

Multi-Epitope Vaccine for Monkeypox Using Pan-Genome and Reverse Vaccinology Approaches




Abstract
1. Introduction
2. Materials and Methods
2.1. Retrieval of Complete Genomes
2.2. Genome Annotation and Pan-Genome Inference
2.3. Target Protein Prediction
2.4. Epitope Prediction
2.4.1. MHC Class-I T-Cell Epitope Prediction
2.4.2. MHC Class-II T-Cell Epitope Prediction
2.4.3. MHC Class-I/II Cluster Analysis
2.4.4. Linear B-Cell Epitope Prediction
2.5. Epitope Selection and Designing Vaccine Construct
2.6. Assessment of Vaccine Constructs
2.7. Modeling and Docking Methods
2.7.1. Modeling and Structural Validation
2.7.2. Target Retrieval and Optimization
2.7.3. Target-Vaccine Constructs Docking
2.8. Coarse-Grained Dynamics Simulation of the ‘Target-Vaccine Complex’ Model
3. Results
3.1. Core vs. Pan-Genome Analysis and Vaccine Target Identification
3.2. Identification of Immunodominant Epitopes from Antigenic Proteins
3.2.1. MHC Class-I T-Cell Epitope Mining
3.2.2. MHC Class-II T-Cell Epitope Mining
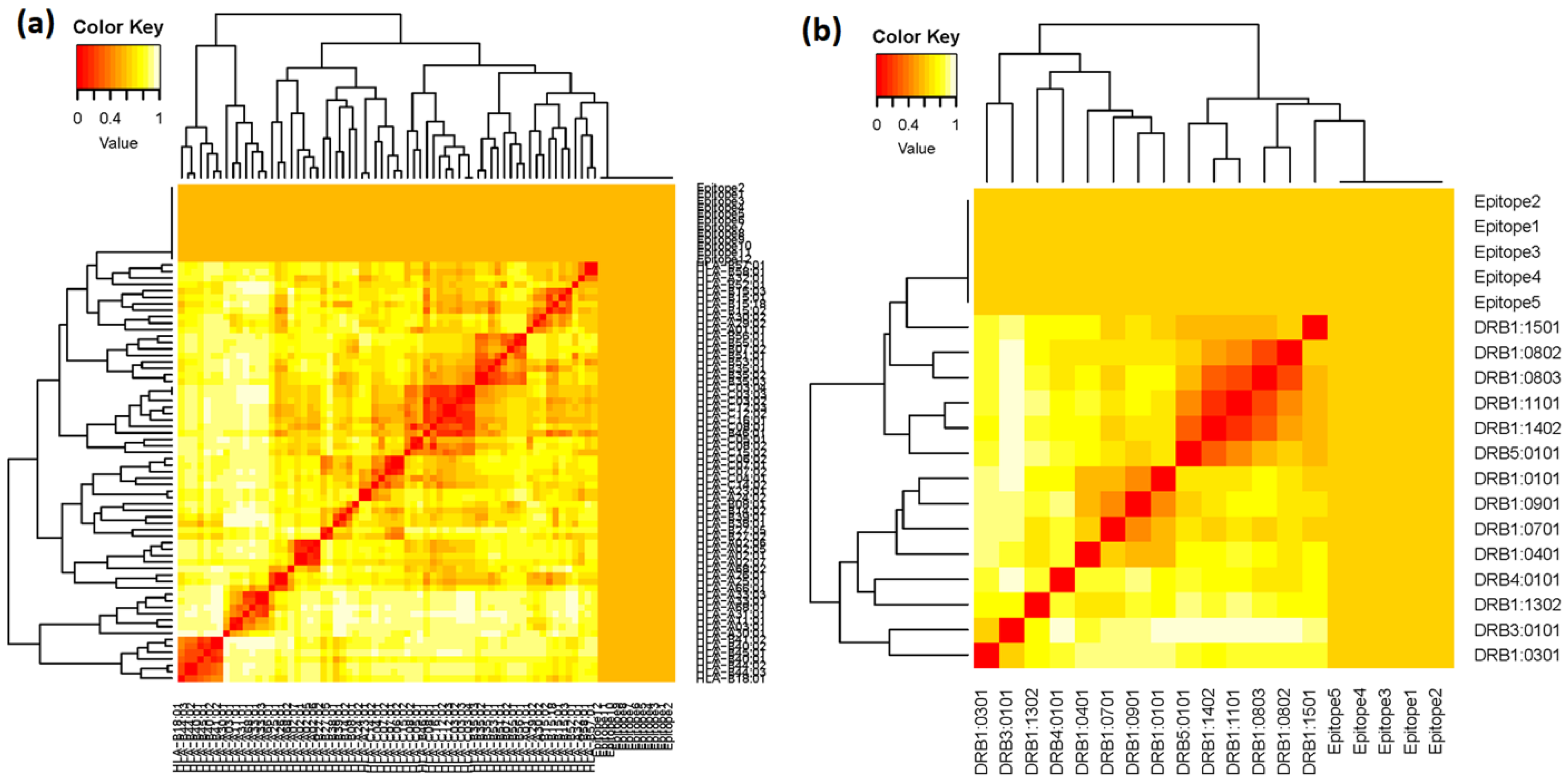
3.2.3. MHC Restricted Alleles Cluster Analysis
3.2.4. Selection of B-Cell Epitope for Vaccine Construction
3.3. Vaccine Construction
3.4. Allergicity, Antigenicity, Solubility and Physio-Chemical Parameters Evaluation of Vaccine Constructs
3.5. Modelling, Docking and Structural Dynamics to Evaluate the Vaccine Constructs
3.5.1. Multi-Epitope Vaccine Construct Models
3.5.2. Docking Studies to Reveal the Binding Affinity of the Suitable Construct
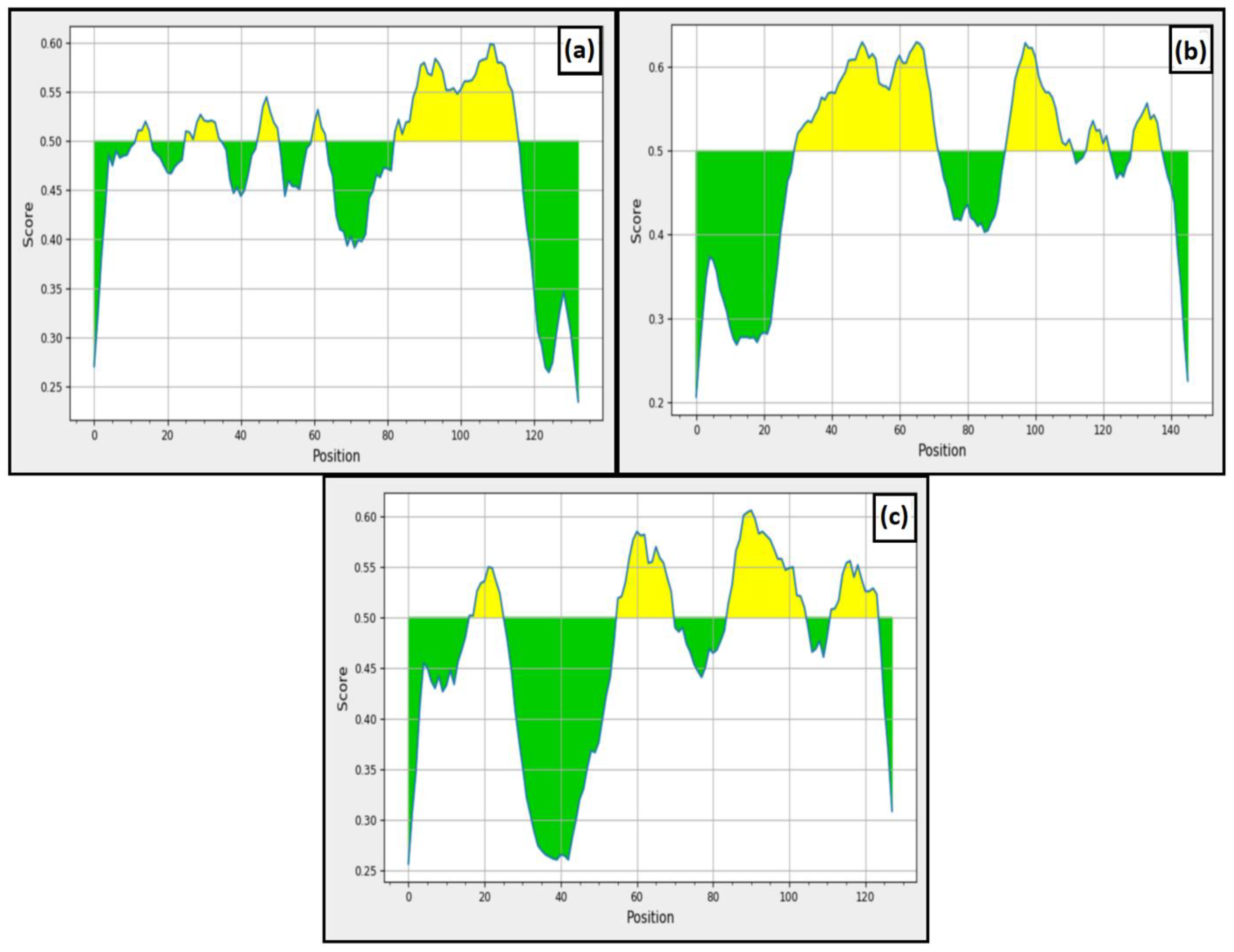
3.5.3. Structural Dynamics to Reveal the Stability of the Complexes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO Multi-Country Outbreak of Monkeypox. World Health Organ. 2022, 1–16.
- Bunge, E.M.; Hoet, B.; Chen, L.; Lienert, F.; Weidenthaler, H.; Baer, L.R.; Steffen, R. The Changing Epidemiology of Human Monkeypox-A Potential Threat? A Systematic Review. PLoS Negl. Trop. Dis. 2022, 16, e0010141. [Google Scholar] [CrossRef] [PubMed]
- Petersen, E.; Kantele, A.; Koopmans, M.; Asogun, D.; Yinka-Ogunleye, A.; Ihekweazu, C.; Zumla, A. Human Monkeypox. Infect. Dis. Clin. N. Am. 2019, 33, 1027–1043. [Google Scholar] [CrossRef] [PubMed]
- Alakunle, E.; Moens, U.; Nchinda, G.; Okeke, M.I. Monkeypox Virus in Nigeria: Infection Biology, Epidemiology, and Evolution. Viruses 2020, 12, 1257. [Google Scholar] [CrossRef] [PubMed]
- Wattal, C.; Datta, S. Monkey Pox Arrives in India. Indian J. Med. Microbiol. 2022, 40, 473–474. [Google Scholar] [CrossRef] [PubMed]
- Rizk, J.G.; Lippi, G.; Henry, B.M.; Forthal, D.N.; Rizk, Y. Prevention and Treatment of Monkeypox. Drugs 2022, 82, 957–963. [Google Scholar] [CrossRef]
- Adler, H.; Gould, S.; Hine, P.; Snell, L.B.; Wong, W.; Houlihan, C.F.; Osborne, J.C.; Rampling, T.; Beadsworth, M.B.; Duncan, C.J.; et al. Clinical Features and Management of Human Monkeypox: A Retrospective Observational Study in the UK. Lancet. Infect. Dis. 2022, 22, 1153–1162. [Google Scholar] [CrossRef]
- Aiman, S.; Alhamhoom, Y.; Ali, F.; Rahman, N.; Rastrelli, L.; Khan, A.; Farooq, Q.u.A.; Ahmed, A.; Khan, A.; Li, C. Multi-Epitope Chimeric Vaccine Design against Emerging Monkeypox Virus via Reverse Vaccinology Techniques—A Bioinformatics and Immunoinformatics Approach. Front. Immunol. 2022, 13. [Google Scholar] [CrossRef]
- Townsend, M.B.; Keckler, M.S.; Patel, N.; Davies, D.H.; Felgner, P.; Damon, I.K.; Karem, K.L. Humoral Immunity to Smallpox Vaccines and Monkeypox Virus Challenge: Proteomic Assessment and Clinical Correlations. J. Virol. 2013, 87, 900–911. [Google Scholar] [CrossRef]
- Fields, F.R.; Lee, S.W.; McConnell, M.J. Using Bacterial Genomes and Essential Genes for the Development of New Antibiotics. Biochem. Pharmacol. 2017, 134, 74–86. [Google Scholar] [CrossRef]
- Rahman, N.; Ali, F.; Basharat, Z.; Shehroz, M.; Khan, M.K.; Jeandet, P.; Nepovimova, E.; Kuca, K.; Khan, H. Vaccine Design from the Ensemble of Surface Glycoprotein Epitopes of SARS-CoV-2: An Immunoinformatics Approach. Vaccines 2020, 8, 423. [Google Scholar] [CrossRef] [PubMed]
