
The International Virus Bioinformatics Meeting 2022

 , , , , , , , , ,
, , , , , , , , ,  ,
,  , , , , , , , , , , ,
, , , , , , , , , , ,  , ,
, ,  and add
Show full author list
and add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Scientific Program
2.1. Satellite Meeting on SARS-CoV-2
2.1.1. SARS-CoV-2 Genomic Epidemiology: Bayesian Phylodynamic Reconstruction, Vaccine Design, and Characterization of Antigenic Evolution (by Philippe Lemey)
2.1.2. The Emergence of SARS-CoV-2 Variants of Concern Is Driven by Acceleration of the Substitution Rate (by Sebastian Duchene)
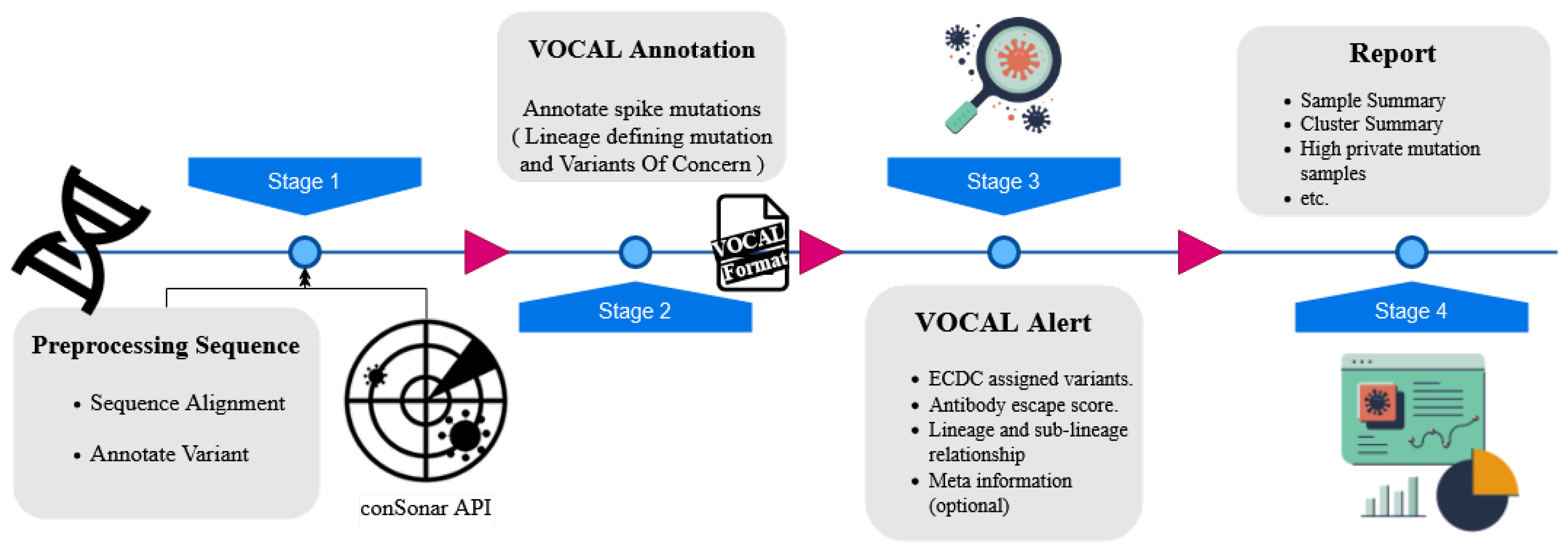
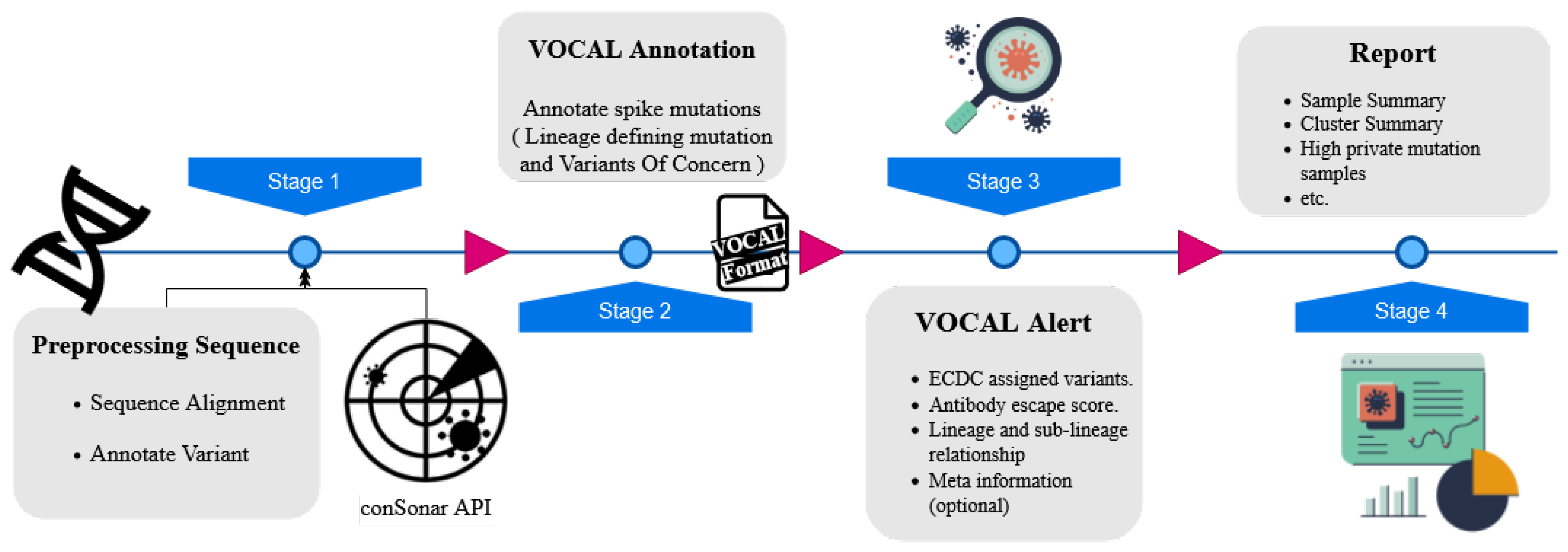
2.1.3. VOCAL: An Early Warning System to Detect Concerning New SARS-CoV-2 Variants from Sequencing Data (by Kunaphas Kongkitimanon)
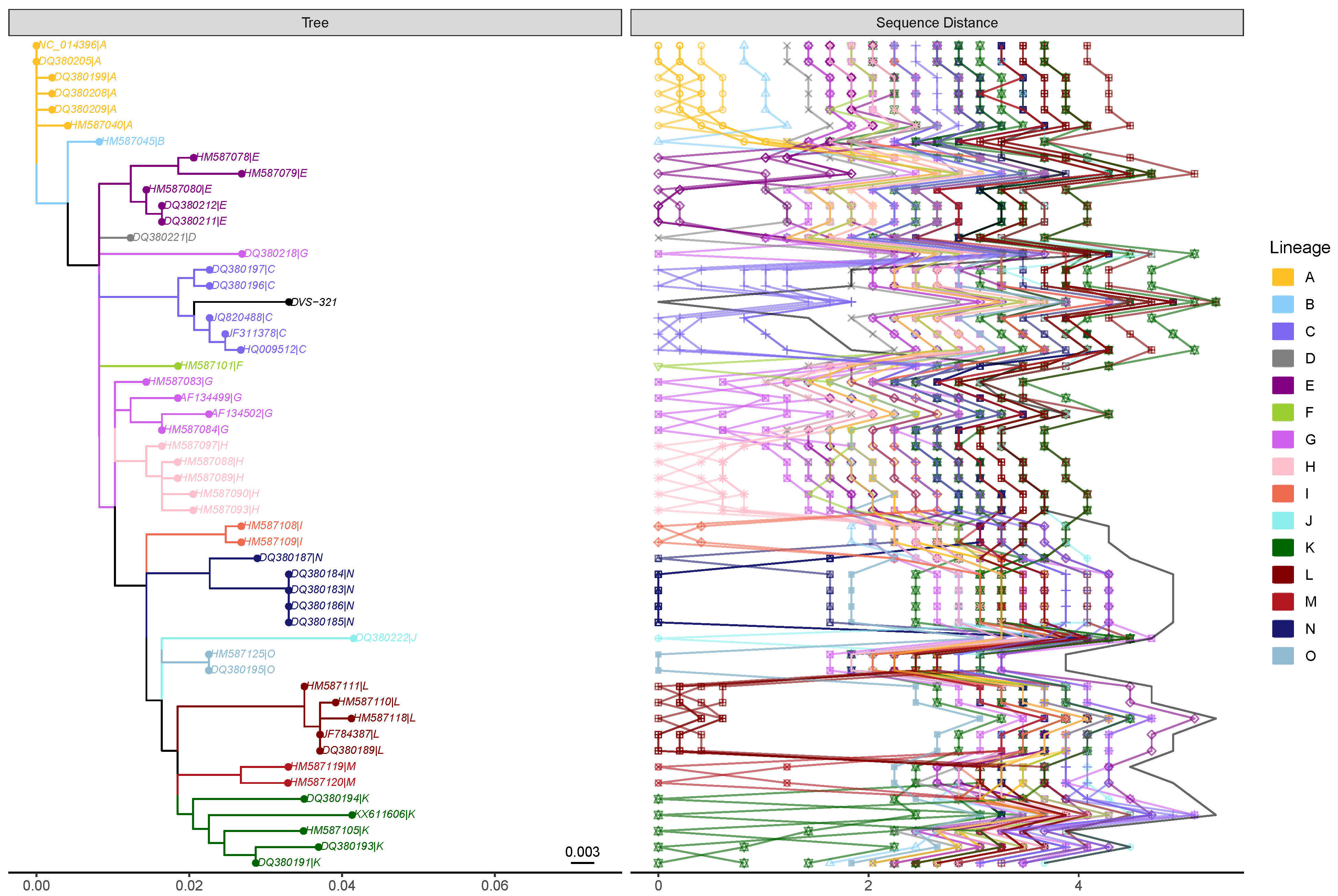
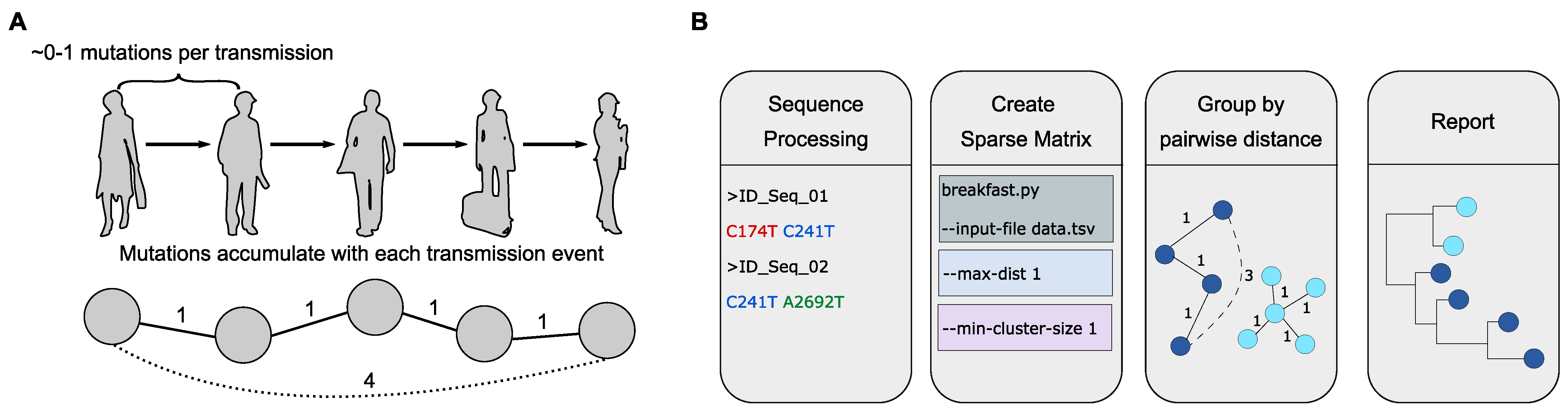
2.1.4. The Power of SARS-CoV-2 Genotyping and SNP-Based Clustering for Contextual Outbreak Assessment (by Denis Beslic)
2.2. Viral Emergence and Surveillance
2.2.1. Real-Time to Real-Life: Phylogenetics, Pandemics, and What Comes Next (by Emma Hodcroft)
2.2.2. Genomic Surveillance of the Rift Valley Fever: From Sequencing to Lineage Assignment (by John Juma)
2.3. Virus–Host Interactions
2.3.1. Diverse Anti-Interferon Strategies by Members of the Genus Phlebovirus (by Friedemann Weber)
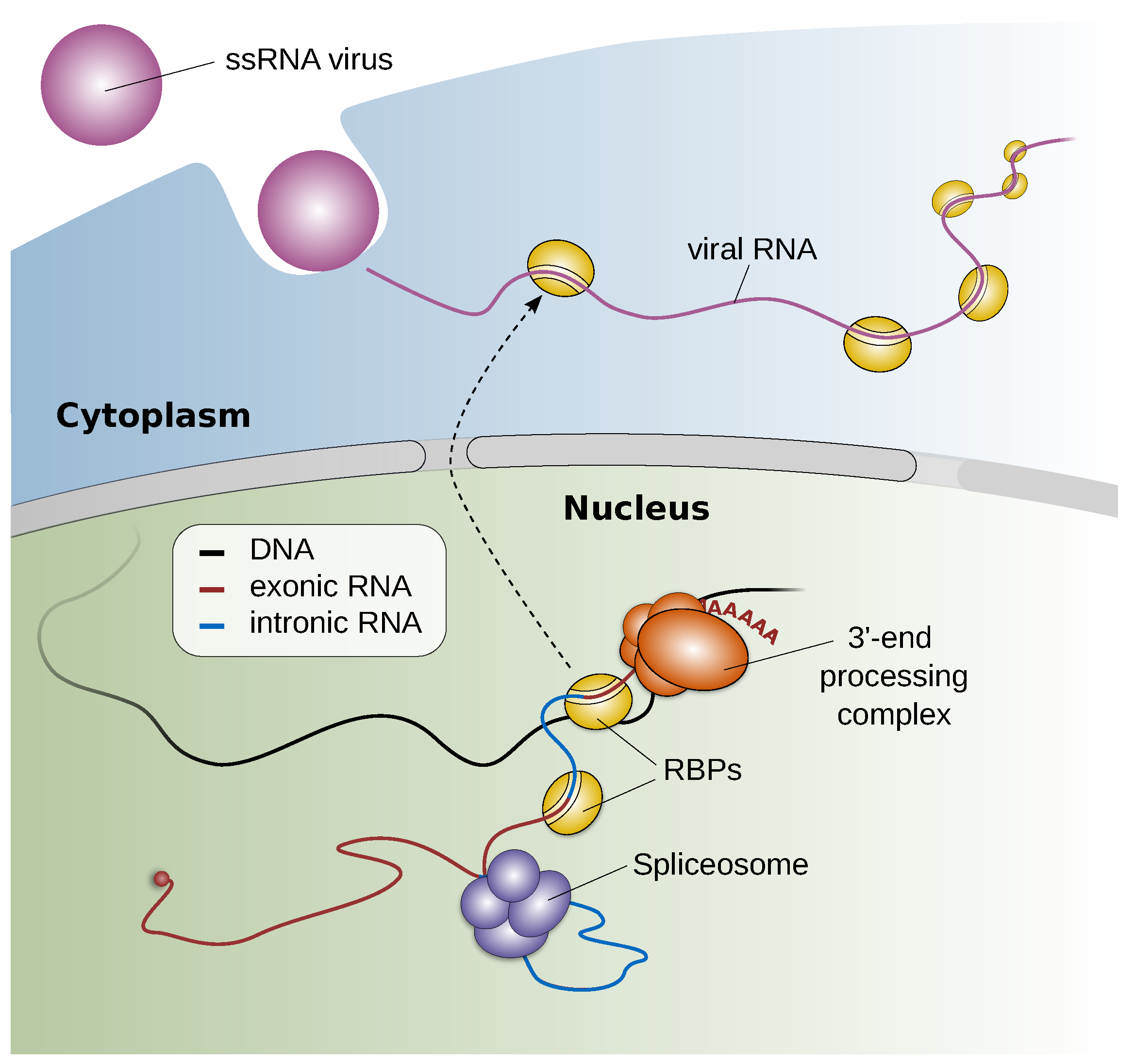
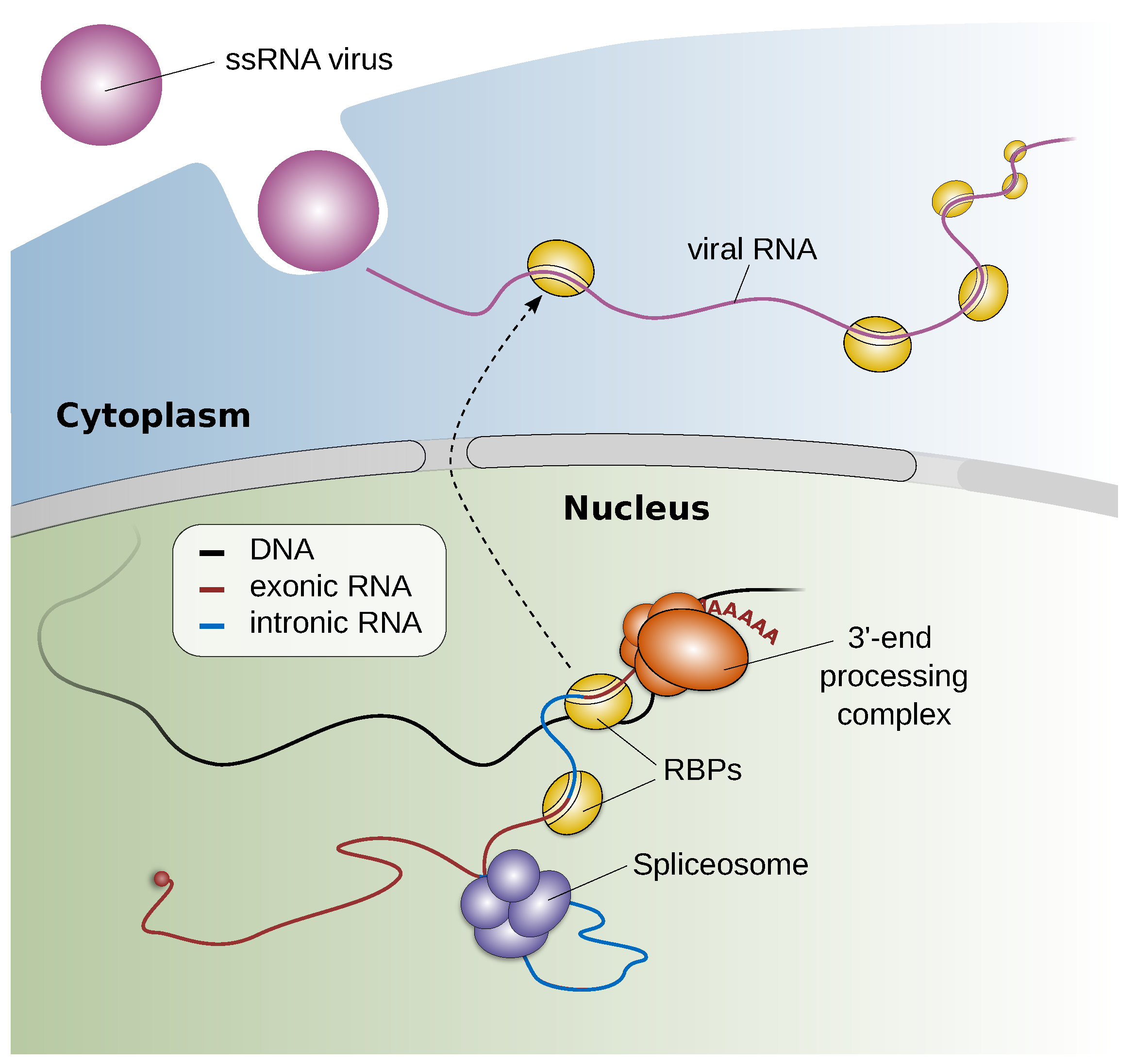
2.3.2. Staying below the Radar and Exploiting the Host—A Toolbox for Studying RNA Virus—Host Factor Interactions (by Andreas J. Gruber)
2.4. Viral Sequence Analysis
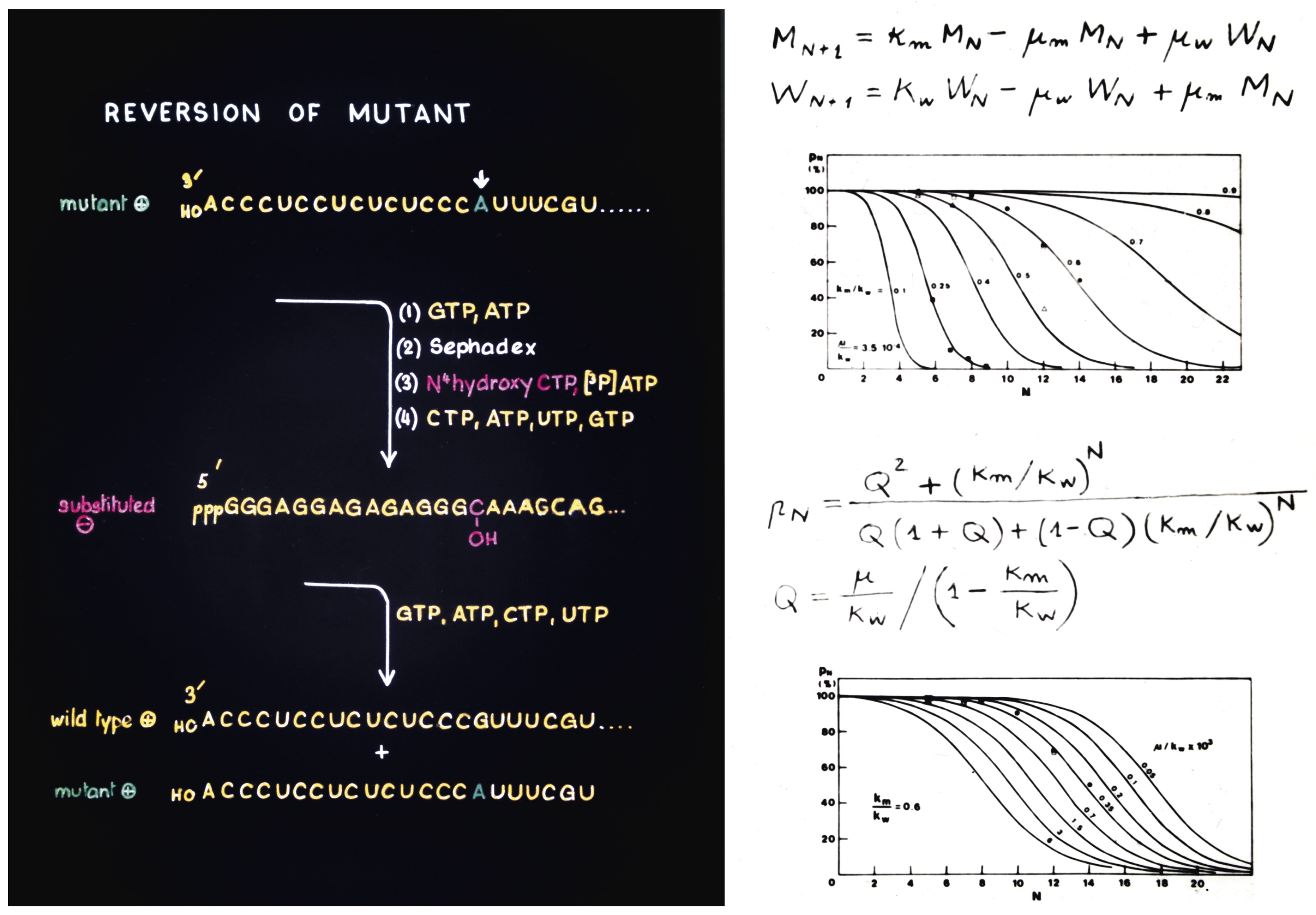
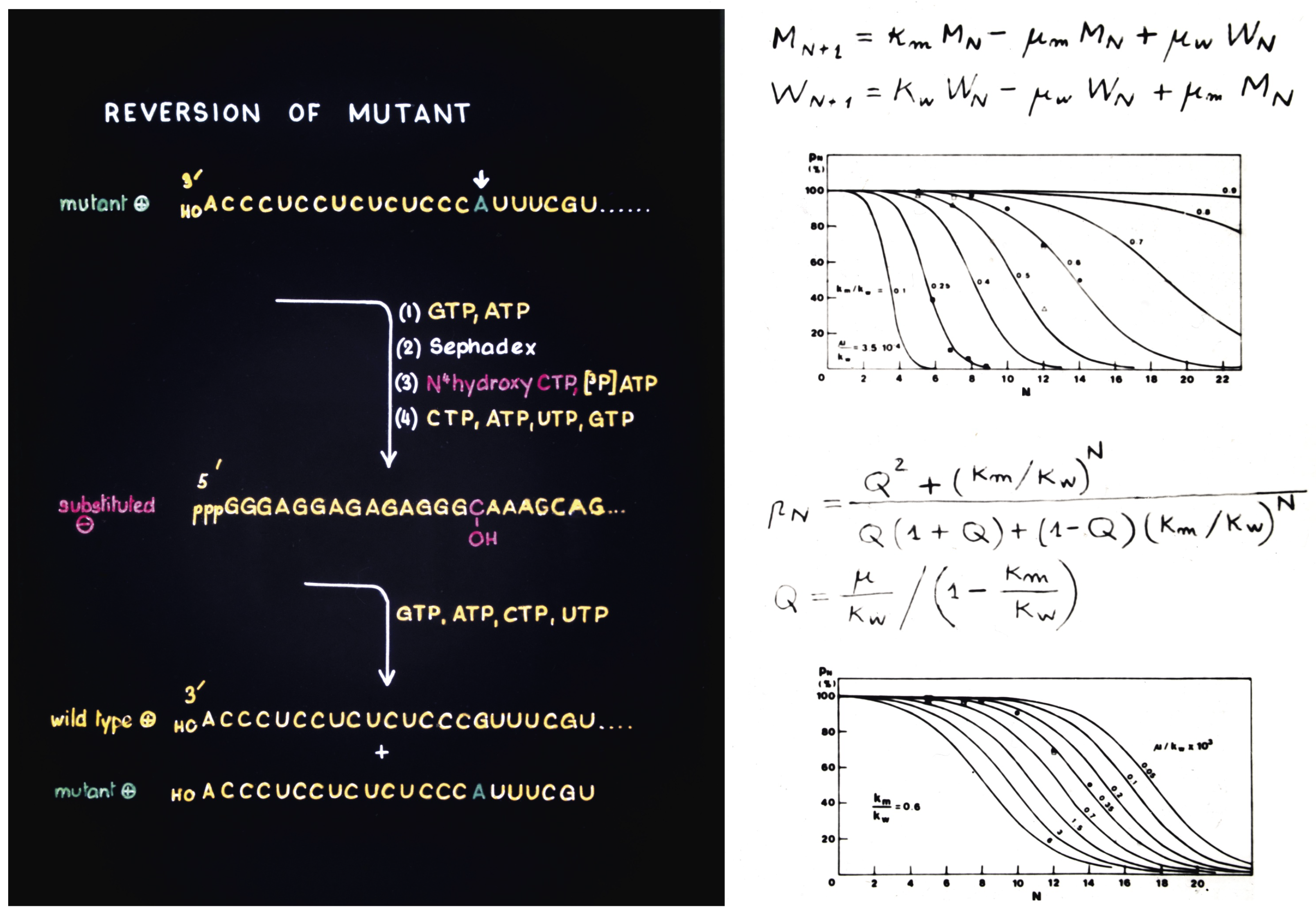
2.4.1. Origins and Implications of the Quasispecies Concept (by Esteban Domingo)
2.4.2. A Guidance to Store Your Virus Sequence and Knowledge (by Muriel Ritsch)
2.4.3. Reproducible RNA–RNA Interaction Probing for RNA Proximity Ligation Data with RNAswarm (by Gabriel Lencioni Lovate)
2.5. Virus Identification and Annotation
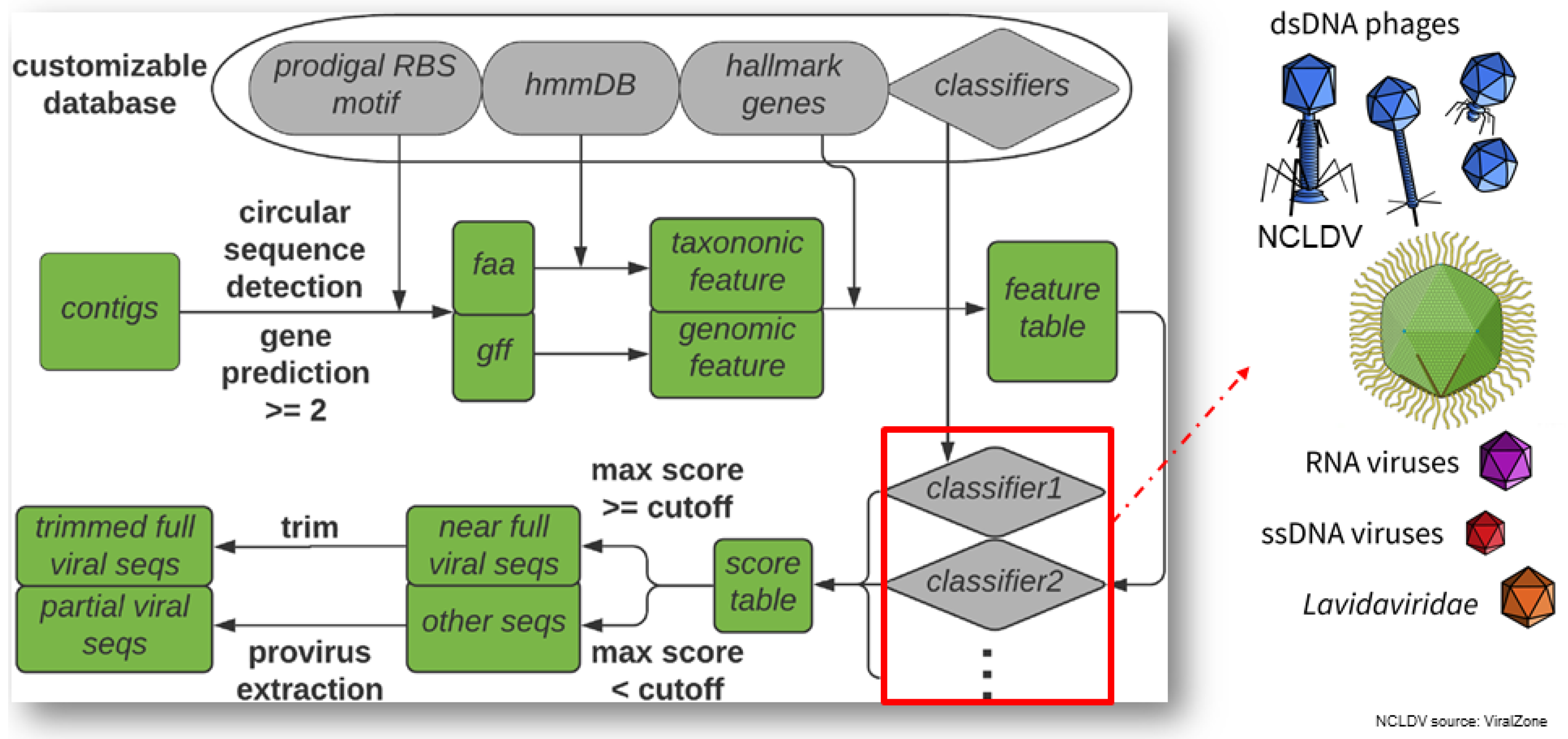
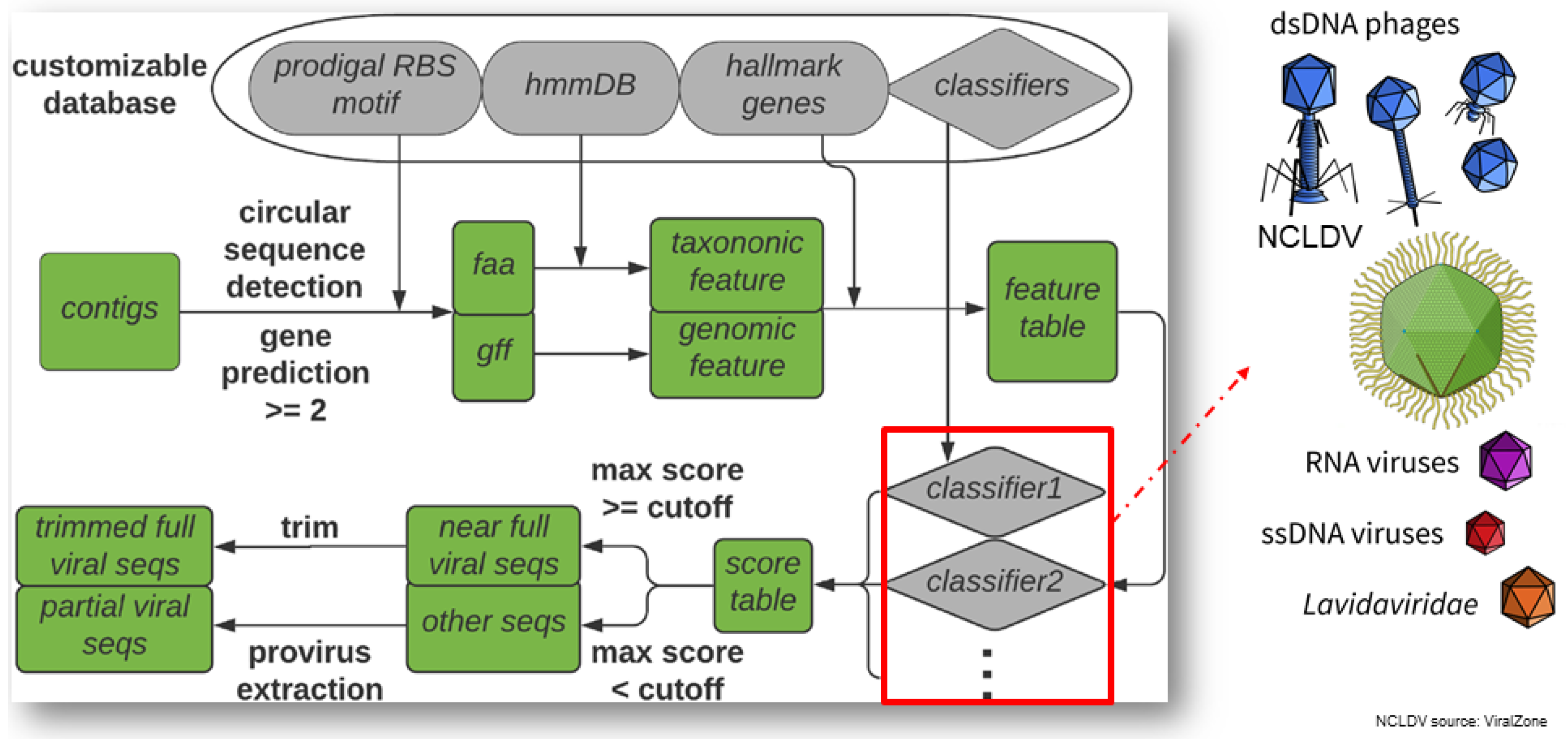
2.5.1. VirSorter2: A Multi-Classifier, Expert-Guided Approach to Detect Diverse DNA and RNA Viruses (by Jiarong Guo)
2.5.2. Evaluation of Gene-Calling Programs for Viral Genome Annotation (by Enrique González-Tortuero)
2.6. Phages
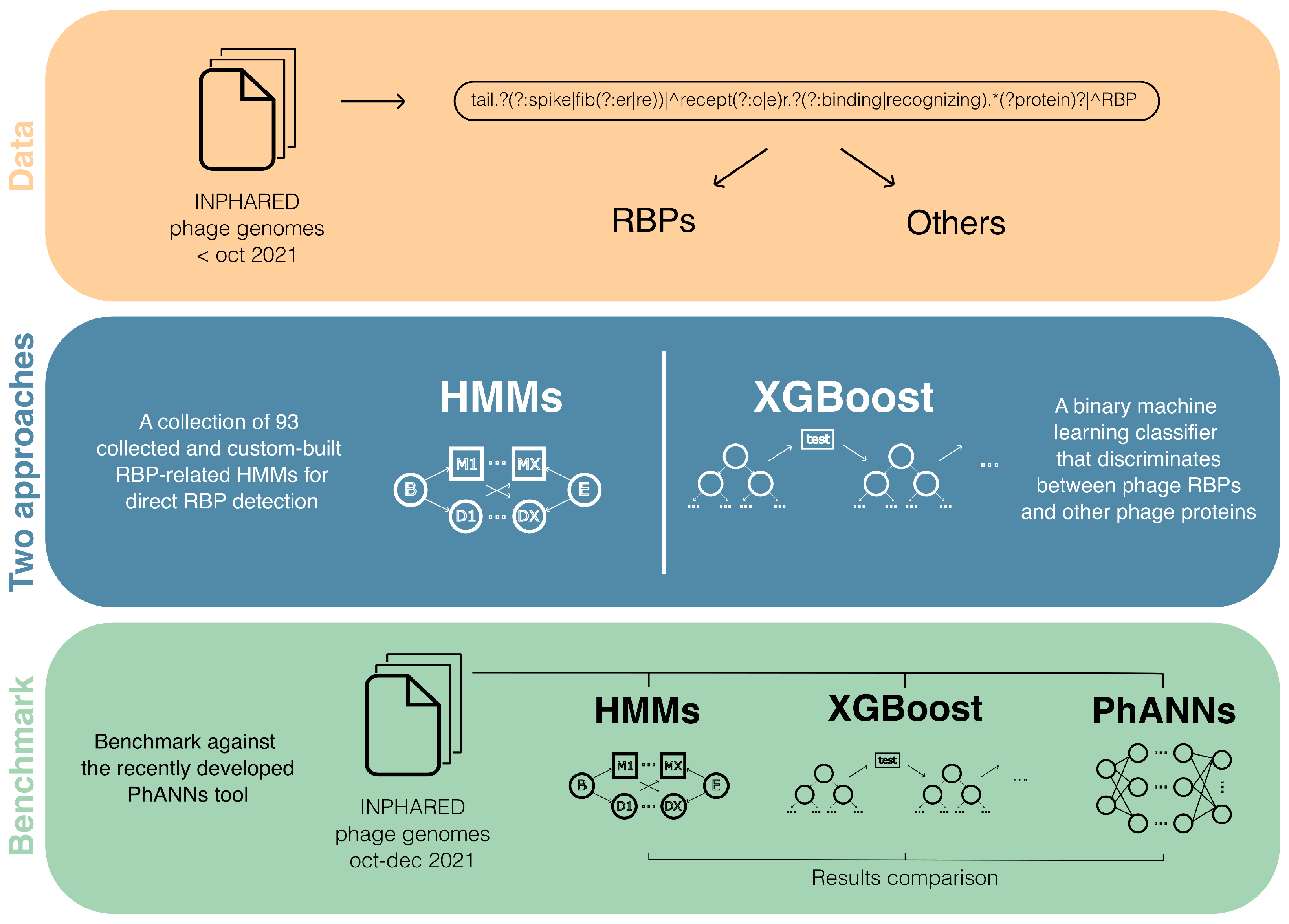
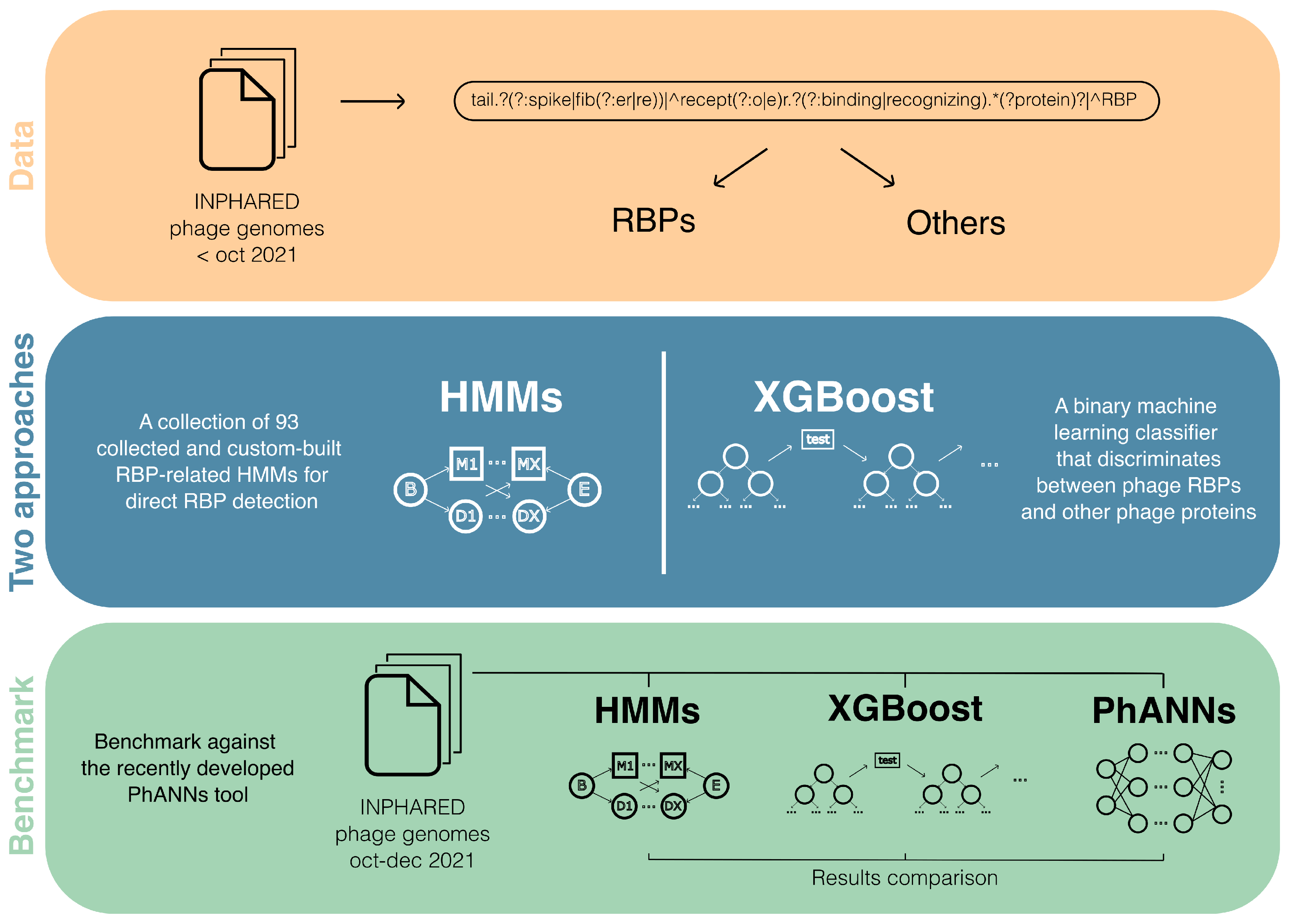
2.6.1. Dual Identification of Novel Phage Receptor-Binding Proteins Based on Protein Domains and Machine Learning (by Dimitri Boeckaerts)
