
ViralVar: A Web Tool for Multilevel Visualization of SARS-CoV-2 Genomes

 , and
, and
Abstract
1. Introduction
2. Methods
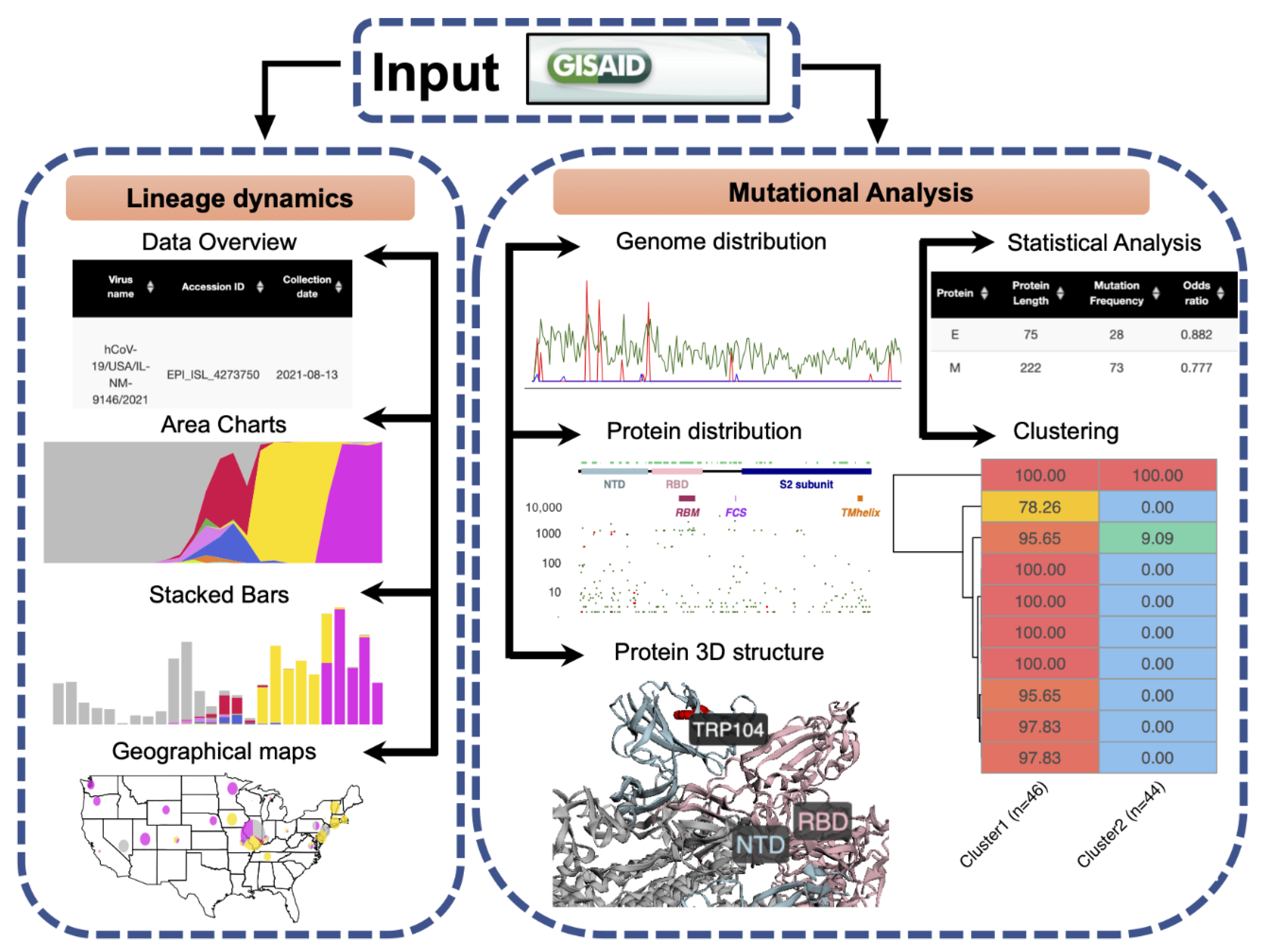
2.1. General Software Workflow
2.2. Data Input
2.3. Lineage Dynamics
2.4. Mutational Analysis
2.5. Applying ViralVar to Assess Dynamics of SARS-CoV-2 Evolution
3. Results and Discussion
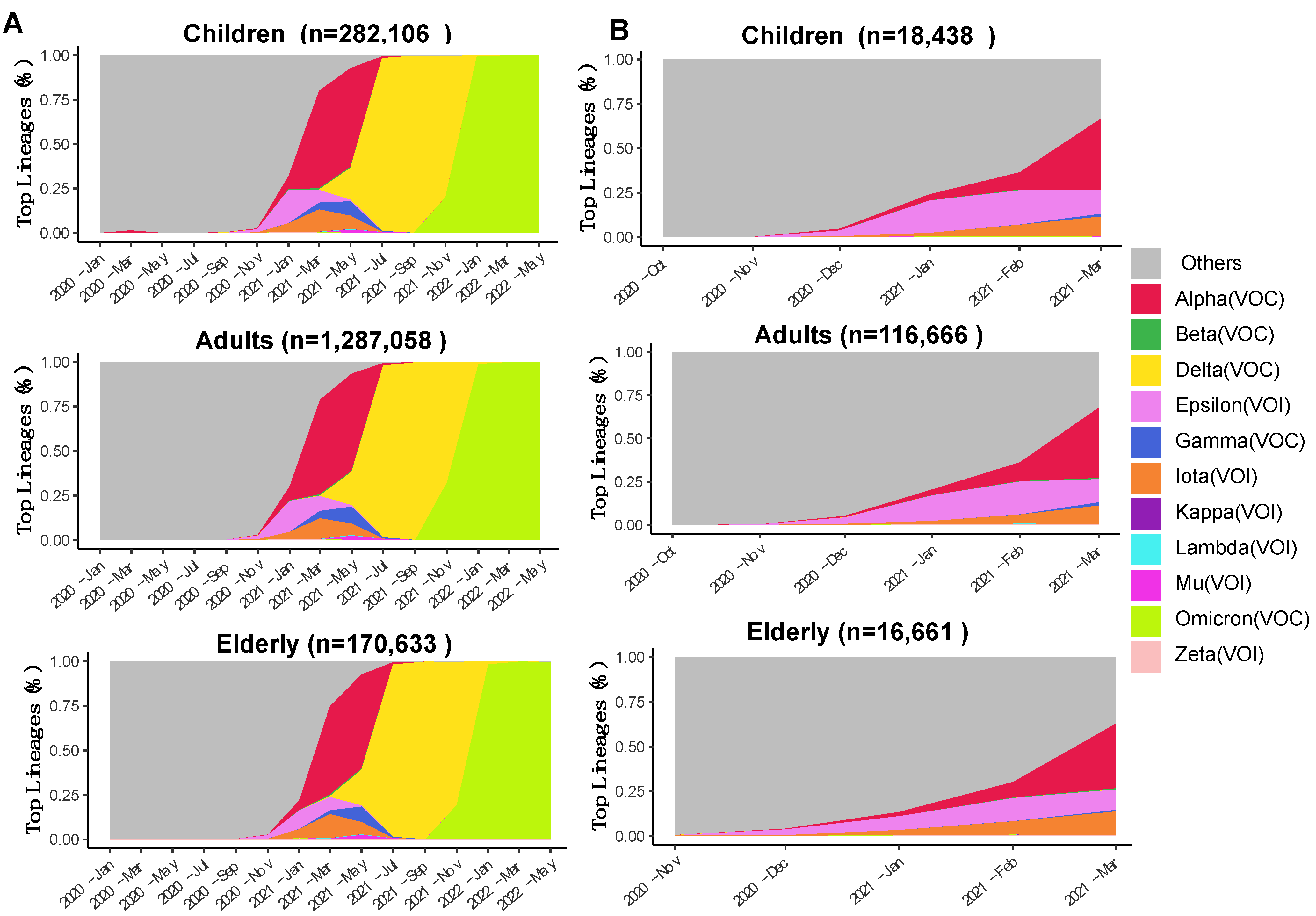
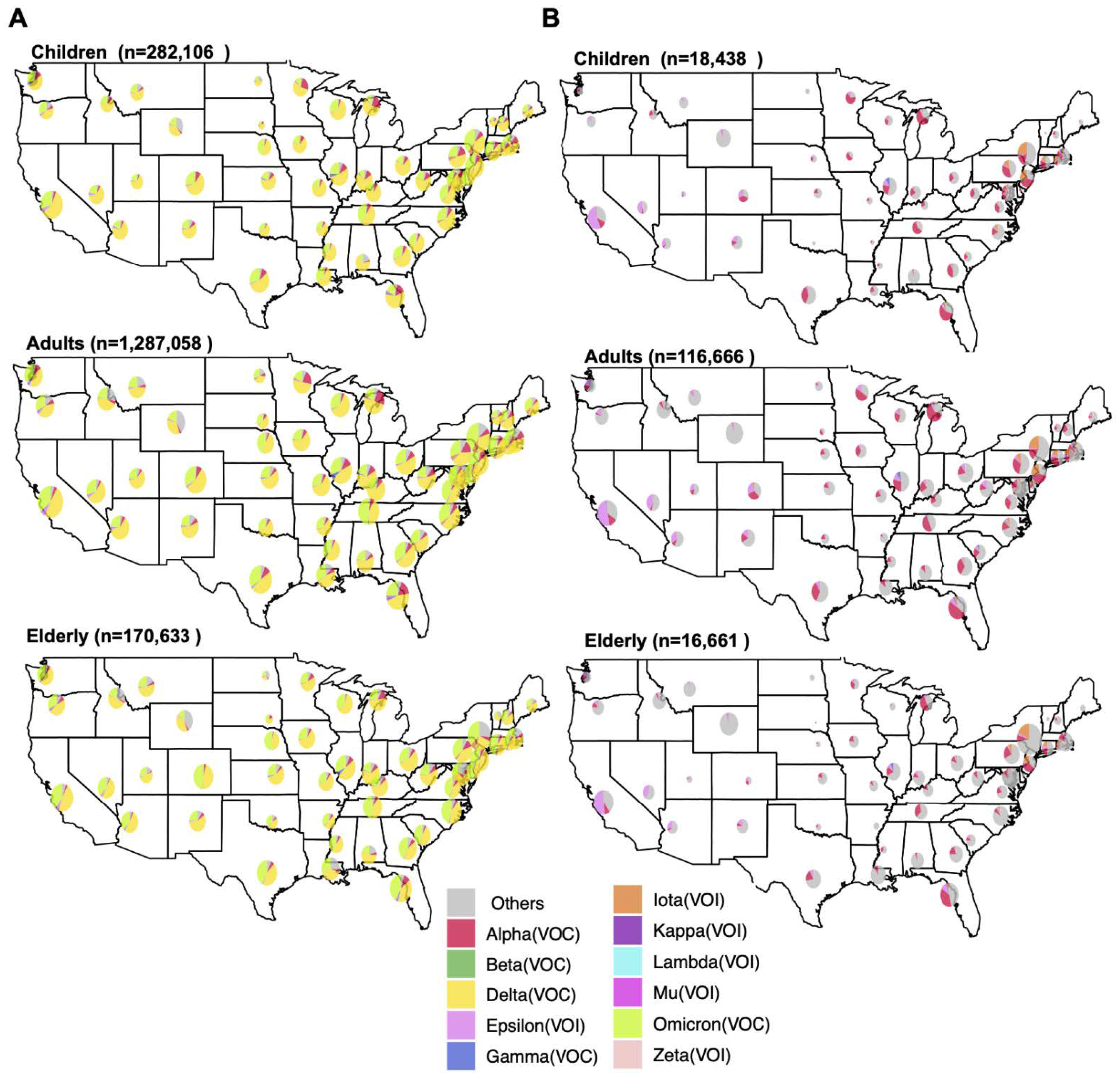
3.1. Spatiotemporal Dynamics of SARS-CoV-2 VOCs in the USA
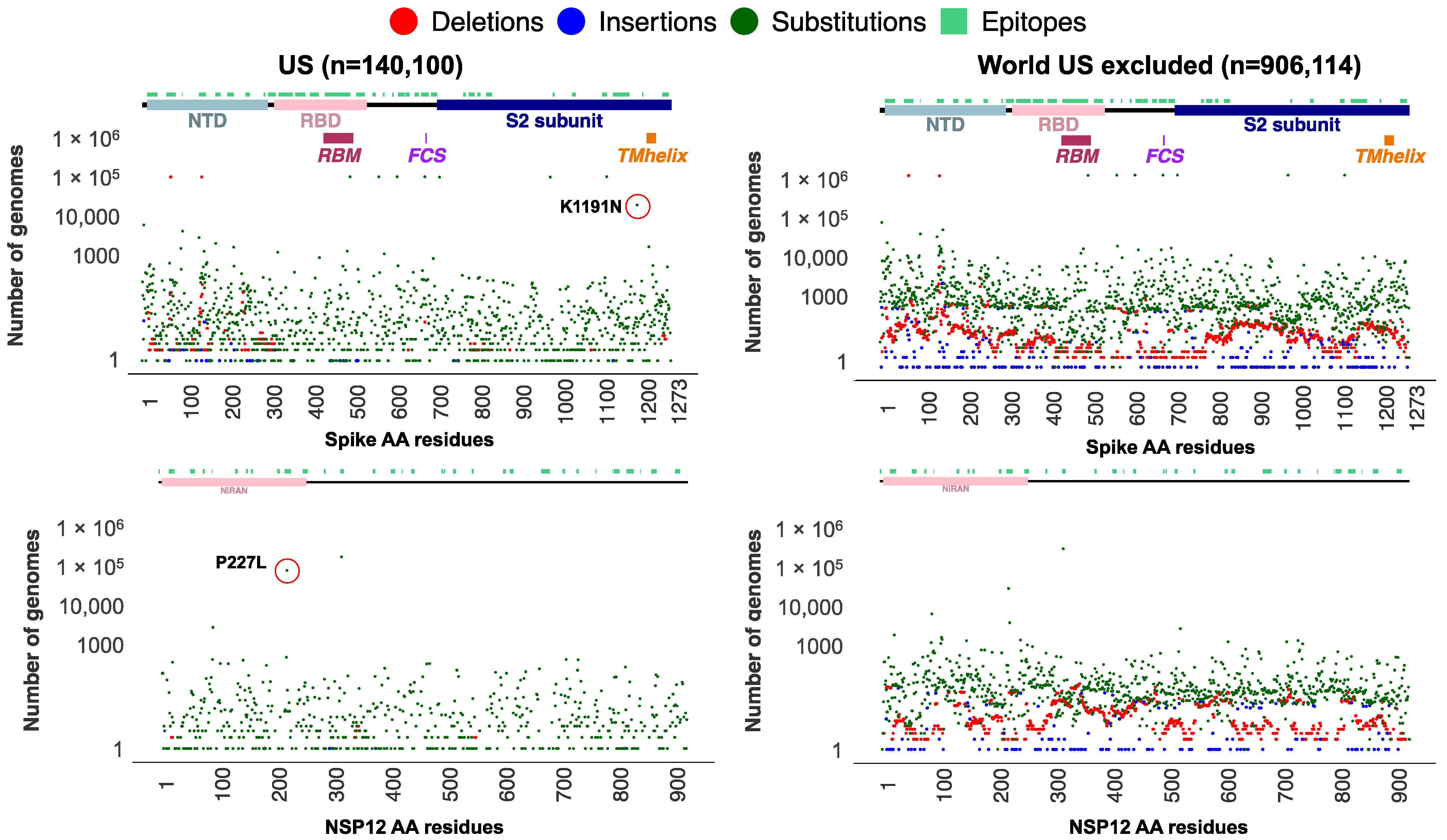
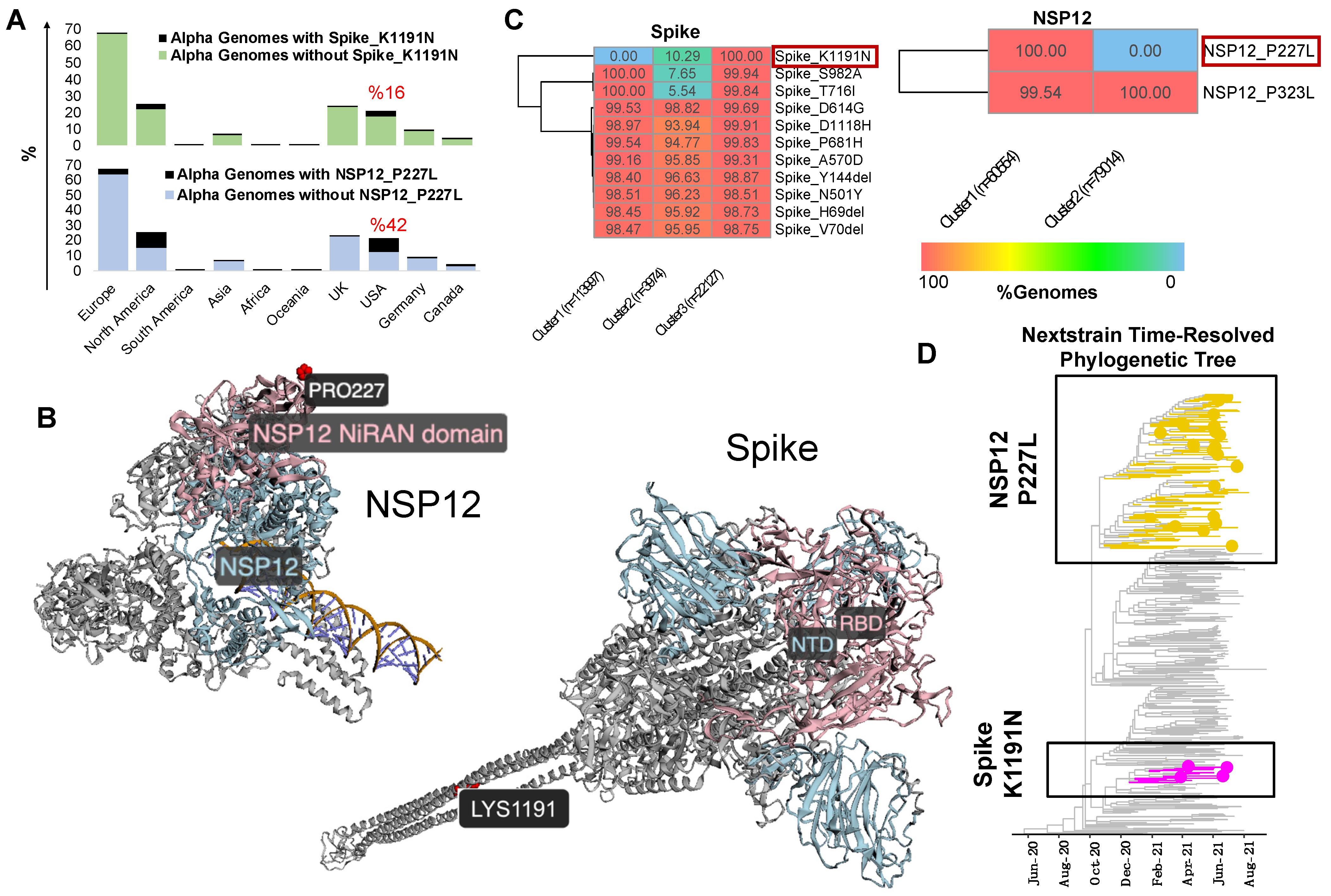
3.2. Mutational Analysis of Alpha Variant Sublineages in the USA
3.3. ViralVar K-Means Clustering Feature Identifies Subclusters of the Alpha Variant in the USA
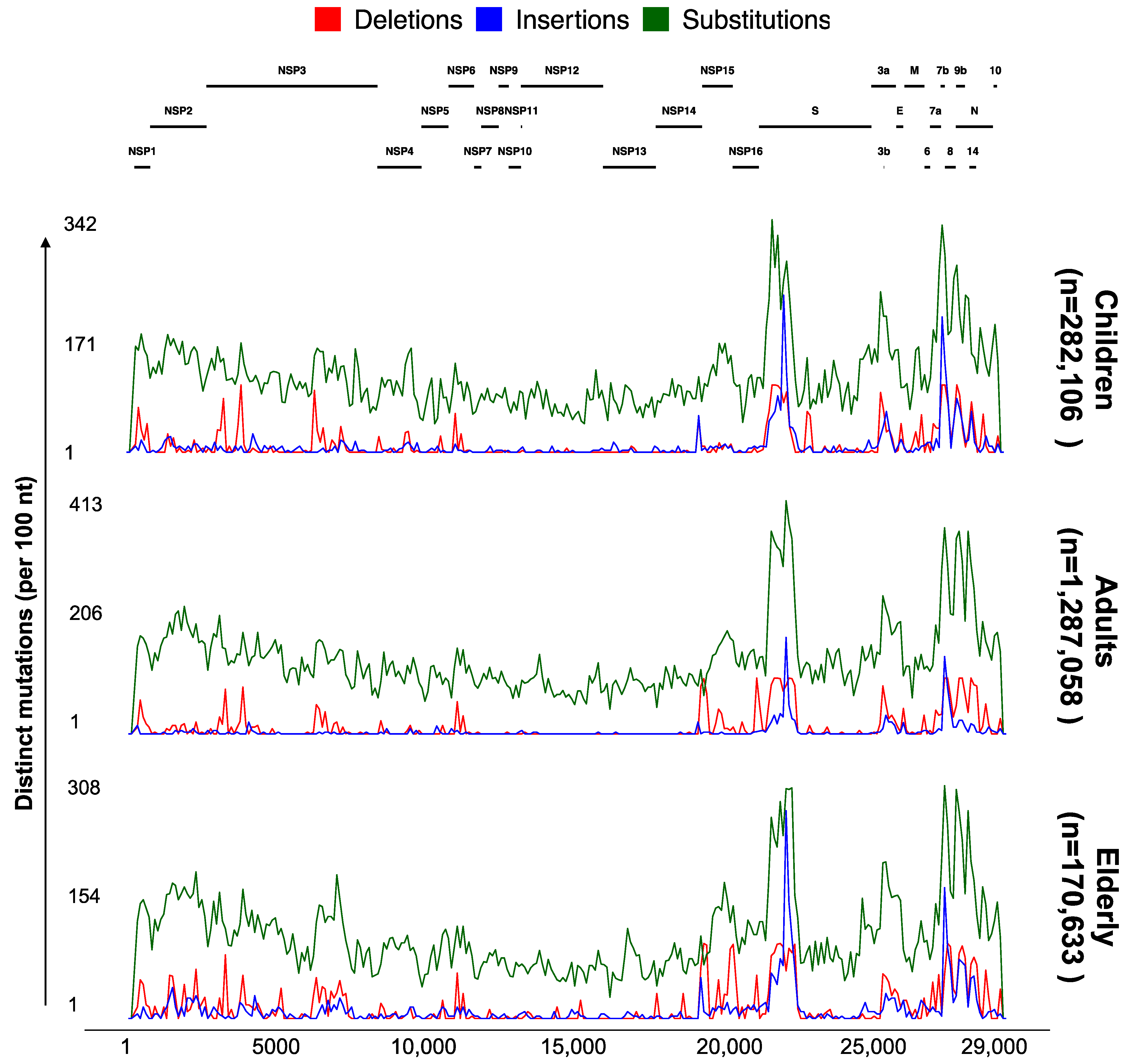
3.4. Significant Nonrandom Distribution of Mutations in SARS-CoV-2 Proteins
3.5. ViralVar Potential in Identifying Novel Variants in Small and Local Cohorts
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Detection of a SARS-CoV-2 variant of concern in South Africa. Nature 2021, 592, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Karim, S.S.A.; Karim, Q.A. Omicron SARS-CoV-2 variant: A new chapter in the COVID-19 pandemic. Lancet 2021, 398, 2126–2128. [Google Scholar] [CrossRef] [PubMed]