- Jalal, K.; Khan, K.; Ahmad, D.; Hayat, A.; Basharat, Z.; Abbas, M.N.; Alghamdi, S.; Almehmadi, M.; Sahibzada, M.U.K. Pan-Genome Reverse Vaccinology Approach for the Design of Multi-Epitope Vaccine Construct against Escherichia Albertii. Int. J. Mol. Sci. 2021, 22, 12814. [Google Scholar] [CrossRef] [PubMed]
- Antonelli, A.C.B.; Almeida, V.P.; de Castro, F.O.F.; Silva, J.M.; Pfrimer, I.A.H.; Cunha-Neto, E.; Maranhão, A.Q.; Brígido, M.M.; Resende, R.O.; Bocca, A.L.; et al. In Silico Construction of a Multiepitope Zika Virus Vaccine Using Immunoinformatics Tools. Sci. Rep. 2022, 12, 53. [Google Scholar] [CrossRef] [PubMed]
- D’Mello, A.; Ahearn, C.P.; Murphy, T.F.; Tettelin, H. ReVac: A Reverse Vaccinology Computational Pipeline for Prioritization of Prokaryotic Protein Vaccine Candidates. BMC Genomics 2019, 20, 981. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Ramaiah, S.; Anbarasu, A. In-Silico Strategies to Combat COVID-19: A Comprehensive Review. Biotechnol. Genet. Eng. Rev. 2021, 37, 64–81. [Google Scholar] [CrossRef]
- Basu, S.; Joshi, S.M.; Ramaiah, S.; Anbarasu, A. Designing Anti-Microbial Peptides Against Major β-Lactamase Enzymes in Clinically Important Gram-Negative Bacterial Pathogens: An In-Silico Study. Probiotics Antimicrob. Proteins 2022, 14, 263–276. [Google Scholar] [CrossRef]
- Swetha, R.G.; Sandhya, M.; Ramaiah, S.; Anbarasu, A. Identification of CD4+ T-Cell Epitope and Investigation of HLA Distribution for the Immunogenic Proteins of Burkholderia Pseudomallei Using in Silico Approaches—A Key Vaccine Development Strategy for Melioidosis. J. Theor. Biol. 2016, 400. [Google Scholar] [CrossRef]
- Basu, S.; Varghese, R.; Debroy, R.; Ramaiah, S.; Veeraraghavan, B.; Anbarasu, A. Non-Steroidal Anti-Inflammatory Drugs Ketorolac and Etodolac Can Augment the Treatment against Pneumococcal Meningitis by Targeting Penicillin-Binding Proteins. Microb. Pathog. 2022, 170, 105694. [Google Scholar] [CrossRef]
- Basu, S.; Veeraraghavan, B.; Ramaiah, S.; Anbarasu, A. Novel Cyclohexanone Compound as a Potential Ligand against SARS-CoV-2 Main-Protease. Microb. Pathog. 2020, 149, 104546. [Google Scholar] [CrossRef]
- Priyamvada, P.; Debroy, R.; Anbarasu, A.; Ramaiah, S. A Comprehensive Review on Genomics, Systems Biology and Structural Biology Approaches for Combating Antimicrobial Resistance in ESKAPE Pathogens: Computational Tools and Recent Advancements. World J. Microbiol. Biotechnol. 2022, 38. [Google Scholar] [CrossRef]
- Debroy, R.; Ramaiah, S. MurC Ligase of Multi-Drug Resistant Salmonella Typhi Can Be Inhibited by Novel Curcumin Derivative: Evidence from Molecular Docking and Dynamics Simulations. Int. J. Biochem. Cell Biol. 2022, 151, 106279. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; Chen, G.-L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic Gene Recognition and Translation Initiation Site Identification. BMC Bioinforma. 2010, 11, 119. [Google Scholar] [CrossRef] [PubMed]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.G.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid Large-Scale Prokaryote Pan Genome Analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Löytynoja, A.; Goldman, N. An Algorithm for Progressive Multiple Alignment of Sequences with Insertions. Proc. Natl. Acad. Sci. USA 2005, 102, 10557–10562. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Shen, H.-B.; Chou, K.-C. Virus-MPLoc: A Fusion Classifier for Viral Protein Subcellular Location Prediction by Incorporating Multiple Sites. J. Biomol. Struct. Dyn. 2010, 28, 175–186. [Google Scholar] [CrossRef]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A Server for Prediction of Protective Antigens, Tumour Antigens and Subunit Vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef]
