
Signaling Pathway Reporter Screen with SARS-CoV-2 Proteins Identifies nsp5 as a Repressor of p53 Activity



Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Plasmids
2.2. Luciferase Reporter Assay
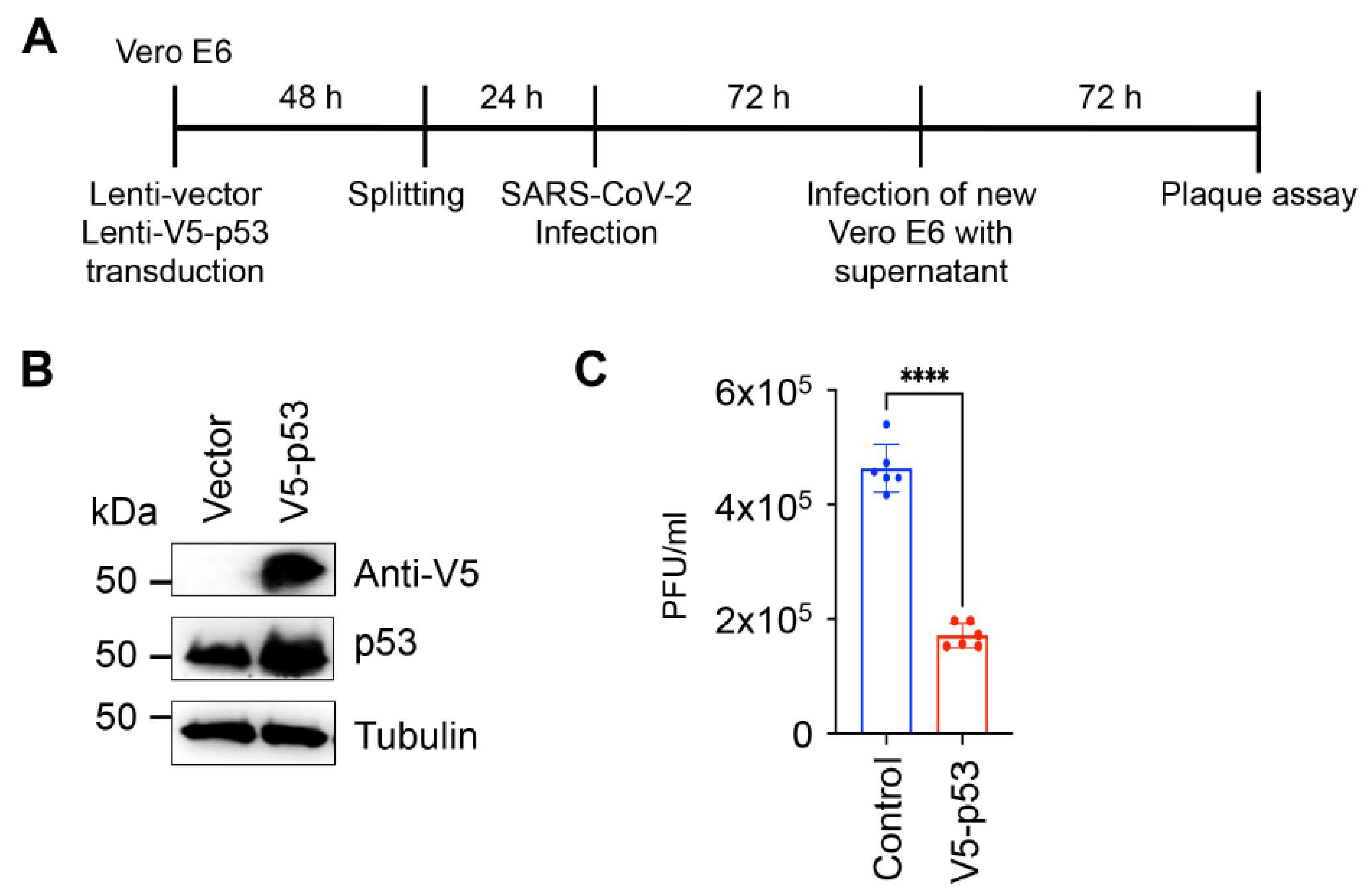
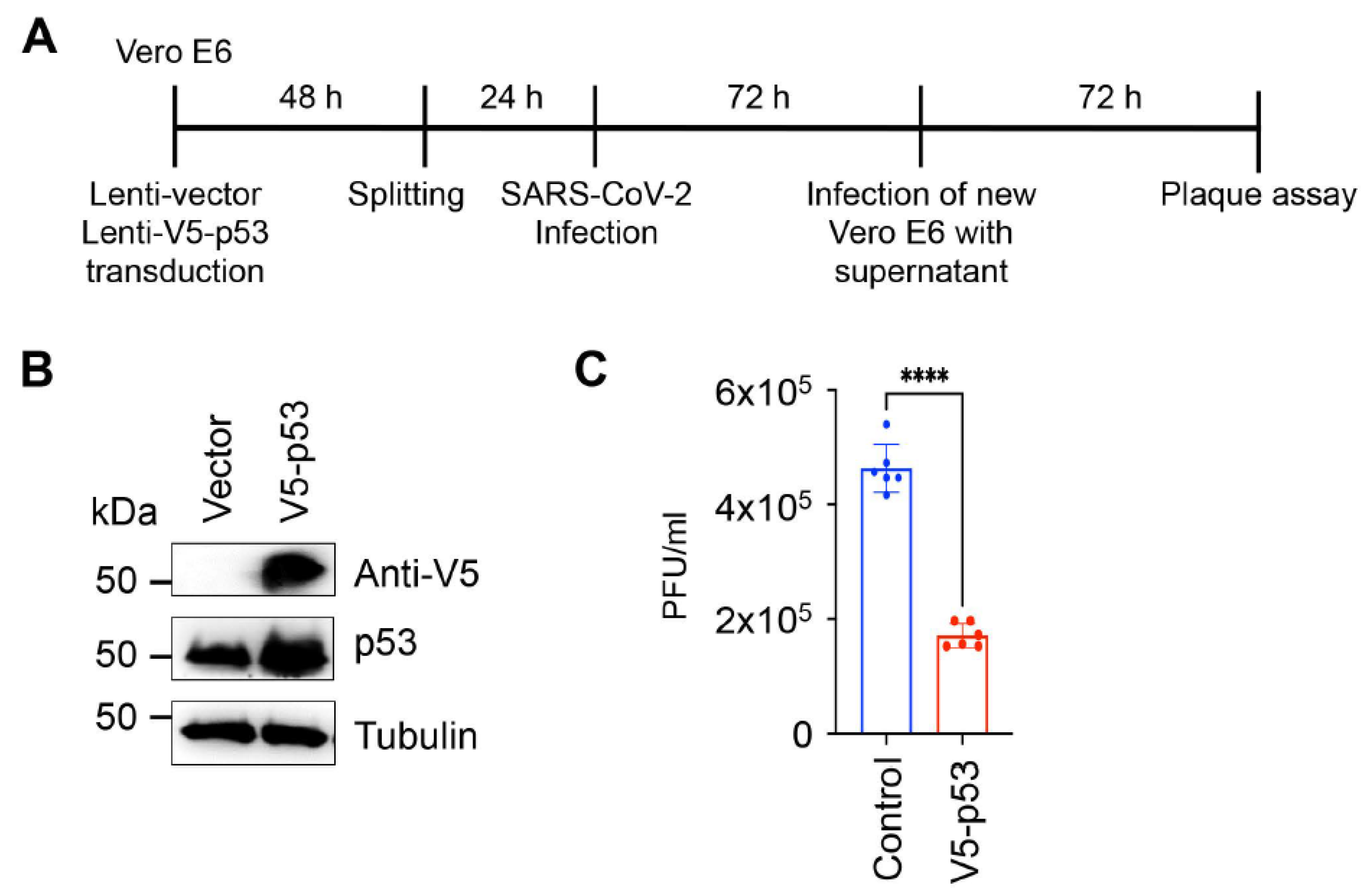
2.3. SARS-CoV-2 Infection and Plaque Assay
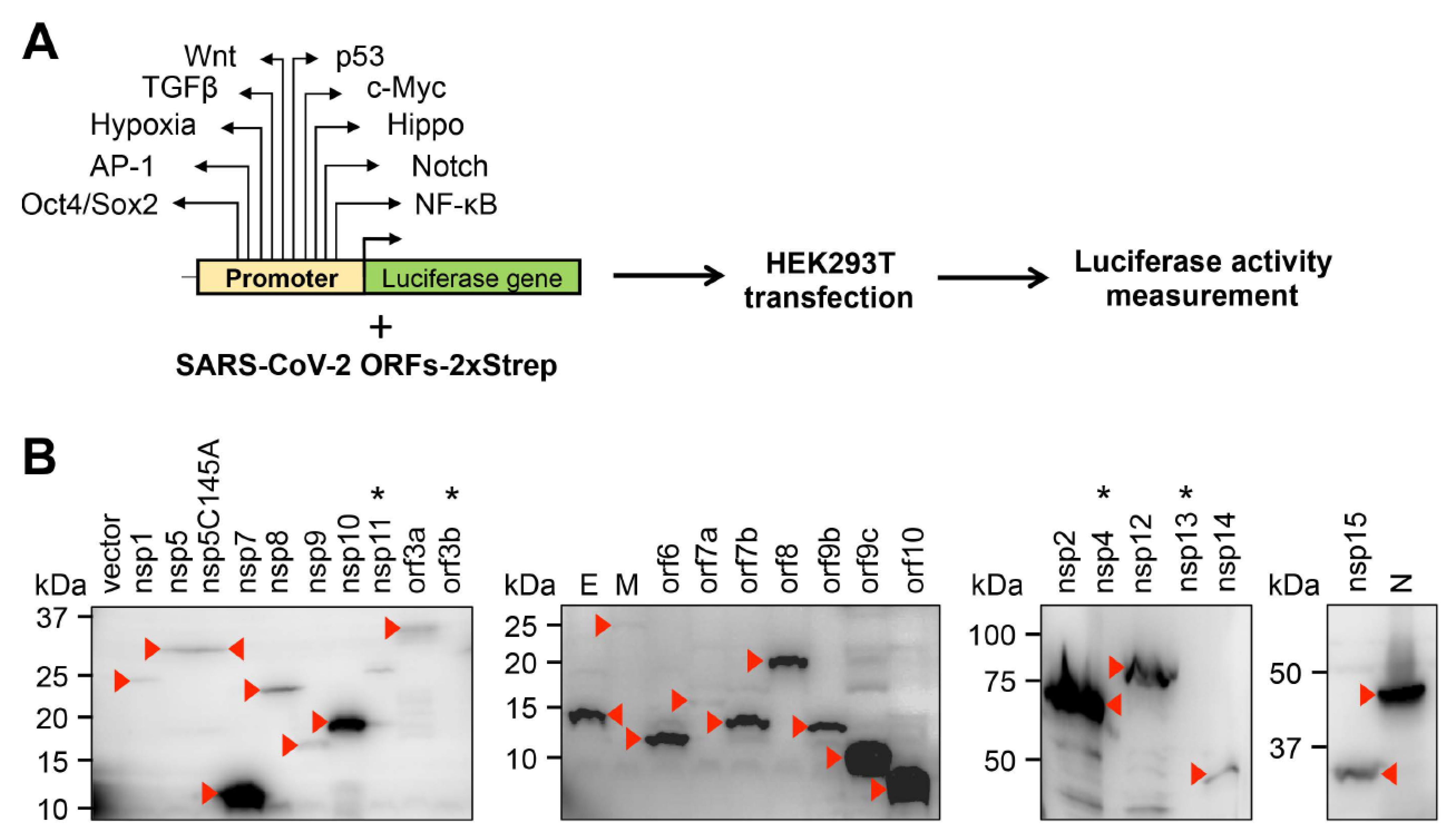
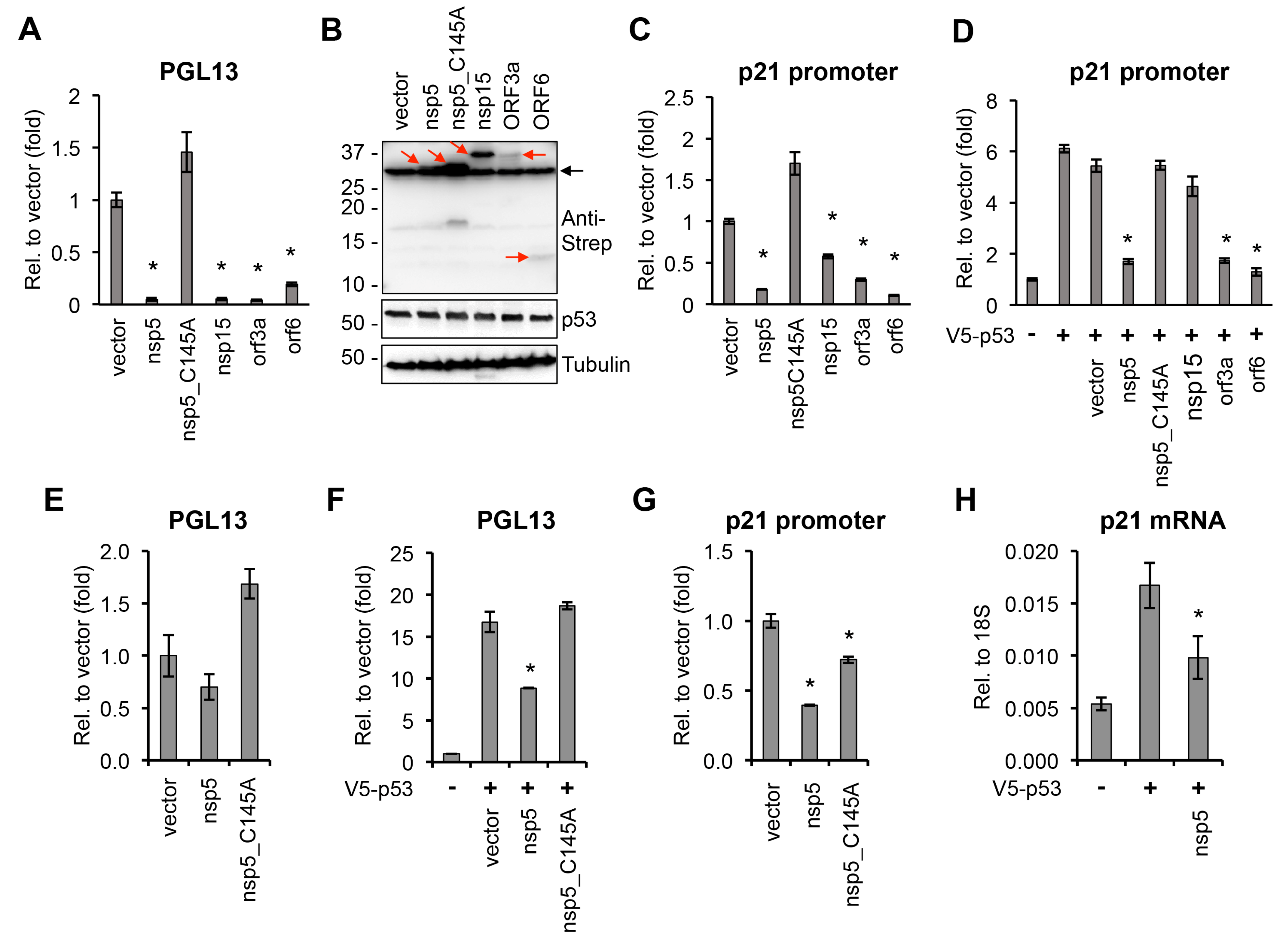
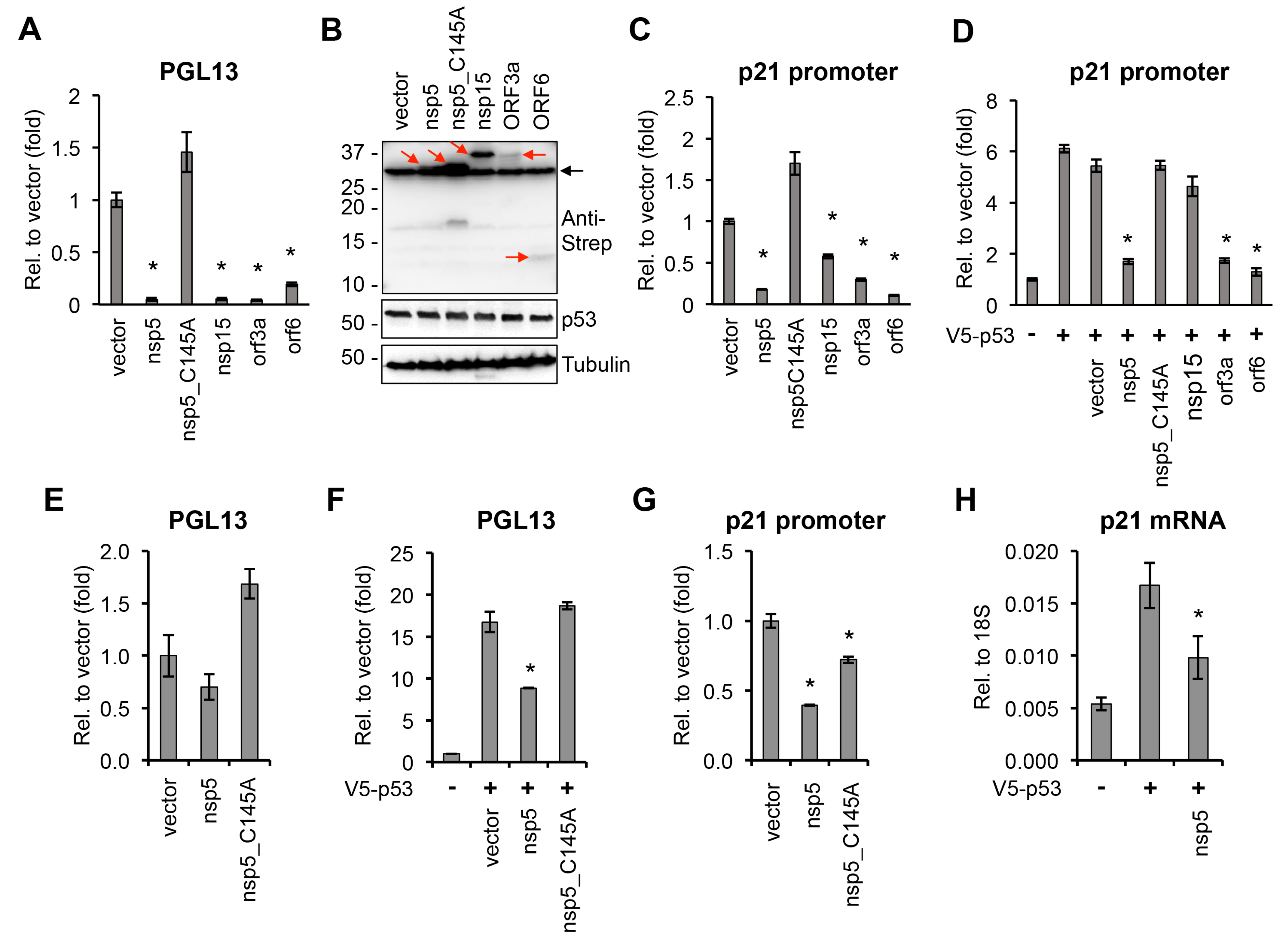
3. Results and Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. China Novel Coronavirus I, Research T. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.X.; Fung, T.S.; Chong, K.K.; Shukla, A.; Hilgenfeld, R. Accessory proteins of SARS-CoV and other coronaviruses. Antivir. Res. 2014, 109, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Yount, B.; Roberts, R.S.; Sims, A.C.; Deming, D.; Frieman, M.B.; Sparks, J.; Denison, M.R.; Davis, N.; Baric, R.S. Severe acute respiratory syndrome coronavirus group-specific open reading frames encode nonessential functions for replication in cell cultures and mice. J. Virol. 2005, 79, 14909–14922. [Google Scholar] [CrossRef] [Green Version]
- Puelles, V.G.; Lutgehetmann, M.; Lindenmeyer, M.T.; Sperhake, J.P.; Wong, M.N.; Allweiss, L.; Chilla, S.; Heinemann, A.; Wanner, N.; Liu, S.; et al. Multiorgan and Renal Tropism of SARS-CoV-2. N. Engl. J. Med. 2020, 383, 590–592. [Google Scholar] [CrossRef]
- Trypsteen, W.; Van Cleemput, J.; Snippenberg, W.V.; Gerlo, S.; Vandekerckhove, L. On the whereabouts of SARS-CoV-2 in the human body: A systematic review. PLoS Pathog. 2020, 16, e1009037. [Google Scholar] [CrossRef]