- Viana, R.; Moyo, S.; Amoako, D.G.; Tegally, H.; Scheepers, C.; Lessells, R.J.; Giandhari, J.; Wolter, N.; Everatt, J.; Rambaut, A. Rapid epidemic expansion of the SARS-CoV-2 Omicron variant in southern Africa. Nature 2022, 603, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Madhi, S.A.; Baillie, V.; Cutland, C.L.; Voysey, M.; Koen, A.L.; Fairlie, L.; Padayachee, S.D.; Dheda, K.; Barnabas, S.L.; Bhorat, Q.E.; et al. Efficacy of the ChAdOx1 nCoV-19 CoV-19 Vaccine against the B.1.351 Variant. N. Engl. J. Med. 2021, 384, 1885–1898. [Google Scholar] [CrossRef] [PubMed]
- Jewell, B.L. Monitoring differences between the SARS-CoV-2 B.1.1.7 variant and other lineages. Lancet Public Health 2021, 6, e267–e268. [Google Scholar] [CrossRef]
- Jassat, W.; Mudara, C.; Ozougwu, L.; Tempia, S.; Blumberg, L.; Davies, M.-A.; Pillay, Y.; Carter, T.; Morewane, R.; Wolmarans, M. Difference in mortality among individuals admitted to hospital with COVID-19 during the first and second waves in South Africa: A cohort study. Lancet Glob. Health 2021, 9, e1216–e1225. [Google Scholar] [CrossRef]