- Magnan, C.N.; Zeller, M.; Kayala, M.A.; Vigil, A.; Randall, A.; Felgner, P.L.; Baldi, P. High-Throughput Prediction of Protein Antigenicity Using Protein Microarray Data. Bioinformatics 2010, 26, 2936–2943. [Google Scholar] [CrossRef]
- Magnan, C.N.; Randall, A.; Baldi, P. SOLpro: Accurate Sequence-Based Prediction of Protein Solubility. Bioinformatics 2009, 25, 2200–2207. [Google Scholar] [CrossRef] [PubMed]
- Seder, R.A.; Hill, A.V.S. Vaccines against Intracellular Infections Requiring Cellular Immunity. Nature 2000, 406, 793–798. [Google Scholar] [CrossRef] [PubMed]
- Singh, H.; Raghava, G.P.S. ProPred1: Prediction of Promiscuous MHC Class-I Binding Sites. Bioinformatics 2003, 19, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- Andreatta, M.; Nielsen, M. Gapped Sequence Alignment Using Artificial Neural Networks: Application to the MHC Class I System. Bioinformatics 2016, 32, 511–517. [Google Scholar] [CrossRef]
- Calis, J.J.A.; Maybeno, M.; Greenbaum, J.A.; Weiskopf, D.; De Silva, A.D.; Sette, A.; Keşmir, C.; Peters, B. Properties of MHC Class I Presented Peptides that Enhance Immunogenicity. PLoS Comput. Biol. 2013, 9, e1003266. [Google Scholar] [CrossRef]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Raghava, G.P.S. In Silico Approach for Predicting Toxicity of Peptides and Proteins. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Wang, P.; Sidney, J.; Kim, Y.; Sette, A.; Lund, O.; Nielsen, M.; Peters, B. Peptide Binding Predictions for HLA DR, DP and DQ Molecules. BMC Bioinform. 2010, 11, 568. [Google Scholar] [CrossRef]
- Dhanda, S.K.; Karosiene, E.; Edwards, L.; Grifoni, A.; Paul, S.; Andreatta, M.; Weiskopf, D.; Sidney, J.; Nielsen, M.; Peters, B.; et al. Predicting HLA CD4 Immunogenicity in Human Populations. Front. Immunol. 2018, 9, 1369. [Google Scholar] [CrossRef]
- Thomsen, M.; Lundegaard, C.; Buus, S.; Lund, O.; Nielsen, M. MHCcluster, a Method for Functional Clustering of MHC Molecules. Immunogenetics 2013, 65, 655–665. [Google Scholar] [CrossRef]
- Jespersen, M.C.; Peters, B.; Nielsen, M.; Marcatili, P. BepiPred-2.0: Improving Sequence-Based B-Cell Epitope Prediction Using Conformational Epitopes. Nucleic Acids Res. 2017, 45, W24–W29. [Google Scholar] [CrossRef]
- Saha, S.; Raghava, G.P.S. Prediction of Continuous B-Cell Epitopes in an Antigen Using Recurrent Neural Network. Proteins 2006, 65, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Dhanda, S.K.; Mahajan, S.; Paul, S.; Yan, Z.; Kim, H.; Jespersen, M.C.; Jurtz, V.; Andreatta, M.; Greenbaum, J.A.; Marcatili, P.; et al. IEDB-AR: Immune Epitope Database-Analysis Resource in 2019. Nucleic Acids Res. 2019, 47, W502–W506. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, M.R.; Gasteiger, E.; Bairoch, A.; Sanchez, J.C.; Williams, K.L.; Appel, R.D.; Hochstrasser, D.F. Protein Identification and Analysis Tools in the ExPASy Server. Methods Mol. Biol. 1999, 112, 531–552. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, I.; Naneva, L.; Doytchinova, I.; Bangov, I. AllergenFP: Allergenicity Prediction by Descriptor Fingerprints. Bioinformatics 2014, 30, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Naha, A.; Veeraraghavan, B.; Ramaiah, S.; Anbarasu, A. In Silico Structure Evaluation of BAG3 and Elucidating Its Association with Bacterial Infections through Protein-Protein and Host-Pathogen Interaction Analysis. J. Cell. Biochem. 2022, 123, 115–127. [Google Scholar] [CrossRef]
- Shankar, C.; Basu, S.; Lal, B.; Shanmugam, S.; Vasudevan, K.; Mathur, P.; Ramaiah, S.; Anbarasu, A.; Veeraraghavan, B. Aerobactin, Seems to Be a Promising Marker Compared to Unstable RmpA2 for the Identification of Hypervirulent Carbapenem-Resistant Klebsiella Pneumoniae: In-Silico and in-vitro Evidence. Front. Cell. Infect. Microbiol. 2021, 776. [Google Scholar] [CrossRef]
- Schwede, T. SWISS-MODEL: An Automated Protein Homology-Modeling Server. Nucleic Acids Res. 2003, 31, 3381–3385. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinforma. 2016, 54, 5–6. [Google Scholar] [CrossRef]