- Kyriakidis, N.C.; Lopez-Cortes, A.; Gonzalez, E.V.; Grimaldos, A.B.; Prado, E.O. SARS-CoV-2 vaccines strategies: A comprehensive review of phase 3 candidates. npj Vaccines 2021, 6, 28. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Bojkova, D.; Klann, K.; Koch, B.; Widera, M.; Krause, D.; Ciesek, S.; Cinatl, J.; Munch, C. Proteomics of SARS-CoV-2-infected host cells reveals therapy targets. Nature 2020, 583, 469–472. [Google Scholar] [CrossRef]
- V’Kovski, P.; Gerber, M.; Kelly, J.; Pfaender, S.; Ebert, N.; Braga Lagache, S.; Simillion, C.; Portmann, J.; Stalder, H.; Gaschen, V.; et al. Determination of host proteins composing the microenvironment of coronavirus replicase complexes by proximity-labeling. eLife 2019, 8, e42037. [Google Scholar] [CrossRef]
- Zhu, Y.; Feng, F.; Hu, G.; Wang, Y.; Yu, Y.; Zhu, Y.; Xu, W.; Cai, X.; Sun, Z.; Han, W.; et al. A genome-wide CRISPR screen identifies host factors that regulate SARS-CoV-2 entry. Nat. Commun. 2021, 12, 961. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Alfajaro, M.M.; DeWeirdt, P.C.; Hanna, R.E.; Lu-Culligan, W.J.; Cai, W.L.; Strine, M.S.; Zhang, S.M.; Graziano, V.R.; Schmitz, C.O.; et al. Genome-wide CRISPR Screens Reveal Host Factors Critical for SARS-CoV-2 Infection. Cell 2021, 184, 76–91.e13. [Google Scholar] [CrossRef] [PubMed]
- Daniloski, Z.; Jordan, T.X.; Wessels, H.H.; Hoagland, D.A.; Kasela, S.; Legut, M.; Maniatis, S.; Mimitou, E.P.; Lu, L.; Geller, E.; et al. Identification of Required Host Factors for SARS-CoV-2 Infection in Human Cells. Cell 2021, 184, 92–105.e16. [Google Scholar] [CrossRef]