- Edward, P.R.; Lorenzo-Redondo, R.; Reyna, M.E.; Simons, L.M.; Hultquist, J.F.; Patel, A.B.; Ozer, E.A.; Muller, W.J.; Heald-Sargent, T.; McHugh, M.; et al. Severity of illness caused by severe acute respiratory syndrome coronavirus 2 variants of concern in children: A single-center retrospective cohort study. J. Pediatr. Infect. Dis. 2022, 11, 440–447. [Google Scholar] [CrossRef]
- Duong, D. Alpha, Beta, Delta, Gamma: What’s important to know about SARS-CoV-2 variants of concern? Can. Med. Assoc. 2021, 193, E1059–E1060. [Google Scholar] [CrossRef]
- Khan, A.; Khan, T.; Ali, S.; Aftab, S.; Wang, Y.; Qiankun, W.; Khan, M.; Suleman, M.; Ali, S.; Heng, W. SARS-CoV-2 new variants: Characteristic features and impact on the efficacy of different vaccines. Biomed. Pharmacother. 2021, 143, 112176. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, R.; Hu, F.; Lan, Y.; Yang, Z.; Zhan, C.; Shi, J.; Deng, X.; Jiang, M.; Zhong, S. Transmission, viral kinetics and clinical characteristics of the emergent SARS-CoV-2 Delta VOC in Guangzhou, China. EClinicalMedicine 2021, 40, 101129. [Google Scholar] [CrossRef]
- Tian, D.; Sun, Y.; Xu, H.; Ye, Q. The emergence and epidemic characteristics of the highly mutated SARS-CoV-2 Omicron variant. J. Med. Virol. 2022, 94, 2376–2383. [Google Scholar] [CrossRef] [PubMed]