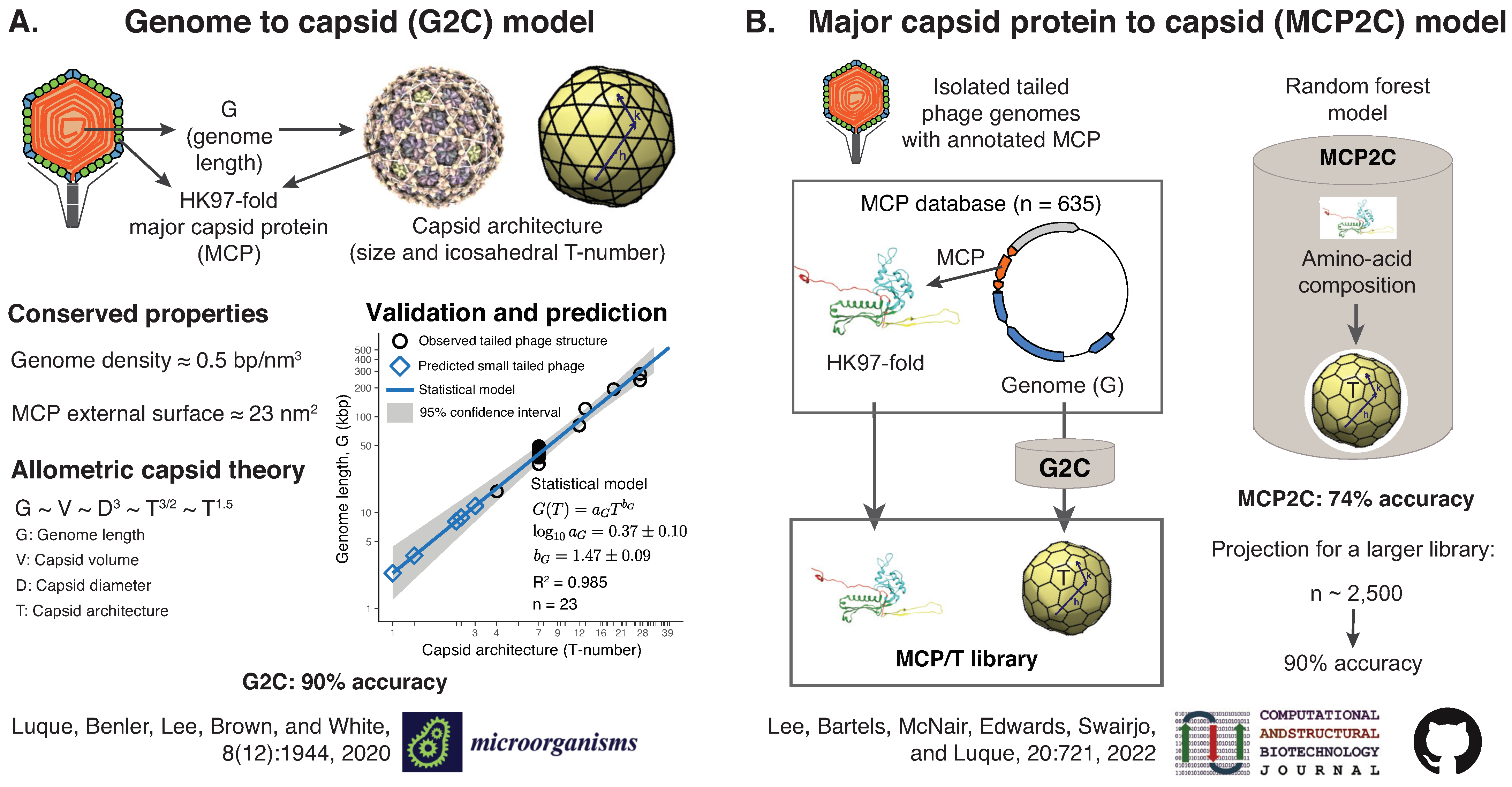
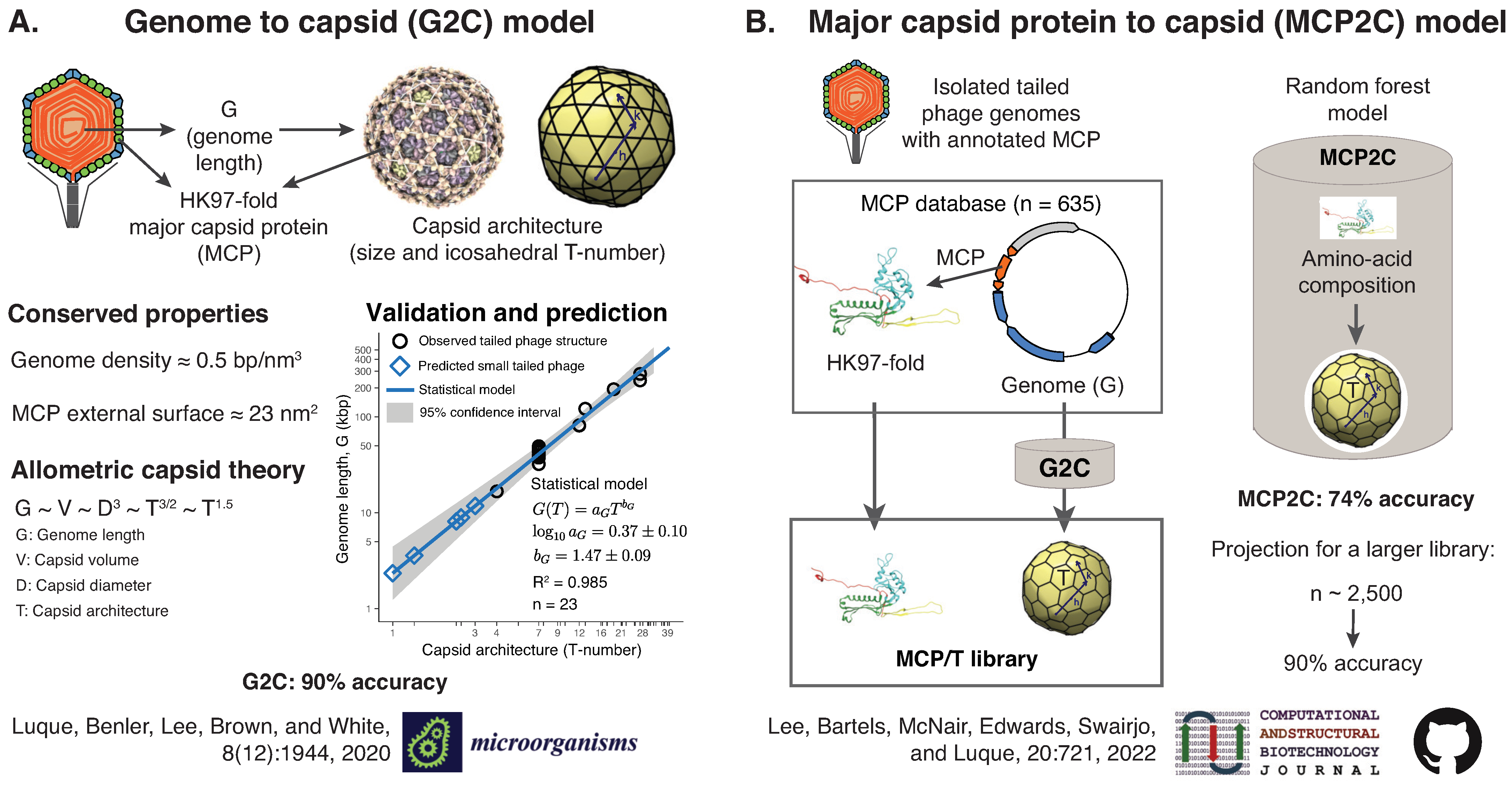
2.6.2. Predicting Viral Capsid Architectures from Metagenomes (by Antoni Luque)
2.6.3. A Blueprint of Tail Fiber Modularity and Its Relationship with Host Specificity for STEC Serovars (by Célia Pas)
2.7. Viral Diversity
2.7.1. Ocean Viruses: Patterns, Processes, and Paradigms on a Planetary Scale (by Matthew Sullivan)
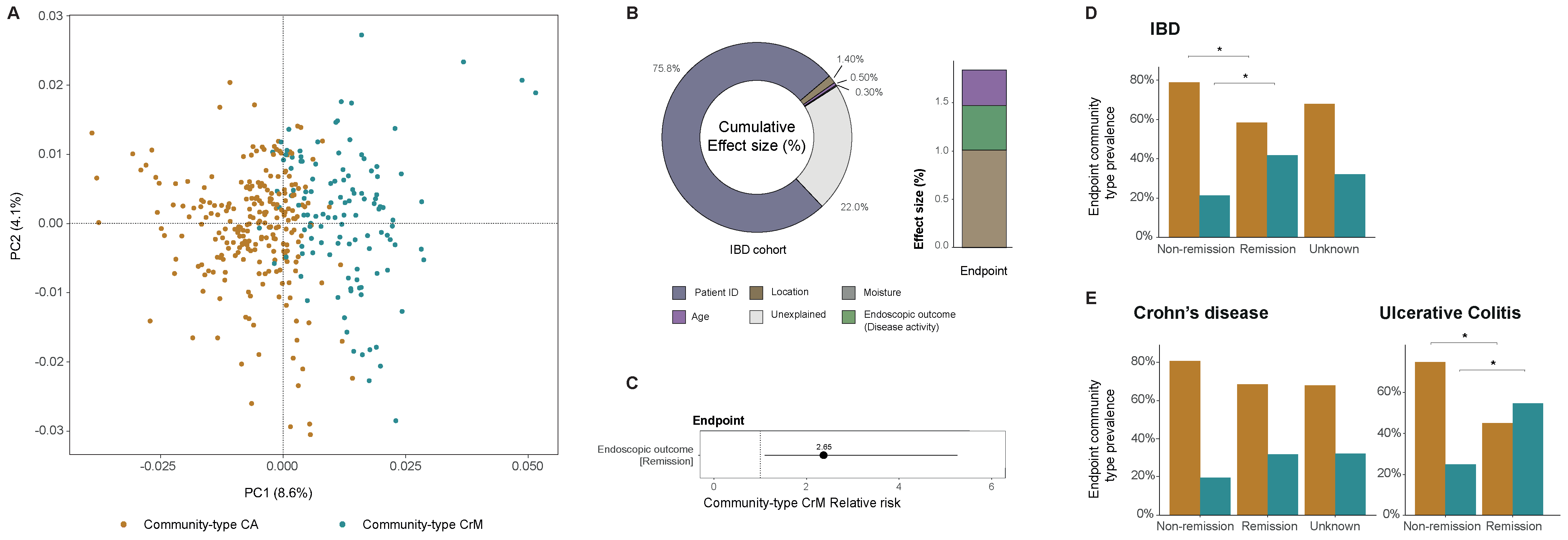
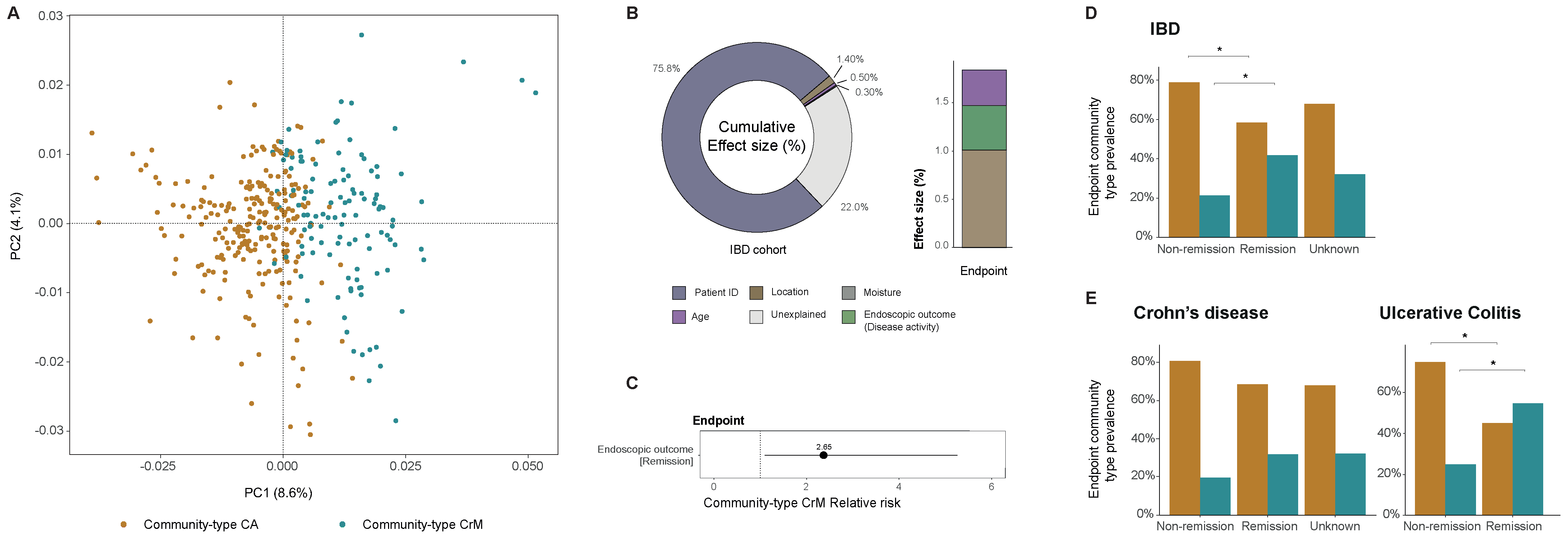
2.7.2. Community-Typing as a Way to Explore Virome Compositional Changes in IBD Patients (by Daan Jansen)
2.7.3. Virome Analyses of the Ancient Individuals Who Lived in the Japanese Archipelago 3000 Years Ago (by Luca Nishimura)
3. EVBC Annual Meeting
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ibrahim, B.; McMahon, D.P.; Hufsky, F.; Beer, M.; Deng, L.; Mercier, P.L.; Palmarini, M.; Thiel, V.; Marz, M. A new era of virus bioinformatics. Virus Res. 2018, 251, 86–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hufsky, F.; Ibrahim, B.; Beer, M.; Deng, L.; Mercier, P.L.; McMahon, D.P.; Palmarini, M.; Thiel, V.; Marz, M. Virologists—Heroes need weapons. PLoS Pathog. 2018, 14, e1006771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hufsky, F.; Beerenwinkel, N.; Meyer, I.M.; Roux, S.; Cook, G.M.; Kinsella, C.M.; Lamkiewicz, K.; Marquet, M.; Nieuwenhuijse, D.F.; Olendraite, I.; et al. The International Virus Bioinformatics Meeting 2020. Viruses 2020, 12, 1398. [Google Scholar] [CrossRef] [PubMed]
- Lemey, P.; Hong, S.L.; Hill, V.; Baele, G.; Poletto, C.; Colizza, V.; O’Toole, A.; McCrone, J.T.; Andersen, K.G.; Worobey, M.; et al. Accommodating individual travel history and unsampled diversity in Bayesian phylogeographic inference of SARS-CoV-2. Nat. Commun. 2020, 11, 5110. [Google Scholar] [CrossRef] [PubMed]
- Worobey, M.; Pekar, J.; Larsen, B.B.; Nelson, M.I.; Hill, V.; Joy, J.B.; Rambaut, A.; Suchard, M.A.; Wertheim, J.O.; Lemey, P. The emergence of SARS-CoV-2 in Europe and North America. Science 2020, 370, 564–570. [Google Scholar] [CrossRef]
- Lemey, P.; Ruktanonchai, N.; Hong, S.L.; Colizza, V.; Poletto, C.; den Broeck, F.V.; Gill, M.S.; Ji, X.; Levasseur, A.; Munnink, B.B.O.; et al. Untangling introductions and persistence in COVID-19 resurgence in Europe. Nature 2021, 595, 713–717. [Google Scholar] [CrossRef]