- Colovos, C.; Yeates, T.O. Verification of Protein Structures: Patterns of Nonbonded Atomic Interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef]
- Gromiha, M.M.; Nagarajan, R.; Selvaraj, S. Protein Structural Bioinformatics: An Overview. In Encyclopedia of Bioinformatics and Computational Biology; Elsevier: Amsterdam, The Netherlands, 2019; pp. 445–459. [Google Scholar]
- Wiltgen, M. Algorithms for Structure Comparison and Analysis: Homology Modelling of Proteins. In Encyclopedia of Bioinformatics and Computational Biology; Elsevier: Amsterdam, The Netherlands, 2019; pp. 38–61. [Google Scholar]
- Cilia, E.; Pancsa, R.; Tompa, P.; Lenaerts, T.; Vranken, W.F. The DynaMine Webserver: Predicting Protein Dynamics from Sequence. Nucleic Acids Res. 2014, 42, 264–270. [Google Scholar] [CrossRef]
- Geourjon, C.; Deléage, G. Sopma: Significant Improvements in Protein Secondary Structure Prediction by Consensus Prediction from Multiple Alignments. Bioinformatics 1995, 11, 681–684. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, W.; Littlejohn, T.G. Swiss-PDB Viewer (Deep View). Brief. Bioinform. 2001, 2, 195–197. [Google Scholar] [CrossRef] [PubMed]
- Heo, L.; Park, H.; Seok, C. GalaxyRefine: Protein Structure Refinement Driven by Side-Chain Repacking. Nucleic Acids Res. 2013, 41, 384–388. [Google Scholar] [CrossRef]
- Varghese, R.; Basu, S.; Neeravi, A.; Pragasam, A.; Aravind, V.; Gupta, R.; Miraclin, A.; Ramaiah, S.; Anbarasu, A.; Veeraraghavan, B. Emergence of Meropenem Resistance among Cefotaxime Non-Susceptible Streptococcus Pneumoniae: Evidence and Challenges. Front. Microbiol. 2022, 12. [Google Scholar] [CrossRef] [PubMed]
- Naha, A.; Banerjee, S.; Debroy, R.; Basu, S.; Ashok, G.; Priyamvada, P.; Kumar, H.; Preethi, A.R.; Singh, H.; Anbarasu, A.; et al. Network Metrics, Structural Dynamics and Density Functional Theory Calculations Identified a Novel Ursodeoxycholic Acid Derivative against Therapeutic Target Parkin for Parkinson’s Disease. Comput. Struct. Biotechnol. J. 2022, 20, 4271–4287. [Google Scholar] [CrossRef]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for Rigid and Symmetric Docking. Nucleic Acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef]
- Mashiach, E.; Schneidman-Duhovny, D.; Andrusier, N.; Nussinov, R.; Wolfson, H.J. FireDock: A Web Server for Fast Interaction Refinement in Molecular Docking. Nucleic Acids Res. 2008, 36, W229–W232. [Google Scholar] [CrossRef]
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. LIGPLOT: A Program to Generate Schematic Diagrams of Protein-Ligand Interactions. Protein Eng. Des. Sel. 1995, 8, 127–134. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera-A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Nabel, G.J. Designing Tomorrow’s Vaccines. N. Engl. J. Med. 2013, 368, 551–560. [Google Scholar] [CrossRef]
- Abdi, S.A.H.; Ali, A.; Sayed, S.F.; Abutahir; Ali, A.; Alam, P. Multi-Epitope-Based Vaccine Candidate for Monkeypox: An in silico Approach. Vaccines 2022, 10, 1564. [Google Scholar] [CrossRef] [PubMed]
- Shantier, S.W.; Mustafa, M.I.; Abdelmoneim, A.H.; Fadl, H.A.; Elbager, S.G.; Makhawi, A.M. Novel Multi Epitope-Based Vaccine against Monkeypox Virus: Vaccinomic Approach. Sci. Rep. 2022, 12, 15983. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, M.; Chatterjee, S.; Nag, S.; Dhama, K.; Chakraborty, C. Designing, Characterization, and Immune Stimulation of a Novel Multi-Epitopic Peptide-Based Potential Vaccine Candidate against Monkeypox Virus through Screening its whole Genome Encoded Proteins: An Immunoinformatics Approach. Travel Med. Infect. Dis. 2022, 50, 102481. [Google Scholar] [CrossRef] [PubMed]
- Dermime, S.; Gilham, D.E.; Shaw, D.M.; Davidson, E.J.; Meziane, E.-K.; Armstrong, A.; Hawkins, R.E.; Stern, P.L. Vaccine and Antibody-Directed T Cell Tumour Immunotherapy. Biochim. Biophys. Acta—Rev. Cancer 2004, 1704, 11–35. [Google Scholar] [CrossRef]