- Post, L.A.; Lorenzo-Redondo, R. Omicron: Fewer adverse outcomes come with new dangers. Lancet 2022, 399, 1280–1281. [Google Scholar] [CrossRef] [PubMed]
- Frampton, D.; Rampling, T.; Cross, A.; Bailey, H.; Heaney, J.; Byott, M.; Scott, R.; Sconza, R.; Price, J.; Margaritis, M. Genomic characteristics and clinical effect of the emergent SARS-CoV-2 B. 1.1.7 lineage in London, UK: A whole-genome sequencing and hospital-based cohort study. Lancet Infect. Dis. 2021, 21, 1246–1256. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, J.; Plante, K.S.; Plante, J.A.; Xie, X.; Zhang, X.; Ku, Z.; An, Z.; Scharton, D.; Schindewolf, C. The N501Y spike substitution enhances SARS-CoV-2 infection and transmission. Nature 2022, 602, 294–299. [Google Scholar] [CrossRef]
- Lusvarghi, S.; Wang, W.; Herrup, R.; Neerukonda, S.N.; Vassell, R.; Bentley, L.; Eakin, A.E.; Erlandson, K.J.; Weiss, C.D. Key substitutions in the spike protein of SARS-CoV-2 variants can predict resistance to monoclonal antibodies, but other substitutions can modify the effects. J. Virol. 2022, 96, e01110–e01121. [Google Scholar] [CrossRef]