- Sanchez-Felipe, L.; Vercruysse, T.; Sharma, S.; Ma, J.; Lemmens, V.; Looveren, D.V.; Javarappa, M.P.A.; Boudewijns, R.; Malengier-Devlies, B.; Liesenborghs, L.; et al. A single-dose live-attenuated YF17D-vectored SARS-CoV-2 vaccine candidate. Nature 2020, 590, 320–325. [Google Scholar] [CrossRef]
- Martin, D.P.; Weaver, S.; Tegally, H.; San, J.E.; Shank, S.D.; Wilkinson, E.; Lucaci, A.G.; Giandhari, J.; Naidoo, S.; Pillay, Y.; et al. The emergence and ongoing convergent evolution of the SARS-CoV-2 N501Y lineages. Cell 2021, 184, 5189–5200.e7. [Google Scholar] [CrossRef]
- Sharma, S.; Vercruysse, T.; Sanchez-Felipe, L.; Kerstens, W.; Rasulova, M.; Abdelnabi, R.; Foo, C.S.; Lemmens, V.; Looveren, D.V.; Maes, P.; et al. Updated vaccine protects from infection with SARS-CoV-2 variants, prevents transmission and is immunogenic against Omicron in hamsters. bioRxiv 2021. [Google Scholar] [CrossRef]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Peacock, S.J.; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef]
- Greaney, A.J.; Starr, T.N.; Bloom, J.D. An antibody-escape calculator for mutations to the SARS-CoV-2 receptor-binding domain. bioRxiv 2021. [Google Scholar] [CrossRef]
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data-from vision to reality. Eurosurveillance 2017, 22, 30494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, E.M.; Jia, H.; Shankar, A.; Hanson, D.; Luo, W.; Masciotra, S.; Owen, S.M.; Oster, A.M.; Galang, R.R.; Spiller, M.W.; et al. Detailed Transmission Network Analysis of a Large Opiate-Driven Outbreak of HIV Infection in the United States. J. Infect. Dis. 2017, 216, 1053–1062. [Google Scholar] [CrossRef] [PubMed]
- Andre, M.; Ijaz, K.; Tillinghast, J.D.; Krebs, V.E.; Diem, L.A.; Metchock, B.; Crisp, T.; McElroy, P.D. Transmission network analysis to complement routine tuberculosis contact investigations. Am. J. Public Health 2007, 97, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Harper, H.; Burridge, A.; Winfield, M.; Finn, A.; Davidson, A.; Matthews, D.; Hutchings, S.; Vipond, B.; Jain, N.; the COVID-19 Genomics UK (COG-UK) Consortium. Detecting SARS-CoV-2 variants with SNP genotyping. PLoS ONE 2021, 16, e0243185. [Google Scholar] [CrossRef] [PubMed]
- Sender, R.; Bar-On, Y.M.; Gleizer, S.; Bernshtein, B.; Flamholz, A.; Phillips, R.; Milo, R. The total number and mass of SARS-CoV-2 virions. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef] [PubMed]
- Aksamentov, I.; Roemer, C.; Hodcroft, E.B.; Neher, R.A. Nextclade: Clade assignment, mutation calling and quality control for viral genomes. J. Open Source Softw. 2021, 6, 3773. [Google Scholar] [CrossRef]