- Meloen, R.H.; Langeveld, J.P.M.; Schaaper, W.M.M.; Slootstra, J.W. Synthetic Peptide Vaccines: Unexpected Fulfillment of Discarded Hope? Biologicals 2001, 29, 233–236. [Google Scholar] [CrossRef]
- Kar, T.; Narsaria, U.; Basak, S.; Deb, D.; Castiglione, F.; Mueller, D.M.; Srivastava, A.P. A Candidate Multi-Epitope Vaccine against SARS-CoV-2. Sci. Rep. 2020, 10, 10895. [Google Scholar] [CrossRef]
- He, Y.; Zhang, J.; Donahue, C.; Falo, L.D. Skin-Derived Dendritic Cells Induce Potent CD8+ T Cell Immunity in Recombinant Lentivector-Mediated Genetic Immunization. Immunity 2006, 24, 643–656. [Google Scholar] [CrossRef]
- Pethe, K.; Alonso, S.; Biet, F.; Delogu, G.; Brennan, M.J.; Locht, C.; Menozzi, F.D. The Heparin-Binding Haemagglutinin of M. Tuberculosis is Required for Extrapulmonary Dissemination. Nature 2001, 412, 190–194. [Google Scholar] [CrossRef]
- Ghaffari-Nazari, H.; Tavakkol-Afshari, J.; Jaafari, M.R.; Tahaghoghi-Hajghorbani, S.; Masoumi, E.; Jalali, S.A. Improving Multi-Epitope Long Peptide Vaccine Potency by Using a Strategy That Enhances CD4+ T Help in BALB/c Mice. PLoS ONE 2015, 10, e0142563. [Google Scholar] [CrossRef]
- George, R.A.; Heringa, J. An Analysis of Protein Domain Linkers: Their Classification and Role in Protein Folding. Protein Eng. 2002, 15, 871–879. [Google Scholar] [CrossRef]
- Aurora, R.; Creamer, T.P.; Srinivasan, R.; Rose, G.D. Local Interactions in Protein Folding: Lessons from the Alpha-Helix. J. Biol. Chem. 1997, 272, 1413–1416. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Name | UniProt ID | AntigenPro | VaxiJen | SolPro | ||
|---|---|---|---|---|---|---|
| Predictefud Probability of Antigenicity | Antigenicity | Overall Protective Probable Antigen Prediction | Solubility upon Overexpression | Probability | ||
| Un-characterized protein | Q3I962 | 0.197732 | Probable Non-Antigen | 0.3865 | Soluble | 0.953785 |
| L5L | Q77HN1 | 0.629557 | Probable Antigen | 0.6787 | Soluble | 0.605143 |
| B20R | Q8V4Q8 | 0.892397 | Probable Antigen | 0.6077 | Insoluble | 0.905405 |
| A35R | Q8V4U4 | 0.972597 | Probable Antigen | 0.5392 | Insoluble | 0.734623 |
| M1R | Q8V502 | 0.940711 | Probable Antigen | 0.6136 | Insoluble | 0.689408 |
| Protein L5 | Q8V4Z8 | 0.352401 | Probable Antigen | 0.4607 | Soluble | 0.531955 |
| C15L | Q8V539 | 0.52584 | Probable Antigen | 0.4806 | Insoluble | 0.850441 |
| A5L | Q8V4 × 1 | 0.942329 | Probable Antigen | 0.4029 | Insoluble | 0.867695 |
| Protein G3 | Q8V509 | 0.523446 | Probable Non-Antigen | 0.356 | Insoluble | 0.828577 |
| A28 | Q8V4U9 | 0.753088 | Probable Antigen | 0.5530 | Soluble | 0.656282 |
| C23R | Q8V531 | 0.637633 | Probable Non-Antigen | 0.3572 | Soluble | 0.792761 |
| C11L | Q8V543 | 0.818476 | Probable Antigen | 0.5360 | Insoluble | 0.68359 |
| A18L | Q8V4W0 | 0.139456 | Probable Non-Antigen | 0.2683 | Insoluble | 0.690301 |
| O2L | Q8V554 | 0.830181 | Probable Antigen | 0.7249 | Insoluble | 0.817524 |
| Protein Name | UniProt ID | Predicted Epitopes | Immunogenicity | Antigenicity | Toxicity |
|---|---|---|---|---|---|
| L5L | Q77HN1 | RVATENIAV | 0.27835 | −0.0904 | Non-Toxin |
| ALAILILAF | 0.2746 | 0.6926 | Non-Toxin | ||
| GLLALAILI | 0.16764 | 0.7314 | Non-Toxin | ||
| ATENIAVRY | 0.2575 | 0.4092 | Non-Toxin | ||
| A28 | Q8V4U9 | NSLSIFFIV | 0.24629 | 0.5713 | Non-Toxin |
| FIVVATAAV | 0.18546 | 0.6997 | Non-Toxin | ||
| KEFNATHAA | 0.15629 | 1.079 | Non-Toxin | ||
| NATHAAFEY | 0.27738 | 1.2587 | Non-Toxin | ||
| KQKWRCVVY | 0.2115 | 1.7426 | Non-Toxin | ||
| SIFGFQAEV | 0.16858 | 0.4227 | Non-Toxin | ||
| MRQCIDFTF | 0.18011 | 1.7234 | Non-Toxin | ||
| L5 | Q8V4Z8 | FEVFVVFIL | 0.38682 | 0.1235 | Non-Toxin |
| IVLFEVFVV | 0.37348 | 0.0325 | Non-Toxin | ||
| NPVFIEPTF | 0.36837 | 1.369 | Non-Toxin | ||
| VFVVFILIY | 0.36262 | 0.2344 | Non-Toxin | ||
| FVVFILIYV | 0.36052 | 0.347 | Non-Toxin | ||
| VLFEVFVVF | 0.34811 | 0.0364 | Non-Toxin | ||