- Jaroszewski, L.; Iyer, M.; Alisoltani, A.; Sedova, M.; Godzik, A. The interplay of SARS-CoV-2 evolution and constraints imposed by the structure and functionality of its proteins. PLoS Comput. Biol. 2021, 17, e1009147. [Google Scholar] [CrossRef]
- Alisoltani, A.; Jaroszewski, L.; Iyer, M.; Iranzadeh, A.; Godzik, A. Increased frequency of indels in hypervariable regions of SARS-CoV-2 proteins–a possible signature of adaptive selection. Front. Genet. 2022, 13, 1019. [Google Scholar] [CrossRef]
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data—From vision to reality. Eurosurveillance 2017, 22, 30494. [Google Scholar] [CrossRef]
- Sedova, M.; Jaroszewski, L.; Alisoltani, A.; Godzik, A. Coronavirus3D: 3D structural visualization of COVID-19 genomic divergence. Bioinformatics 2020, 36, 4360–4362. [Google Scholar] [CrossRef]
- Mercatelli, D.; Holding, A.N.; Giorgi, F.M. Web tools to fight pandemics: The COVID-19 experience. Brief. Bioinform. 2021, 22, 690–700. [Google Scholar] [CrossRef]
- Portelli, S.; Olshansky, M.; Rodrigues, C.H.; D’Souza, E.N.; Myung, Y.; Silk, M.; Alavi, A.; Pires, D.E.; Ascher, D.B. Exploring the structural distribution of genetic variation in SARS-CoV-2 with the COVID-3D online resource. Nat. Genet. 2020, 52, 999–1001. [Google Scholar] [CrossRef] [PubMed]