- Wuerth, J.D.; Weber, F. Phleboviruses and the Type I Interferon Response. Viruses 2016, 8, 174. [Google Scholar] [CrossRef] [Green Version]
- Kainulainen, M.; Habjan, M.; Hubel, P.; Busch, L.; Lau, S.; Colinge, J.; Superti-Furga, G.; Pichlmair, A.; Weber, F. Virulence factor NSs of rift valley fever virus recruits the F-box protein FBXO3 to degrade subunit p62 of general transcription factor TFIIH. J. Virol. 2014, 88, 3464–3473. [Google Scholar] [CrossRef] [Green Version]
- Kainulainen, M.; Lau, S.; Samuel, C.E.; Hornung, V.; Weber, F. NSs Virulence Factor of Rift Valley Fever Virus Engages the F-Box Proteins FBXW11 and β-TRCP1 To Degrade the Antiviral Protein Kinase PKR. J. Virol. 2016, 90, 6140–6147. [Google Scholar] [CrossRef] [Green Version]
- Wuerth, J.D.; Habjan, M.; Wulle, J.; Superti-Furga, G.; Pichlmair, A.; Weber, F. NSs Protein of Sandfly Fever Sicilian Phlebovirus Counteracts Interferon (IFN) Induction by Masking the DNA-Binding Domain of IFN Regulatory Factor 3. J. Virol. 2018, 92, e01202-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wuerth, J.D.; Weber, F. NSs of the mildly virulent sandfly fever Sicilian virus is unable to inhibit interferon signaling and upregulation of interferon-stimulated genes. J. Gen. Virol. 2021, 102, 001676. [Google Scholar] [CrossRef] [PubMed]
- Wuerth, J.D.; Habjan, M.; Kainulainen, M.; Berisha, B.; Bertheloot, D.; Superti-Furga, G.; Pichlmair, A.; Weber, F. eIF2B as a Target for Viral Evasion of PKR-Mediated Translation Inhibition. mBio 2020, 11, e00976-20. [Google Scholar] [CrossRef] [PubMed]
- Kashiwagi, K.; Shichino, Y.; Osaki, T.; Sakamoto, A.; Nishimoto, M.; Takahashi, M.; Mito, M.; Weber, F.; Ikeuchi, Y.; Iwasaki, S.; et al. eIF2B-capturing viral protein NSs suppresses the integrated stress response. Nat. Commun. 2021, 12, 7102. [Google Scholar] [CrossRef]
- Barnhart, M.D.; Moon, S.L.; Emch, A.W.; Wilusz, C.J.; Wilusz, J. Changes in Cellular mRNA Stability, Splicing, and Polyadenylation through HuR Protein Sequestration by a Cytoplasmic RNA Virus. Cell. Rep. 2013, 5, 909–917. [Google Scholar] [CrossRef] [Green Version]
- Lal, A.; Ferrarini, M.G.; Gruber, A.J. Investigating the human host-ssRNA virus interaction landscape using the SMEAGOL toolbox. bioRxiv 2021. [Google Scholar] [CrossRef]
- Bak, M.; van Nimwegen, E.; Schmidt, R.; Zavolan, M.; Gruber, A.J. Frequent co-regulation of splicing and polyadenylation by RNA-binding proteins inferred with MAPP. bioRxiv 2022. [Google Scholar] [CrossRef]