- Baggen, J.; Persoons, L.; Vanstreels, E.; Jansen, S.; Van Looveren, D.; Boeckx, B.; Geudens, V.; De Man, J.; Jochmans, D.; Wauters, J.; et al. Genome-wide CRISPR screening identifies TMEM106B as a proviral host factor for SARS-CoV-2. Nat. Genet. 2021, 53, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Shou, S.; Liu, M.; Yang, Y.; Kang, N.; Song, Y.; Tan, D.; Liu, N.; Wang, F.; Liu, J.; Xie, Y. Animal Models for COVID-19: Hamsters, Mouse, Ferret, Mink, Tree Shrew, and Non-human Primates. Front. Microbiol. 2021, 12, 626553. [Google Scholar] [CrossRef] [PubMed]
- Lednicky, J.; Salemi, M.; Subramaniam, K.; Waltzek, T.B.; Sabo-Attwood, T.; Loeb, J.C.; Hentschel, S.; Tagliamonte, M.S.; Marini, S.; Alam, M.M.; et al. Earliest detection to date of SARS-CoV-2 in Florida: Identification together with influenza virus on the main entry door of a university building, February 2020. PLoS ONE 2021, 16, e0245352. [Google Scholar] [CrossRef] [PubMed]
- Redondo, N.; Zaldivar-Lopez, S.; Garrido, J.J.; Montoya, M. SARS-CoV-2 Accessory Proteins in Viral Pathogenesis: Knowns and Unknowns. Front. Immunol. 2021, 12, 708264. [Google Scholar] [CrossRef] [PubMed]
- Dominguez Andres, A.; Feng, Y.; Campos, A.R.; Yin, J.; Yang, C.C.; James, B.; Murad, R.; Kim, H.; Deshpande, A.J.; Gordon, D.E.; et al. SARS-CoV-2 ORF9c Is a Membrane-Associated Protein that Suppresses Antiviral Responses in Cells. bioRxiv 2020. [Google Scholar] [CrossRef]
- Li, X.; Hou, P.; Ma, W.; Wang, X.; Wang, H.; Yu, Z.; Chang, H.; Wang, T.; Jin, S.; Wang, X.; et al. SARS-CoV-2 ORF10 suppresses the antiviral innate immune response by degrading MAVS through mitophagy. Cell Mol. Immunol. 2022, 19, 67–78. [Google Scholar] [CrossRef]
