveals the Fate of a MicroRNA Following Target Regulation in Mammalian Cells. Curr. Biol. 2011, 21, 369–376. [Google Scholar] [CrossRef]
- Greber, U.F.; Webster, P.; Weber, J.; Helenius, A. The Role of the Adenovirus Protease on Virus Entry into Cells. EMBO J. 1996, 15, 1766–1777. [Google Scholar] [CrossRef]
- Yang, Y.; Nunes, F.A.; Berencsi, K.; Furth, E.E.; Gönczöl, E.; Wilson, J.M. Cellular Immunity to Viral Antigens Limits E1-Deleted Adenoviruses for Gene Therapy. Proc. Natl. Acad. Sci. USA 1994, 91, 4407–4411. [Google Scholar] [CrossRef]
- Martina, Y.; Avitabile, D.; Piersanti, S.; Cherubini, G.; Saggio, I. Different Modulation of Cellular Transcription by Adenovirus 5, DeltaE1/E3 Adenovirus and Helper-Dependent Vectors. Virus Res. 2007, 130, 71–84. [Google Scholar] [CrossRef]
- Komatsu, T.; Dacheux, D.; Kreppel, F.; Nagata, K.; Wodrich, H. A Method for Visualization of Incoming Adenovirus Chromatin Complexes in Fixed and Living Cells. PLoS ONE 2015, 10, e0137102. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, T.; Maekawa, A.; Suzuki, M.; Tabata, H.; Sato, K.; Mori, M.; Saito, I. Construction of Adenovirus Vectors Simultaneously Expressing Four Multiplex, Double-Nicking Guide RNAs of CRISPR/Cas9 and in Vivo Genome Editing. Sci. Rep. 2021, 11, 3961. [Google Scholar] [CrossRef] [PubMed]
- Stripecke, R.; Carmen Villacres, M.; Skelton, D.; Satake, N.; Halene, S.; Kohn, D. Immune Response to Green Fluorescent Protein: Implications for Gene Therapy. Gene Ther. 1999, 6, 1305–1312. [Google Scholar] [CrossRef] [PubMed]
- Ansari, A.M.; Ahmed, A.K.; Matsangos, A.E.; Lay, F.; Born, L.J.; Marti, G.; Harmon, J.W.; Sun, Z. Cellular GFP Toxicity and Immunogenicity: Potential Confounders in in Vivo Cell Tracking Experiments. Stem. Cell Rev. 2016, 12, 553–559. [Google Scholar] [CrossRef]
- Weklak, D.; Pembaur, D.; Koukou, G.; Jönsson, F.; Hagedorn, C.; Kreppel, F. Genetic and Chemical Capsid Modifications of Adenovirus Vectors to Modulate Vector–Host Interactions. Viruses 2021, 13, 1300. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clone # | |||||
|---|---|---|---|---|---|
| miRNA | Analysed | Positive | Negative | Other | Efficiency [%] |
| miR1 | 2 | 2 | 0 | 0 | 100 |
| miR2 | 2 | 2 | 0 | 0 | 100 |
| miR3 | 14 | 13 | 0 | 1 | 92.9 |
| miR4 | 14 | 12 | 1 | 1 | 85.7 |
| miR5 | 14 | 12 | 1 | 1 | 85.7 |
| miR6 | 14 | 8 | 1 | 5 | 57.1 |
| miR7 | 14 | 11 | 1 | 2 | 78.6 |
| miR8 | 10 | 8 | 1 | 1 | 80.0 |
| miR9 | 10 | 7 | 3 | 0 | 70.0 |
| miR10 | 10 | 7 | 2 | 1 | 70.0 |
| miR11 | 10 | 10 | 0 | 0 | 100.0 |
| miR12 | 11 | 5 | 6 | 0 | 45.5 |
| miR13 | 11 | 9 | 1 | 1 | 81.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scholz, J.; Weil, P.P.; Pembaur, D.; Koukou, G.; Aydin, M.; Hauert, D.; Postberg, J.; Kreppel, F.; Hagedorn, C. An Adenoviral Vector as a Versatile Tool for Delivery and Expression of miRNAs. Viruses 2022, 14, 1952. https://doi.org/10.3390/v14091952
Scholz J, Weil PP, Pembaur D, Koukou G, Aydin M, Hauert D, Postberg J, Kreppel F, Hagedorn C. An Adenoviral Vector as a Versatile Tool for Delivery and Expression of miRNAs. Viruses. 2022; 14(9):1952. https://doi.org/10.3390/v14091952
Chicago/Turabian StyleScholz, Jonas, Patrick Philipp Weil, Daniel Pembaur, Georgia Koukou, Malik Aydin, Dorota Hauert, Jan Postberg, Florian Kreppel, and Claudia Hagedorn. 2022. "An Adenoviral Vector as a Versatile Tool for Delivery and Expression of miRNAs" Viruses 14, no. 9: 1952. https://doi.org/10.3390/v14091952
APA StyleScholz, J., Weil, P. P., Pembaur, D., Koukou, G., Aydin, M., Hauert, D., Postberg, J., Kreppel, F., & Hagedorn, C. (2022). An Adenoviral Vector as a Versatile Tool for Delivery and Expression of miRNAs. Viruses, 14(9), 1952. https://doi.org/10.3390/v14091952