| EVFVVFILI | 0.33578 | 0.1919 | Non-Toxin | ||
| FILIYVFFR | 0.33278 | 0.21 | Non-Toxin | ||
| RLIVLFEVF | 0.29276 | −0.2760 | Non-Toxin | ||
| VFILIYVFF | 0.2614 | 0.0066 | Non-Toxin | ||
| NVYFNPVFI | 0.2031 | 1.4789 | Non-Toxin | ||
| LMIYGLPWI | 0.1821 | 0.388 | Non-Toxin |
| Protein Name | UniProt ID | Predicted Epitopes | Immunogenicity | Antigenicity | Toxicity |
|---|---|---|---|---|---|
| L5 | Q8V4Z8 | FFRSELNMFFMPKRK | 56.1564 | 0.7973 | Non-Toxin |
| FRSELNMFFMPKRKI | 58.0094 | 0.5608 | Non-Toxin | ||
| SELNMFFMPKRKIPD | 51.2572 | 0.5064 | Non-Toxin | ||
| ELNMFFMPKRKIPDP | 56.1681 | 0.6630 | Non-Toxin | ||
| LNMFFMPKRKIPDPI | 53.3717 | 0.6685 | Non-Toxin |
| Protein Name | MHC Class-I T-Cell | MHC Class-II T-Cell | B-Cell | |||
|---|---|---|---|---|---|---|
| Predicted Epitope | Position | Predicted Epitope | Position | Predicted Epitope | Position | |
| L5L | ALAILILAF | 124–132 | - | - | NKDD | 13–16 |
| GLLALAILI | 121–129 | - | - | KSIVRIGIDTR | 26–36 | |
| ATENIAVRY | 95–103 | - | - | ISRC | 61–64 | |
| - | - | - | - | LDVNNVCDSKRVATENIAVRYLNQEIRYPIIDIK | 83–116 | |
| A28 | NSLSIFFIV | 2–10 | - | - | QAEVGPNNTRSIRKFNTMRQ | 92–111 |
| FIVVATAAV | 8–16 | - | - | FSDVINI | 117–123 | |
| KEFNATHAA | 34–42 | - | - | YGNIKEFNATHAAFEYSKSIGGTPALDRRVQDVNDTISDVKQ | 30–71 | |
| NATHAAFEY | 37–45 | - | - | - | - | |
| KQKWRCVVY | 70–78 | - | - | - | - | |
| SIFGFQAEV | 87–95 | - | - | - | - | |
| MRQCIDFTF | 109–117 | - | - | - | - | |
| L5 | NVYFNPVFI | 6–14 | FFRSELNMFFMPKRK | 46–60 | KHSLLSVY | 19–26 |
| NPVFIEPTF | 10–18 | FRSELNMFFMPKRKI | 47–61 | MPKRKIPDPIDRLRR | 56–70 | |
| - | - | SELNMFFMPKRKIPD | 49–63 | LPWITTQTSALSINSKPIVYK | 85–105 | |
| - | - | ELNMFFMPKRKIPDP | 50–64 | - | - | |
| - | - | LNMFFMPKRKIPDPI | 51–65 | - | - | |
| Vaccine Construct | Vaccine Composition | Sequence |
|---|---|---|
| V1 | HBHA, E1, E2, E3, E4, linkers, PADRE, His Tag | EAAKMAENPNIDDLPAPLLAALGAADLALATVNDLIANLRERA EETRAETRTRVEERRARLTKFQEDLPEQFIELRDKFTTEELRKAAE GYLEAATNRYNELVERGEAALQRLRSQTAFEDASARAEGYVDQA VELTQEALGTVASQTRAVGERAAKLVGIELEAAKGPGPGAKFVA AWTLKAAAGPGPGLDVNNVCDSKRVATENIAVRYLNQEIRYPII DIKRVRRYGNIKEFNATHAAFEYSKSIGGTPALDRRVQDVNDTIS DVKQRVRRQAEVGPNNTRSIRKFNTMRQCIDFTFSDVINIRVRRM PKRKIPDPIDRLRRGPGPGAKFVAAWTLKAAAGPGPGHHHHHH |
| V2 | HBHA conserved, E1, E2, E3, E4, linkers, PADRE, His Tag | EAAAKMAENSNIDDIKAPLLAALGAADLALATVNELITNLRERA EETRRSRVEESRARLTKLQEDLPEQLTELREKFTAEELRKAAEGYL EAATSELVERGEAALERLRSQQSFEEVSARAEGYVDQAVELTQEA LGTVASQVEGRAAKLVGIELEAAAKGPGPGAKFVAAWTLKAAA GPGPGLDVNNVCDSKRVATENIAVRYLNQEIRYPIIDIKRVRRYG NIKEFNATHAAFEYSKSIGGTPALDRRVQDVNDTISDVKQRVRRQ AEVGPNNTRSIRKFNTMRQCIDFTFSDVINIRVRRMPKRKIPDPID RLRRGPGPGAKFVAAWTLKAAAGPGPGHHHHHH |
| V3 | β-Defensin, E1, E2, E3, E4, linkers, PADRE, His Tag | EAAKGIINTLQKYYCRVRGGRCAVLSCLPKEEQIGKCSTRGRKCC RRKKEAAKGPGPGAKFVAAWTLKAAAGPGPGLDVNNVCDSKR VATENIAVRYLNQEIRYPIIDIKRVRRYGNIKEFNATHAAFEYSKSI GGTPALDRRVQDVNDTISDVKQRVRRQAEVGPNNTRSIRKFNTM RQCIDFTFSDVINIRVRRMPKRKIPDPIDRLRRGPGPGAKFVAAWT LKAAAGPGPGHHHHHH |
| V4 | L7/L12 Ribosomal protein, E1, E2, E3, E4, linkers, PADRE, His Tag | EAAKMAKLSTDELLDAFKEMTLLELSDFVKKFEETFEVTAAAPVA VAAAGAAPAGAAVEAAEEQSEFDVILEAAGDKKIGVIKVVREIVS GLGLKEAKDLVDGAPKPLLEKVAKEAADEAKAKLEAAGATVTV KEAAAKGPGPGAKFVAAWTLKAAAGPGPGLDVNNVCDSKRVA TENIAVRYLNQEIRYPIIDIKRVRRYGNIKEFNATHAAFEYSKSIGG TPALDRRVQDVNDTISDVKQRVRRQAEVGPNNTRSIRKFNTMRQ CIDFTFSDVINIRVRRMPKRKIPDPIDRLRRGPGPGAKFVAAWTLK AAAGPGPGHHHHHH |
| V5 | HBHA, E2, E1, E3, E4, linkers, PADRE, His Tag | EAAKMAENPNIDDLPAPLLAALGAADLALATVNDLIANLRERA EETRAETRTRVEERRARLTKFQEDLPEQFIELRDKFTTEELRKAAE GYLEAATNRYNELVERGEAALQRLRSQTAFEDASARAEGYVDQ AVELTQEALGTVASQTRAVGERAAKLVGIELEAAKGPGPGAKFV AAWTLKAAAGPGPGYGNIKEFNATHAAFEYSKSIGGTPALDRRV QDVNDTISDVKRVRRLDVNNVCDSKRVATENIAVRYLNQEIRYPI IDIKRVRRQAEVGPNNTRSIRKFNTMRQCIDFTFSDVINIRVRRMPK RKIPDPIDRLRRGPGPGAKFVAAWTLKAAAGPGPGHHHHHH |