- Pancer, K.; Milewska, A.; Owczarek, K.; Dabrowska, A.; Kowalski, M.; Labaj, P.P.; Branicki, W.; Sanak, M.; Pyrc, K. The SARS-CoV-2 ORF10 is not essential in vitro or in vivo in humans. PLoS Pathog. 2020, 16, e1008959. [Google Scholar] [CrossRef]
- Su, C.M.; Wang, L.; Yoo, D. Activation of NF-kappaB and induction of proinflammatory cytokine expressions mediated by ORF7a protein of SARS-CoV-2. Sci. Rep. 2021, 11, 13464. [Google Scholar] [CrossRef] [PubMed]
- Ferreira-Gomes, M.; Kruglov, A.; Durek, P.; Heinrich, F.; Tizian, C.; Heinz, G.A.; Pascual-Reguant, A.; Du, W.; Mothes, R.; Fan, C.; et al. SARS-CoV-2 in severe COVID-19 induces a TGF-beta-dominated chronic immune response that does not target itself. Nat. Commun. 2021, 12, 1961. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Liu, W.; Li, X.; Zhao, P.; Shereen, M.A.; Zhu, C.; Huang, S.; Liu, S.; Yu, X.; Yue, M.; et al. HIF-1alpha promotes SARS-CoV-2 infection and aggravates inflammatory responses to COVID-19. Signal Transduct. Target. Ther. 2021, 6, 308. [Google Scholar] [CrossRef] [PubMed]
- Breikaa, R.M.; Lilly, B. The Notch Pathway: A Link Between COVID-19 Pathophysiology and Its Cardiovascular Complications. Front. Cardiovasc. 2021, 8, 681948. [Google Scholar] [CrossRef] [PubMed]
- Quillard, T.; Charreau, B. Impact of notch signaling on inflammatory responses in cardiovascular disorders. Int. J. Mol. Sci. 2013, 14, 6863–6888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaim, S.; Chong, J.H.; Sankaranarayanan, V.; Harky, A. COVID-19 and Multiorgan Response. Curr. Probl. Cardiol. 2020, 45, 100618. [Google Scholar] [CrossRef]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef] [Green Version]