- Mei, L.-C.; Jin, Y.; Wang, Z.; Hao, G.-F.; Yang, G.-F. Web resources facilitate drug discovery in treatment of COVID-19. Drug Discov. Today 2021, 26, 2358–2366. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.T.; Altschuler, K.; Zhan, S.H.; Chan, Y.A.; Deverman, B.E. COVID-19 CG enables SARS-CoV-2 mutation and lineage tracking by locations and dates of interest. Elife 2021, 10, e63409. [Google Scholar] [CrossRef] [PubMed]
- Gangavarapu, K.; Latif, A.A.; Mullen, J.L.; Alkuzweny, M.; Hufbauer, E.; Tsueng, G.; Haag, E.; Zeller, M.; Aceves, C.M.; Zaiets, K. Outbreak. info genomic reports: Scalable and dynamic surveillance of SARS-CoV-2 variants and mutations. Res. Sq. 2022, rs. 3, rs-1723829. [Google Scholar]
- Hodcroft, E. CoVariants: SARS-CoV-2 Mutations and Variants of Interest. 2021. Available online: https://covariants.org (accessed on 4 October 2022).
- Lu, G.; Moriyama, N.E. 2019nCoVR—A comprehensive genomic resource for SARS-CoV-2 variant surveillance. Innovation 2021, 2, 100150. [Google Scholar] [CrossRef]
- Singer, J.; Gifford, R.; Cotten, M.; Robertson, D. CoV-GLUE: A Web Application for Tracking SARS-CoV-2 Genomic Variation. 2020. Available online: http://cov-glue.cvr.gla.ac.uk (accessed on 4 October 2022).