| V6 | HBHA conserved, E2, E1, E3, E4, linkers, PADRE, His Tag | EAAAKMAENSNIDDIKAPLLAALGAADLALATVNELITNLRERA EETRRSRVEESRARLTKLQEDLPEQLTELREKFTAEELRKAAEGYL EAATSELVERGEAALERLRSQQSFEEVSARAEGYVDQAVELTQEA LGTVASQVEGRAAKLVGIELEAAAKGPGPGAKFVAAWTLKAAA GPGPGYGNIKEFNATHAAFEYSKSIGGTPALDRRVQDVNDTISDV KRVRRLDVNNVCDSKRVATENIAVRYLNQEIRYPIIDIKRVRRQA EVGPNNTRSIRKFNTMRQCIDFTFSDVINIRVRRMPKRKIPDPIDRLR RGPGPGAKFVAAWTLKAAAGPGPGHHHHHH |
| V7 | β-Defensin, E2, E1, E3, E4, linkers, PADRE, His Tag | EAAKGIINTLQKYYCRVRGGRCAVLSCLPKEEQIGKCSTRGRKCC RRKKEAAKGPGPGAKFVAAWTLKAAAGPGPGYGNIKEFNATH AAFEYSKSIGGTPALDRRVQDVNDTISDVKRVRRLDVNNVCDSK RVATENIAVRYLNQEIRYPIIDIKRVRRQAEVGPNNTRSIRKFNTM RQCIDFTFSDVINIRVRRMPKRKIPDPIDRLRRGPGPGAKFVAAWT LKAAAGPGPGHHHHHH |
| V8 | L7/L12 Ribosomal protein, E2, E1, E3, E4, linkers, PADRE, His Tag | EAAKMAKLSTDELLDAFKEMTLLELSDFVKKFEETFEVTAAAPVA VAAAGAAPAGAAVEAAEEQSEFDVILEAAGDKKIGVIKVVREIVS GLGLKEAKDLVDGAPKPLLEKVAKEAADEAKAKLEAAGATVTV KEAAAKGPGPGAKFVAAWTLKAAAGPGPGYGNIKEFNATHAA FEYSKSIGGTPALDRRVQDVNDTISDVKRVRRLDVNNVCDSKRVA TENIAVRYLNQEIRYPIIDIKRVRRQAEVGPNNTRSIRKFNTMRQCI DFTFSDVINIRVRRMPKRKIPDPIDRLRRGPGPGAKFVAAWTLKA AAGPGPGHHHHHH |
| V9 | HBHA, E3, E2, E1, E4, linkers, PADRE, His Tag | EAAKMAENPNIDDLPAPLLAALGAADLALATVNDLIANLRERA EETRAETRTRVEERRARLTKFQEDLPEQFIELRDKFTTEELRKAAE GYLEAATNRYNELVERGEAALQRLRSQTAFEDASARAEGYVDQ AVELTQEALGTVASQTRAVGERAAKLVGIELEAAKGPGPGAKFV AAWTLKAAAGPGPGQAEVGPNNTRSIRKFNTMRQCIDFTFSDVI NIRVRRYGNIKEFNATHAAFEYSKSIGGTPALDRRVQDVNDTISD VKRVRRLDVNNVCDSKRVATENIAVRYLNQEIRYPIIDIKRVRRM PKRKIPDPIDRLRRGPGPGAKFVAAWTLKAAAGPGPGHHHHHH |
| V10 | HBHA conserved, E3, E2, E1, E4, linkers, PADRE, His Tag | EAAAKMAENSNIDDIKAPLLAALGAADLALATVNELITNLRERA EETRRSRVEESRARLTKLQEDLPEQLTELREKFTAEELRKAAEGYL EAATSELVERGEAALERLRSQQSFEEVSARAEGYVDQAVELTQEA LGTVASQVEGRAAKLVGIELEAAAKGPGPGAKFVAAWTLKAAA GPGPGQAEVGPNNTRSIRKFNTMRQCIDFTFSDVINIRVRRYGNIK EFNATHAAFEYSKSIGGTPALDRRVQDVNDTISDVKRVRRLDVN NVCDSKRVATENIAVRYLNQEIRYPIIDIKRVRRMPKRKIPDPIDRL RRGPGPGAKFVAAWTLKAAAGPGPGHHHHHH |
| V11 | β-Defensin, E3, E2, E1, E4, linkers, PADRE, His Tag | EAAKGIINTLQKYYCRVRGGRCAVLSCLPKEEQIGKCSTRGRKCC RRKKEAAKGPGPGAKFVAAWTLKAAAGPGPGQAEVGPNNTRSI RKFNTMRQCIDFTFSDVINIRVRRYGNIKEFNATHAAFEYSKSIGG TPALDRRVQDVNDTISDVKRVRRLDVNNVCDSKRVATENIAVRY LNQEIRYPIIDIKRVRRMPKRKIPDPIDRLRRGPGPGAKFVAAWTL KAAAGPGPGHHHHHH |
| V12 | L7/L12 Ribosomal protein, E3, E2, E1, E4, linkers, PADRE, His Tag | EAAKMAKLSTDELLDAFKEMTLLELSDFVKKFEETFEVTAAAPVA VAAAGAAPAGAAVEAAEEQSEFDVILEAAGDKKIGVIKVVREIVS GLGLKEAKDLVDGAPKPLLEKVAKEAADEAKAKLEAAGATVTV KEAAAKGPGPGAKFVAAWTLKAAAGPGPGQAEVGPNNTRSIRK FNTMRQCIDFTFSDVINIRVRRYGNIKEFNATHAAFEYSKSIGGTP ALDRRVQDVNDTISDVKRVRRLDVNNVCDSKRVATENIAVRYL NQEIRYPIIDIKRVRRMPKRKIPDPIDRLRRGPGPGAKFVAAWTLK AAAGPGPGHHHHHH |
| V13 | HBHA, E4, E3, E2, E1, linkers, PADRE, His Tag | EAAKMAENPNIDDLPAPLLAALGAADLALATVNDLIANLRERA EETRAETRTRVEERRARLTKFQEDLPEQFIELRDKFTTEELRKAAE GYLEAATNRYNELVERGEAALQRLRSQTAFEDASARAEGYVDQ AVELTQEALGTVASQTRAVGERAAKLVGIELEAAKGPGPGAKFV AAWTLKAAAGPGPGMPKRKIPDPIDRLRRRVRRQAEVGPNNTRS IRKFNTMRQCIDFTFSDVINIRVRRYGNIKEFNATHAAFEYSKSIGG TPALDRRVQDVNDTISDVKRVRRLDVNNVCDSKRVATENIAVRY LNQEIRYPIIDIKGPGPGAKFVAAWTLKAAAGPGPGHHHHHH |
| V14 | HBHA conserved, E4, E3, E2, E1, linkers, PADRE, His Tag | EAAAKMAENSNIDDIKAPLLAALGAADLALATVNELITNLRERA EETRRSRVEESRARLTKLQEDLPEQLTELREKFTAEELRKAAEGYL EAATSELVERGEAALERLRSQQSFEEVSARAEGYVDQAVELTQEA LGTVASQVEGRAAKLVGIELEAAAKGPGPGAKFVAAWTLKAAA GPGPGMPKRKIPDPIDRLRRRVRRQAEVGPNNTRSIRKFNTMRQC IDFTFSDVINIRVRRYGNIKEFNATHAAFEYSKSIGGTPALDRRVQ DVNDTISDVKRVRRLDVNNVCDSKRVATENIAVRYLNQEIRYPII DIKGPGPGAKFVAAWTLKAAAGPGPGHHHHHH |
V15 | Defensin, E4, E3, E2, E1, linkers, PADRE, His Tag | EAAKGIINTLQKYYCRVRGGRCAVLSCLPKEEQIGKCSTRGRKCC RRKKEAAKGPGPGAKFVAAWTLKAAAGPGPGMPKRKIPDPIDR LRRRVRRQAEVGPNNTRSIRKFNTMRQCIDFTFSDVINIRVRRYG NIKEFNATHAAFEYSKSIGGTPALDRRVQDVNDTISDVKRVRRLD VNNVCDSKRVATENIAVRYLNQEIRYPIIDIKGPGPGAKFVAAWT LKAAAGPGPGHHHHHH |