- Munoz-Fontela, C.; Macip, S.; Martinez-Sobrido, L.; Brown, L.; Ashour, J.; Garcia-Sastre, A.; Lee, S.W.; Aaronson, S.A. Transcriptional role of p53 in interferon-mediated antiviral immunity. J. Exp. Med. 2008, 205, 1929–1938. [Google Scholar] [CrossRef] [Green Version]
- Rivas, C.; Aaronson, S.A.; Munoz-Fontela, C. Dual Role of p53 in Innate Antiviral Immunity. Viruses 2010, 2, 298–313. [Google Scholar] [CrossRef] [Green Version]
- el-Deiry, W.S.; Tokino, T.; Velculescu, V.E.; Levy, D.B.; Parsons, R.; Trent, J.M.; Lin, D.; Mercer, W.E.; Kinzler, K.W.; Vogelstein, B. WAF1, a potential mediator of p53 tumor suppression. Cell 1993, 75, 817–825. [Google Scholar] [CrossRef]
- Royds, J.A.; Hibma, M.; Dix, B.R.; Hananeia, L.; Russell, I.A.; Wiles, A.; Wynford-Thomas, D.; Braithwaite, A.W. p53 promotes adenoviral replication and increases late viral gene expression. Oncogene 2006, 25, 1509–1520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma-Lauer, Y.; Carbajo-Lozoya, J.; Hein, M.Y.; Muller, M.A.; Deng, W.; Lei, J.; Meyer, B.; Kusov, Y.; von Brunn, B.; Bairad, D.R.; et al. p53 down-regulates SARS coronavirus replication and is targeted by the SARS-unique domain and PLpro via E3 ubiquitin ligase RCHY1. Proc. Natl. Acad. Sci. USA 2016, 113, E5192–E5201. [Google Scholar] [CrossRef] [PubMed]
- Meyers, J.M.; Ramanathan, M.; Shanderson, R.L.; Beck, A.; Donohue, L.; Ferguson, I.; Guo, M.G.; Rao, D.S.; Miao, W.; Reynolds, D.; et al. The proximal proteome of 17 SARS-CoV-2 proteins links to disrupted antiviral signaling and host translation. PLoS Pathog. 2021, 17, e1009412. [Google Scholar] [CrossRef] [PubMed]