- Lytras, S.; Hughes, J. Synonymous Dinucleotide Usage: A Codon-Aware Metric for Quantifying Dinucleotide Representation in Viruses. Viruses 2020, 12, 462. [Google Scholar] [CrossRef] [Green Version]
- Domingo, E.; Perales, C. Viral quasispecies. PLoS Genet. 2019, 15, e1008271. [Google Scholar] [CrossRef] [Green Version]
- Eigen, M.; Schuster, P. The Hypercycle; Springer: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Batschelet, E.; Domingo, E.; Weissmann, C. The proportion of revertant and mutant phage in a growing population, as a function of mutation and growth rate. Gene 1976, 1, 27–32. [Google Scholar] [CrossRef]
- Domingo, E.; Sabo, D.; Taniguchi, T.; Weissmann, C. Nucleotide sequence heterogeneity of an RNA phage population. Cell 1978, 13, 735–744. [Google Scholar] [CrossRef]
- Flavell, R.; Sabo, D.; Bandle, E.; Weissmann, C. Site-directed mutagenesis: Generation of an extracistronic mutation in bacteriophage Qβ RNA. J. Mol. Biol. 1974, 89, 255–272. [Google Scholar] [CrossRef]
- Domingo, E.; Flavell, R.; Weissmann, C. In vitro site-directed mutagenesis: Generation and properties of an infectious extracistronic mutant of bacteriophage Qβ. Gene 1976, 1, 3–25. [Google Scholar] [CrossRef]
- Domingo, E.; García-Crespo, C.; Perales, C. Historical Perspective on the Discovery of the Quasispecies Concept. Annu. Rev. Virol. 2021, 8, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Priyadarshini, P.; Vrati, S. Unraveling the web of viroinformatics: Computational tools and databases in virus research. J. Virol. 2015, 89, 1489–1501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuiken, C.; Korber, B.; Shafer, R.W. HIV sequence databases. AIDS Rev. 2003, 5, 52–61. [Google Scholar]
- Paez-Espino, D.; Eloe-Fadrosh, E.A.; Pavlopoulos, G.A.; Thomas, A.D.; Huntemann, M.; Mikhailova, N.; Rubin, E.; Ivanova, N.N.; Kyrpides, N.C. Uncovering Earth’s virome. Nature 2016, 536, 425–430. [Google Scholar] [CrossRef]
- Roux, S.; Adriaenssens, E.M.; Dutilh, B.E.; Koonin, E.V.; Kropinski, A.M.; Krupovic, M.; Kuhn, J.H.; Lavigne, R.; Brister, J.R.; Varsani, A.; et al. Minimum Information about an Uncultivated Virus Genome (MIUViG). Nat. Biotechnol. 2019, 37, 29–37. [Google Scholar] [CrossRef]
- Guo, J.; Bolduc, B.; Zayed, A.A.; Varsani, A.; Dominguez-Huerta, G.; Delmont, T.O.; Pratama, A.A.; Gazitúa, M.C.; Vik, D.; Sullivan, M.B.; et al. VirSorter2: A multi-classifier, expert-guided approach to detect diverse DNA and RNA viruses. Microbiome 2021, 9, 37. [Google Scholar] [CrossRef]