| V16 | L7/L12 Ribosomal protein, E4, E3, E2, E1, linkers, PADRE, His Tag | EAAKMAKLSTDELLDAFKEMTLLELSDFVKKFEETFEVTAAAPVA VAAAGAAPAGAAVEAAEEQSEFDVILEAAGDKKIGVIKVVREIVS GLGLKEAKDLVDGAPKPLLEKVAKEAADEAKAKLEAAGATVTV KEAAAKGPGPGAKFVAAWTLKAAAGPGPGMPKRKIPDPIDRLR RRVRRQAEVGPNNTRSIRKFNTMRQCIDFTFSDVINIRVRRYGNIK EFNATHAAFEYSKSIGGTPALDRRVQDVNDTISDVKRVRRLDVN NVCDSKRVATENIAVRYLNQEIRYPIIDIKGPGPGAKFVAAWTLK AAAGPGPGHHHHHH |
| Vaccine | Probable Non-Allergen | Probable Antigen | Solubility Probability | Molecular Weight | Theoretical pI | Instability Index | Aliphatic Index | GRAVY | |
|---|---|---|---|---|---|---|---|---|---|
| Server Used | AlgPred, AllergenFP 2.0 | VaxiJen | AntigenPro | ProtParam | ProtParam | ProtParam | ProtParam | ProtParam | ProtParam |
| V1 | + | +(0.4936) | +(0.668389) | +(0.871680) | 39243.35 | 9.35 | −(43.12) | 80.90 | −0.570 |
| V2 | + | +(0.4957) | +(0.602766) | +(0.920375) | 38267.29 | 9.11 | −(47.62) | 83.92 | −0.530 |
| V3 | + | +(0.5062) | +(0.732167) | +(0.979442) | 26775.90 | 10.40 | −(42.88) | 71.58 | −0.644 |
| V4 | + | +(0.4583) | +(0.719020) | +(0.857865) | 35126.27 | 9.31 | +(33.47) | 85.12 | −0.300 |
| V5 | + | +(0.5092) | +(0.521264) | +(0.857733) | 39115.22 | 9.35 | −(43.33) | 81.13 | −0.562 |
| V6 | + | +(0.5116) | −(0.428664) | +(0.919505) | 38139.16 | 9.11 | −(47.96) | 84.16 | −0.521 |
| V7 | + | +(0.5295) | +(0.702073) | +(0.980207) | 26647.77 | 10.40 | −(43.35) | 71.88 | −0.632 |
| V8 | + | +(0.4751) | +(0.717110) | +(0.859654) | 34998.14 | 9.31 | +(33.79) | 85.38 | −0.290 |
| V9 | + | +(0.4976) | +(0.500000) | +(0.850564) | 39115.22 | 9.35 | −(42.92) | 81.13 | −0.562 |
| V10 | + | +(0.4999) | −(0.420452) | +(0.919082) | 38139.16 | 9.11 | −(47.43) | 84.16 | −0.521 |
| V11 | + | +(0.5124) | +(0.670082) | +(0.978970) | 26647.77 | 10.4 | −(42.59) | 71.88 | −0.632 |
| V12 | + | +(0.4626) | +(0.721238) | +(0.846745) | 34998.14 | 9.31 | +(33.22) | 85.38 | −0.290 |
| V13 | + | +(0.4928) | +(0.500000) | +(0.863467) | 39115.22 | 9.35 | −(44.17) | 81.13 | −0.562 |
| V14 | + | +(0.4950) | −(0.406867) | +(0.917314) | 38139.16 | 9.11 | −(48.70) | 84.16 | −0.521 |
| V15 | + | +(0.5053) | +(0.668008) | +(0.979064) | 26647.77 | 10.40 | −(44.43) | 71.88 | −0.632 |
| V16 | + | +(0.4574) | +(0.686732) | +(0.850063) | 34998.14 | 9.31 | +(34.57) | 85.38 | −0.290 |
| Properties | V4 | V8 | V12 | V16 |
|---|---|---|---|---|
| Quality factor | 99.05 | 94.32 | 95.87 | 95.06 |
| Ramachandran favored | 96.06% | 93.5% | 95.05% | 92.88% |
| Verify3D structural violations | Pass | Pass | Fail | Pass |
| Distribution Z-score | −0.11 ± 0.43 | −0.16 ± 0.43 | 1.47 ± 0.45 | −0.35 ± 0.45 |
| Backbone dynamics (Average S2-parameter scores) | 0.79 | 0.81 | 0.81 | 0.81 |
| Vaccine Construct | Target (PDB IDs) | Global Energy (kcal/mol) | Attractive vdW | Atomic Contact Energy (kcal/mol) | Hydrogen Bond Contribution (kcal/mol) |
|---|---|---|---|---|---|
| Vaccine Construct-4 (V4) | 1H15 | −23.62 | −42.33 | 14.93 | −5.68 |
| 2FSE | −17.38 | −34.59 | 12.27 | −3.15 | |
| 2Z65 | −1.98 | −2.30 | 2.60 | 0.00 | |
| 2Q6W | −0.97 | −6.92 | −0.01 | −0.76 | |
| 2SEB | −0.61 | −25.23 | 4.33 | −1.63 | |
| 1A6A | 1.45 | −35.06 | 6.69 | −2.41 | |
| 3C5J | 2.74 | −21.38 | 9.68 | −4.05 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Swetha, R.G.; Basu, S.; Ramaiah, S.; Anbarasu, A. Multi-Epitope Vaccine for Monkeypox Using Pan-Genome and Reverse Vaccinology Approaches. Viruses 2022, 14, 2504. https://doi.org/10.3390/v14112504
Swetha RG, Basu S, Ramaiah S, Anbarasu A. Multi-Epitope Vaccine for Monkeypox Using Pan-Genome and Reverse Vaccinology Approaches. Viruses. 2022; 14(11):2504. https://doi.org/10.3390/v14112504
Chicago/Turabian StyleSwetha, Rayapadi G., Soumya Basu, Sudha Ramaiah, and Anand Anbarasu. 2022. "Multi-Epitope Vaccine for Monkeypox Using Pan-Genome and Reverse Vaccinology Approaches" Viruses 14, no. 11: 2504. https://doi.org/10.3390/v14112504
APA StyleSwetha, R. G., Basu, S., Ramaiah, S., & Anbarasu, A. (2022). Multi-Epitope Vaccine for Monkeypox Using Pan-Genome and Reverse Vaccinology Approaches. Viruses, 14(11), 2504. https://doi.org/10.3390/v14112504