- Sims, A.C.; Tilton, S.C.; Menachery, V.D.; Gralinski, L.E.; Schafer, A.; Matzke, M.M.; Webb-Robertson, B.J.; Chang, J.; Luna, M.L.; Long, C.E.; et al. Release of severe acute respiratory syndrome coronavirus nuclear import block enhances host transcription in human lung cells. J. Virol. 2013, 87, 3885–3902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, X.; Dong, X.; Ma, R.; Wang, W.; Xiao, X.; Tian, Z.; Wang, C.; Wang, Y.; Li, L.; Ren, L. Activation and evasion of type I interferon responses by SARS-CoV-2. Nat. Commun. 2020, 11, 3810. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Host Signaling Pathways | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Wnt | p53 | TGFβ | c-Myc | Hypoxia | Hippo | AP-1 | Notch | Oct4/Sox2 | NFkB | ||
| SARS-CoV-2 proteins | vector | 1.0 ± 0.1 | 1.0 ± 0.1 | 1.0 ± 0.4 | 1.0 ± 0.2 | 1.0 ± 0.03 | 1.0 ± 0.04 | 1.0 ± 0.1 | 1.0 ± 0.3 | 1.0 ± 0.1 | 1.0 ± 0.1 |
| nsp1 | 0.7 ± 0.1 | 2.2 ± 0.1 | 0.2 ± 0.02 | 0.1 ± 0.002 | 1.6 ± 0.2 | 0.5 ± 0.04 | 0.03 ± 0.002 | 5.6 ± 2.0 | 1.3 ± 0.1 | 11.1 ± 0.9 | |
| nsp2 | 1.0 ± 0.2 | 0.8 ± 0.1 | 2.4 ± 0.5 | 2.0 ± 0.1 | 3.1 ± 0.5 | 0.9 ± 0.3 | 1.3 ± 0.03 | 2.3 ± 0.8 | 0.8 ± 0.1 | 1.7 ± 0.7 | |
| nsp5 | 1.1 ± 0.3 | 0.05 ± 0.01 | 0.4 ± 0.1 | 0.5 ± 0.02 | 0.5 ± 0.1 | 0.4 ± 0.1 | 0.1 ± 0.001 | 1.3 ± 0.4 | 0.7 ± 0.1 | 0.7 ± 0.1 | |
| nsp5 C145A | 0.7 ± 0.2 | 1.5 ± 0.2 | 2.2 ± 0.4 | 1.4 ± 0.1 | 2.3 ± 0.2 | 0.8 ± 0.2 | 0.4 ± 0.1 | 1.9 ± 0.4 | 0.7 ± 0.1 | 1.2 ± 0.2 | |
| nsp7 | 0.8 ± 0.1 | 0.7 ± 0.1 | 3.6 ± 0.5 | 1.7 ± 0.2 | 3.2 ± 0.3 | 1.0 ± 0.1 | 0.5 ± 0.1 | 1.1 ± 0.4 | 0.5 ± 0.03 | 1.0 ± 0.1 | |