- Roux, S.; Enault, F.; Hurwitz, B.L.; Sullivan, M.B. VirSorter: Mining viral signal from microbial genomic data. PeerJ 2015, 3, e985. [Google Scholar] [CrossRef]
- Zayed, A.A.; Lücking, D.; Mohssen, M.; Cronin, D.; Bolduc, B.; Gregory, A.C.; Hargreaves, K.R.; Piehowski, P.D.; III, R.A.W.; Huang, E.L.; et al. efam: An expanded, metaproteome-supported HMM profile database of viral protein families. Bioinformatics 2021, 37, 4202–4208. [Google Scholar] [CrossRef] [PubMed]
- Koster, J.; Rahmann, S. Snakemake–a scalable bioinformatics workflow engine. Bioinformatics 2012, 28, 2520–2522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyatt, D.; LoCascio, P.F.; Hauser, L.J.; Uberbacher, E.C. Gene and translation initiation site prediction in metagenomic sequences. Bioinformatics 2012, 28, 2223–2230. [Google Scholar] [CrossRef]
- Rho, M.; Tang, H.; Ye, Y. FragGeneScan: Predicting genes in short and error-prone reads. Nucleic Acids Res. 2010, 38, e191. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; Chen, G.L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Besemer, J.; Lomsadze, A.; Borodovsky, M. GeneMarkS: A self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001, 29, 2607–2618. [Google Scholar] [CrossRef] [Green Version]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Dunne, M.; Prokhorov, N.S.; Loessner, M.J.; Leiman, P.G. Reprogramming bacteriophage host range: Design principles and strategies for engineering receptor binding proteins. Curr. Opin. Biotechnol. 2021, 68, 272–281, Systems Biology Nanobiotechnology. [Google Scholar] [CrossRef]
- Cantu, V.A.; Salamon, P.; Seguritan, V.; Redfield, J.; Salamon, D.; Edwards, R.A.; Segall, A.M. PhANNs, a fast and accurate tool and web server to classify phage structural proteins. PLoS Comput. Biol. 2020, 16, 1–18. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2020, 49, D412–D419. [Google Scholar] [CrossRef]
- Chen, T.; Guestrin, C. Xgboost: A scalable tree boosting system. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016. [Google Scholar]
- Ackermann, H.W. 5500 Phages examined in the electron microscope. Arch. Virol. 2006, 152, 227–243. [Google Scholar] [CrossRef] [PubMed]