- Wright, D.W.; Harvey, W.T.; Hughes, J.; Cox, M.; Peacock, T.P.; Colquhoun, R.; Jackson, B.; Orton, R.; Nielsen, M.; Hsu, N.S. Tracking SARS-CoV-2 mutations and variants through the COG-UK-Mutation Explorer. Virus Evol. 2022, 8, veac023. [Google Scholar] [CrossRef]
- Tzou, P.L.; Tao, K.; Sahoo, M.K.; Pond, S.L.K.; Pinsky, B.A.; Shafer, R.W. Sierra SARS-CoV-2 sequence and antiviral resistance analysis program. 2022. J. Clin. Virol. 2022, 157, 105323. [Google Scholar] [CrossRef]
- Mercatelli, D.; Triboli, L.; Fornasari, E.; Ray, F.; Giorgi, F.M. Coronapp: A web application to annotate and monitor SARS-CoV-2 mutations. J. Med. Virol. 2021, 93, 3238–3245. [Google Scholar] [CrossRef]
- Bernasconi, A.; Gulino, A.; Alfonsi, T.; Canakoglu, A.; Pinoli, P.; Sandionigi, A.; Ceri, S. VirusViz: Comparative analysis and effective visualization of viral nucleotide and amino acid variants. Nucleic Acids Res. 2021, 49, e90. [Google Scholar] [CrossRef]
- Alsulami, A.F.; Thomas, S.E.; Jamasb, A.R.; Beaudoin, C.A.; Moghul, I.; Bannerman, B.; Copoiu, L.; Vedithi, S.C.; Torres, P.; Blundell, T.L. SARS-CoV-2 3D database: Understanding the coronavirus proteome and evaluating possible drug targets. Brief. Bioinform. 2021, 22, 769–780. [Google Scholar] [CrossRef]
- Gowthaman, R.; Guest, J.D.; Yin, R.; Adolf-Bryfogle, J.; Schief, W.R.; Pierce, B.G. CoV3D: A database of high resolution coronavirus protein structures. Nucleic Acids Res. 2021, 49, D282–D287. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. Data Analysis. In Ggplot2; Springer: Cham, Germany, 2016. [Google Scholar] [CrossRef]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Jespersen, M.C.; Peters, B.; Nielsen, M.; Marcatili, P. BepiPred-2.0: Improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res. 2017, 45, W24–W29. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- DeepMind. Computational Predictions of Protein Structures Associated with COVID-19. Available online: https://www.deepmind.com/open-source/computational-predictions-of-protein-structures-associated-with-COVID-19 (accessed on 31 October 2021).
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef]
- Hozumi, Y.; Wang, R.; Yin, C.; Wei, G.W. UMAP-assisted K-means clustering of large-scale SARS-CoV-2 mutation datasets. Comput. Biol. Med. 2021, 131, 104264. [Google Scholar] [CrossRef]
- Mullick, B.; Magar, R.; Jhunjhunwala, A.; Farimani, A.B. Understanding mutation hotspots for the SARS-CoV-2 spike protein using Shannon Entropy and K-means clustering. Comput. Biol. Med. 2021, 138, 104915. [Google Scholar] [CrossRef]
- Charrad, M.; Ghazzali, N.; Boiteau, V.; Niknafs, A. NbClust: An R package for determining the relevant number of clusters in a data set. J. Stat. Softw. 2014, 61, 1–36. [Google Scholar] [CrossRef]
- Deng, X.; Gu, W.; Federman, S.; Du Plessis, L.; Pybus, O.G.; Faria, N.R.; Wang, C.; Yu, G.; Bushnell, B.; Pan, C.-Y. Genomic surveillance reveals multiple introductions of SARS-CoV-2 into Northern California. Science 2020, 369, 582–587. [Google Scholar] [CrossRef]
- Russell, A.; O’Connor, C.; Lasek-Nesselquist, E.; Plitnick, J.; Kelly, J.P.; Lamson, D.M.; George, K.S. Spatiotemporal Analyses of 2 Co-Circulating SARS-CoV-2 Variants, New York State, USA. Emerg. Infect. Dis. 2022, 28, 650. [Google Scholar] [CrossRef]
- Alpert, T.; Brito, A.F.; Lasek-Nesselquist, E.; Rothman, J.; Valesano, A.L.; MacKay, M.J.; Petrone, M.E.; Breban, M.I.; Watkins, A.E.; Vogels, C.B. Early introductions and transmission of SARS-CoV-2 variant B. 1.1. 7 in the United States. Cell 2021, 184, 2595–2604. [Google Scholar] [CrossRef] [PubMed]
- Stevens, L.J.; Pruijssers, A.J.; Lee, H.W.; Gordon, C.J.; Tchesnokov, E.P.; Gribble, J.; George, A.S.; Hughes, T.M.; Lu, X.; Li, J. Mutations in the SARS-CoV-2 RNA dependent RNA polymerase confer resistance to remdesivir by distinct mechanisms. Sci. Transl. Med. 2022, 14, eabo0718. [Google Scholar] [CrossRef] [PubMed]
- Posthuma, C.C.; Te Velthuis, A.J.; Snijder, E.J. Nidovirus RNA polymerases: Complex enzymes handling exceptional RNA genomes. Virus Res. 2017, 234, 58–73. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yang, C.; Xu, X.-F.; Xu, W.; Liu, S.-W. Structural and functional properties of SARS-CoV-2 spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Nagy, Á.; Pongor, S.; Győrffy, B. Different mutations in SARS-CoV-2 associate with severe and mild outcome. Int. J. Antimicrob. Agents 2021, 57, 106272. [Google Scholar] [CrossRef]
- Farkas, C.; Mella, A.; Turgeon, M.; Haigh, J.J. A novel SARS-CoV-2 viral sequence bioinformatic pipeline has found genetic evidence that the viral 3′ untranslated region (UTR) is evolving and generating increased viral diversity. Front. Microbiol. 2021, 12, 665041. [Google Scholar] [CrossRef]