| nsp8 | 0.8 ± 0.2 | 1.0 ± 0.1 | 2.9 ± 0.3 | 1.4 ± 0.2 | 4.0 ± 0.6 | 0.8 ± 0.1 | 0.6 ± 0.02 | 1.5 ± 0.6 | 0.6 ± 0.04 | 0.8 ± 0.1 | |
| nsp9 | 1.0 ± 0.1 | 0.9 ± 0.1 | 5.1 ± 0.4 | 1.6 ± 0.3 | 3.3 ± 0.3 | 1.1 ± 0.1 | 0.6 ± 0.1 | 2.0 ± 0.3 | 0.9 ± 0.1 | 1.6 ± 0.3 | |
| nsp10 | 1.0 ± 0.1 | 0.3 ± 0.03 | 5.1 ± 0.7 | 2.0 ± 0.1 | 2.7 ± 0.8 | 1.0 ± 0.1 | 0.7 ± 0.1 | 2.2 ± 0.4 | 0.8 ± 0.04 | 1.5 ± 0.3 | |
| nsp12 | 0.8 ± 0.05 | 0.9 ± 0.1 | 4.2 ± 0.7 | 3.1 ± 0.6 | 3.3 ± 0.8 | 0.8 ± 0.04 | 0.7 ± 0.01 | 3.3 ± 0.5 | 0.7 ± 0.1 | 2.0 ± 0.3 | |
| nsp14 | 1.4 ± 0.3 | 2.3 ± 0.3 | 2.6 ± 0.4 | 1.9 ± 0.2 | 5.4 ± 0.3 | 3.3 ± 0.3 | 3.7 ± 0.1 | 17.7 ± 3.2 | 2.6 ± 0.1 | 12.6 ± 0.3 | |
| nsp15 | 0.8 ± 0.1 | 0.1 ± 0.01 | 1.2 ± 0.2 | 1.0 ± 0.01 | 2.3 ± 0.2 | 0.6 ± 0.01 | 0.2 ± 0.03 | 1.4 ± 0.2 | 0.5 ± 0.1 | 1.5 ± 0.1 | |
| E | 1.1 ± 0.1 | 0.4 ± 0.1 | 4.4 ± 0.1 | 0.7 ± 0.1 | 3.1 ± 0.5 | 0.7 ± 0.04 | 0. ± 0.2 | 2.7 ± 0.6 | 1.0 ± 0.01 | 10.4 ± 1.5 | |
| M | 0.9 ± 0.1 | 0.5 ± 0.02 | 1.0 ± 0.3 | 0.6 ± 0.1 | 2.5 ± 0.1 | 0.3 ± 0.03 | 0.3 ± 0.1 | 1.8 ± 0.2 | 0.8 ± 0.1 | 7.3 ± 0.8 | |
| N | 1.6 ± 0.4 | 11.7 ± 1.2 | 6.3 ± 0.4 | 3.1 ± 0.4 | 4.4 ± 0.5 | 1.5 ± 0.2 | 1.4 ± 0.3 | 4.6 ± 0.3 | 0.9 ± 0.1 | 3.9 ± 0.6 | |
| orf3a | 0.8 ± 0.1 | 0.04 ± 0.01 | 1.1 ± 0.2 | 0.6 ± 0.1 | 1.8 ± 0.1 | 0.4 ± 0.1 | 0.4 ± 0.02 | 1.6 ± 0.6 | 0.6 ± 0.02 | 11.1 ± 0.9 | |
| orf6 | 0.8 ± 0.2 | 0.2 ± 0.02 | 0.2 ± 0.01 | 0.3 ± 0.1 | 0.8 ± 0.2 | 0.7 ± 0.1 | 0.1 ± 0.02 | 1.4 ± 0.3 | 0.8 ± 0.1 | 0.9 ± 0.1 | |
| orf7a | 1.2 ± 0.1 | 1.7 ± 0.1 | 2.8 ± 0.1 | 1.2 ± 0.1 | 5.4 ± 0.8 | 0.8 ± 0.1 | 3.3 ± 0.4 | 3.1 ± 0.2 | 0.7 ± 0.1 | 59.2 ± 7.1 | |