- Brum, J.R.; Schenck, R.O.; Sullivan, M.B. Global morphological analysis of marine viruses shows minimal regional variation and dominance of non-tailed viruses. ISME J. 2013, 7, 1738–1751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montiel-Garcia, D.; Santoyo-Rivera, N.; Ho, P.; Carrillo-Tripp, M.; Brooks, C.L., III; Johnson, J.E.; Reddy, V.S. VIPERdb v3.0: A structure-based data analytics platform for viral capsids. Nucleic Acids Res. 2020, 49, D809–D816. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Dolja, V.V.; Krupovic, M.; Varsani, A.; Wolf, Y.I.; Yutin, N.; Zerbini, F.M.; Kuhn, J.H. Global Organization and Proposed Megataxonomy of the Virus World. Microbiol. Mol. Biol. Rev. 2020, 84, e00061-19. [Google Scholar] [CrossRef] [PubMed]
- Twarock, R.; Luque, A. Structural puzzles in virology solved with an overarching icosahedral design principle. Nat. Commun. 2019, 10, 4414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendrix, R.W.; Johnson, J.E. Bacteriophage HK97 Capsid Assembly and Maturation. In Viral Molecular Machines; Springer: Boston, MA, USA, 2011; pp. 351–363. [Google Scholar] [CrossRef]
- Suhanovsky, M.M.; Teschke, C.M. Nature’s favorite building block: Deciphering folding and capsid assembly of proteins with the HK97-fold. Virology 2015, 479–480, 487–497. [Google Scholar] [CrossRef] [Green Version]
- Luque, A.; Benler, S.; Lee, D.Y.; Brown, C.; White, S. The Missing Tailed Phages: Prediction of Small Capsid Candidates. Microorganisms 2020, 8, 1944. [Google Scholar] [CrossRef]
- Benler, S.; Yutin, N.; Antipov, D.; Rayko, M.; Shmakov, S.; Gussow, A.B.; Pevzner, P.; Koonin, E.V. Thousands of previously unknown phages discovered in whole-community human gut metagenomes. Microbiome 2021, 9, 78. [Google Scholar] [CrossRef]
- Nayfach, S.; Camargo, A.P.; Schulz, F.; Eloe-Fadrosh, E.; Roux, S.; Kyrpides, N.C. CheckV assesses the quality and completeness of metagenome-assembled viral genomes. Nat. Biotechnol. 2020, 39, 578–585. [Google Scholar] [CrossRef]
- Lee, D.Y.; Bartels, C.; McNair, K.; Edwards, R.A.; Swairjo, M.A.; Luque, A. Predicting the capsid architecture of phages from metagenomic data. Comput. Struct. Biotechnol. J. 2022, 20, 721–732. [Google Scholar] [CrossRef]
- Krupovic, M.; Koonin, E.V. Multiple origins of viral capsid proteins from cellular ancestors. Proc. Natl. Acad. Sci. USA 2017, 114, E2401–E2410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costea, P.I.; Hildebrand, F.; Arumugam, M.; Bäckhed, F.; Blaser, M.J.; Bushman, F.D.; de Vos, W.M.; Ehrlich, S.D.; Fraser, C.M.; Hattori, M.; et al. Enterotypes in the landscape of gut microbial community composition. Nat. Microbiol. 