- Yang, K.; Wang, C.; White, K.I.; Pfuetzner, R.A.; Esquivies, L.; Brunger, A.T. Structural conservation among variants of the SARS-CoV-2 spike postfusion bundle. Proc. Natl. Acad. Sci. USA 2022, 119, e2119467119. [Google Scholar] [CrossRef]
- Tao, K.; Tzou, P.L.; Nouhin, J.; Gupta, R.K.; de Oliveira, T.; Kosakovsky Pond, S.L.; Fera, D.; Shafer, R.W. The biological and clinical significance of emerging SARS-CoV-2 variants. Nat. Rev. Genet. 2021, 22, 757–773. [Google Scholar] [CrossRef]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Du, P.; Ding, N.; Li, J.; Zhang, F.; Wang, Q.; Chen, Z.; Song, C.; Han, K.; Xie, W.; Liu, J.; et al. Genomic surveillance of COVID-19 cases in Beijing. Nat. Commun. 2020, 11, 5503. [Google Scholar] [CrossRef]
- Wang, R.; Chen, J.; Gao, K.; Hozumi, Y.; Yin, C.; Wei, G.W. Author Correction: Analysis of SARS-CoV-2 mutations in the United States suggests presence of four substrains and novel variants. Commun. Biol. 2021, 4, 300. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, K.R.; Rennick, L.J.; Nambulli, S.; Robinson-McCarthy, L.R.; Bain, W.G.; Haidar, G.; Duprex, W.P. Recurrent deletions in the SARS-CoV-2 spike glycoprotein drive antibody escape. Science 2021, 371, 1139–1142. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Weaver, S.; Tegally, H.; San, J.E.; Shank, S.D.; Wilkinson, E.; Lucaci, A.G.; Giandhari, J.; Naidoo, S.; Pillay, Y. The emergence and ongoing convergent evolution of the SARS-CoV-2 N501Y lineages. Cell 2021, 184, 5189–5200.e7. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Dong, X.; Ma, R.; Wang, W.; Xiao, X.; Tian, Z.; Wang, C.; Wang, Y.; Li, L.; Ren, L.; et al. Activation and evasion of type I interferon responses by SARS-CoV-2. Nat. Commun. 2020, 11, 3810. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.; Cheng, M.; Teng, F.; Wang, H.; Deng, Y.; Zhang, J.; Qin, C.; Guo, S.; Zhao, H.; Yu, X. Proteome-wide epitope mapping identifies a resource of antibodies for SARS-CoV-2 detection and neutralization. Signal Transduct. Target. Ther. 2021, 6, 166. [Google Scholar] [CrossRef]
- Smith, C.C.; Olsen, K.S.; Gentry, K.M.; Sambade, M.; Beck, W.; Garness, J.; Entwistle, S.; Willis, C.; Vensko, S.; Woods, A. Landscape and selection of vaccine epitopes in SARS-CoV-2. Genome Med. 2021, 13, 101. [Google Scholar] [CrossRef]
- Mou, K.; Mukhtar, F.; Khan, M.T.; Darwish, D.B.; Peng, S.; Muhammad, S.; Al-Sehemi, A.G.; Wei, D.-Q. Emerging Mutations in Nsp1 of SARS-CoV-2 and Their Effect on the Structural Stability. Pathogens 2021, 10, 1285. [Google Scholar] [CrossRef]
- Lin, J.W.; Tang, C.; Wei, H.C.; Du, B.; Chen, C.; Wang, M.; Zhou, Y.; Yu, M.X.; Cheng, L.; Kuivanen, S.; et al. Genomic monitoring of SARS-CoV-2 uncovers an Nsp1 deletion variant that modulates type I interferon response. Cell Host Microbe 2021, 29, 489–502.e8. [Google Scholar] [CrossRef]
- Ozer, E.A.; Simons, L.M.; Adewumi, O.M.; Fowotade, A.A.; Omoruyi, E.C.; Adeniji, J.A.; Olayinka, O.A.; Dean, T.J.; Zayas, J.; Bhimalli, P.P. Multiple expansions of globally uncommon SARS-CoV-2 lineages in Nigeria. Nat. Commun. 2022, 13, 688. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequences | Mean Age | Median Age | |
|---|---|---|---|
| Children (<18) | 282,106 | 10.22 | 10.5 |
| Adults (18–65) | 1,287,058 | 38.92 | 37.5 |
| Elderly (>65) | 170,633 | 74.42 | 72.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alisoltani, A.; Jaroszewski, L.; Godzik, A.; Iranzadeh, A.; Simons, L.M.; Dean, T.J.; Lorenzo-Redondo, R.; Hultquist, J.F.; Ozer, E.A. ViralVar: A Web Tool for Multilevel Visualization of SARS-CoV-2 Genomes. Viruses 2022, 14, 2714. https://doi.org/10.3390/v14122714
Alisoltani A, Jaroszewski L, Godzik A, Iranzadeh A, Simons LM, Dean TJ, Lorenzo-Redondo R, Hultquist JF, Ozer EA. ViralVar: A Web Tool for Multilevel Visualization of SARS-CoV-2 Genomes. Viruses. 2022; 14(12):2714. https://doi.org/10.3390/v14122714
Chicago/Turabian StyleAlisoltani, Arghavan, Lukasz Jaroszewski, Adam Godzik, Arash Iranzadeh, Lacy M. Simons, Taylor J. Dean, Ramon Lorenzo-Redondo, Judd F. Hultquist, and Egon A. Ozer. 2022. "ViralVar: A Web Tool for Multilevel Visualization of SARS-CoV-2 Genomes" Viruses 14, no. 12: 2714. https://doi.org/10.3390/v14122714
APA StyleAlisoltani, A., Jaroszewski, L., Godzik, A., Iranzadeh, A., Simons, L. M., Dean, T. J., Lorenzo-Redondo, R., Hultquist, J. F., & Ozer, E. A. (2022). ViralVar: A Web Tool for Multilevel Visualization of SARS-CoV-2 Genomes. Viruses, 14(12), 2714. https://doi.org/10.3390/v14122714