| orf7b | 1.0 ± 0.3 | 0.4 ± 0.03 | 0.3 ± 0.01 | 0.4 ± 0.04 | 2.9 ± 0.5 | 0.7 ± 0.05 | 0.4 ± 0.1 | 2.5 ± 0.9 | 0.7 ± 0.1 | 3.5 ± 0.8 | |
| orf8 | 0.8 ± 0.1 | 2.1 ± 0.04 | 1.6 ± 0.3 | 0.8 ± 0.1 | 2.5 ± 0.3 | 0.8 ± 0.1 | 0.4 ± 0.1 | 1.7 ± 0.4 | 0.6 ± 0.1 | 2.1 ± 0.2 | |
| orf9b | 4.8 ± 0.7 | 19.9 ± 2.2 | 16.0 ± 1.4 | 2.4 ± 0.4 | 20.8 ± 0.5 | 4.0 ± 0.4 | 15.4 ± 1.0 | 13.3 ± 1.8 | 2.9 ± 0.3 | 10.6 ± 0.8 | |
| orf9c | 3.5 ± 0.1 | 8.3 ± 0.4 | 14.5 ± 2.0 | 2.2 ± 0.3 | 13.7 ± 1.0 | 2.9 ± 0.1 | 3.9 ± 0.6 | 11.1 ± 0.9 | 1.8 ± 0.5 | 10.0 ± 0.5 | |
| orf10 | 3.6 ± 0.3 | 9.9 ± 0.4 | 14.9 ± 1.2 | 2.2 ± 0.1 | 15.6 ± 0.4 | 3.0 ± 0.1 | 5.0 ± 0.5 | 11.1 ± 2.7 | 2.0 ± 0.1 | 12.3 ± 0.7 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, A.; Grams, T.R.; Bloom, D.C.; Toth, Z. Signaling Pathway Reporter Screen with SARS-CoV-2 Proteins Identifies nsp5 as a Repressor of p53 Activity. Viruses 2022, 14, 1039. https://doi.org/10.3390/v14051039
Kumar A, Grams TR, Bloom DC, Toth Z. Signaling Pathway Reporter Screen with SARS-CoV-2 Proteins Identifies nsp5 as a Repressor of p53 Activity. Viruses. 2022; 14(5):1039. https://doi.org/10.3390/v14051039
Chicago/Turabian StyleKumar, Abhishek, Tristan R. Grams, David C. Bloom, and Zsolt Toth. 2022. "Signaling Pathway Reporter Screen with SARS-CoV-2 Proteins Identifies nsp5 as a Repressor of p53 Activity" Viruses 14, no. 5: 1039. https://doi.org/10.3390/v14051039
APA StyleKumar, A., Grams, T. R., Bloom, D. C., & Toth, Z. (2022). Signaling Pathway Reporter Screen with SARS-CoV-2 Proteins Identifies nsp5 as a Repressor of p53 Activity. Viruses, 14(5), 1039. https://doi.org/10.3390/v14051039