2017, 3, 8–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmes, I.; Harris, K.; Quince, C. Dirichlet Multinomial Mixtures: Generative Models for Microbial Metagenomics. PLoS ONE 2012, 7, e30126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conceição-Neto, N.; Zeller, M.; Lefrère, H.; Bruyn, P.D.; Beller, L.; Deboutte, W.; Yinda, C.K.; Lavigne, R.; Maes, P.; Ranst, M.V.; et al. Modular approach to customise sample preparation procedures for viral metagenomics: A reproducible protocol for virome analysis. Sci. Rep. 2015, 5, 16532. [Google Scholar] [CrossRef] [Green Version]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. metaSPAdes: A new versatile metagenomic assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef] [Green Version]
- Md, V.; Misra, S.; Li, H.; Aluru, S. Efficient Architecture-Aware Acceleration of BWA-MEM for Multicore Systems. arXiv 2019, arXiv:1907.12931. [Google Scholar]
- Nayfach, S.; Páez-Espino, D.; Call, L.; Low, S.J.; Sberro, H.; Ivanova, N.N.; Proal, A.D.; Fischbach, M.A.; Bhatt, A.S.; Hugenholtz, P.; et al. Metagenomic compendium of 189,680 DNA viruses from the human gut microbiome. Nat. Microbiol. 2021, 6, 960–970. [Google Scholar] [CrossRef]
- Nishimura, L.; Sugimoto, R.; Inoue, J.; Nakaoka, H.; Kanzawa-Kiriyama, H.; Shinoda, K.-i.; Inoue, I. Identification of ancient viruses from metagenomic data of the Jomon people. J. Hum. Genet. 2020, 66, 287–296. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hufsky, F.; Beslic, D.; Boeckaerts, D.; Duchene, S.; González-Tortuero, E.; Gruber, A.J.; Guo, J.; Jansen, D.; Juma, J.; Kongkitimanon, K.; et al. The International Virus Bioinformatics Meeting 2022. Viruses 2022, 14, 973. https://doi.org/10.3390/v14050973
Hufsky F, Beslic D, Boeckaerts D, Duchene S, González-Tortuero E, Gruber AJ, Guo J, Jansen D, Juma J, Kongkitimanon K, et al. The International Virus Bioinformatics Meeting 2022. Viruses. 2022; 14(5):973. https://doi.org/10.3390/v14050973
Chicago/Turabian StyleHufsky, Franziska, Denis Beslic, Dimitri Boeckaerts, Sebastian Duchene, Enrique González-Tortuero, Andreas J. Gruber, Jiarong Guo, Daan Jansen, John Juma, Kunaphas Kongkitimanon, and et al. 2022. "The International Virus Bioinformatics Meeting 2022" Viruses 14, no. 5: 973. https://doi.org/10.3390/v14050973
APA StyleHufsky, F., Beslic, D., Boeckaerts, D., Duchene, S., González-Tortuero, E., Gruber, A. J., Guo, J., Jansen, D., Juma, J., Kongkitimanon, K., Luque, A., Ritsch, M., Lencioni Lovate, G., Nishimura, L., Pas, C., Domingo, E., Hodcroft, E., Lemey, P., Sullivan, M. B., ... Marz, M. (2022). The International Virus Bioinformatics Meeting 2022. Viruses, 14(5), 973. https://doi.org/10.3390/v14050973