
H1N1 Influenza A Virus Protein NS2 Inhibits Innate Immune Response by Targeting IRF7



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Biosafety and Ethical Statements
2.2. Cells, Viruses, and Plasmids
2.3. Reagents and Antibodies
2.4. Dual-Luciferase Reporter Assays
2.5. RNA Isolation and Quantitative PCR
2.6. Western Blot
2.7. Co-Immunoprecipitation
2.8. Confocal Microscopy
2.9. VSV-GFP Bioassay
2.10. Statistical Analysis
3. Results
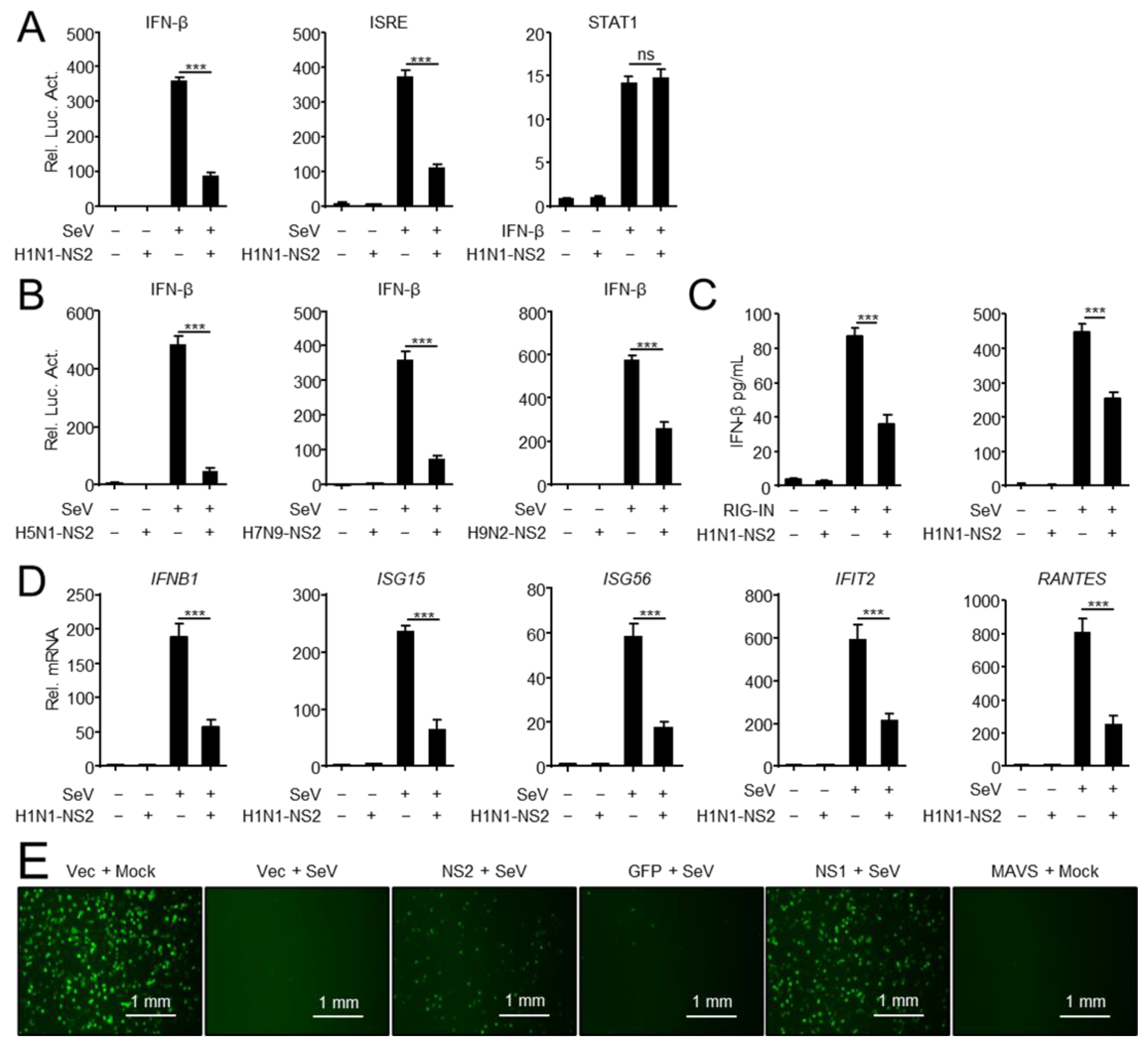
3.1. NS2 Inhibits IFN-I Induction
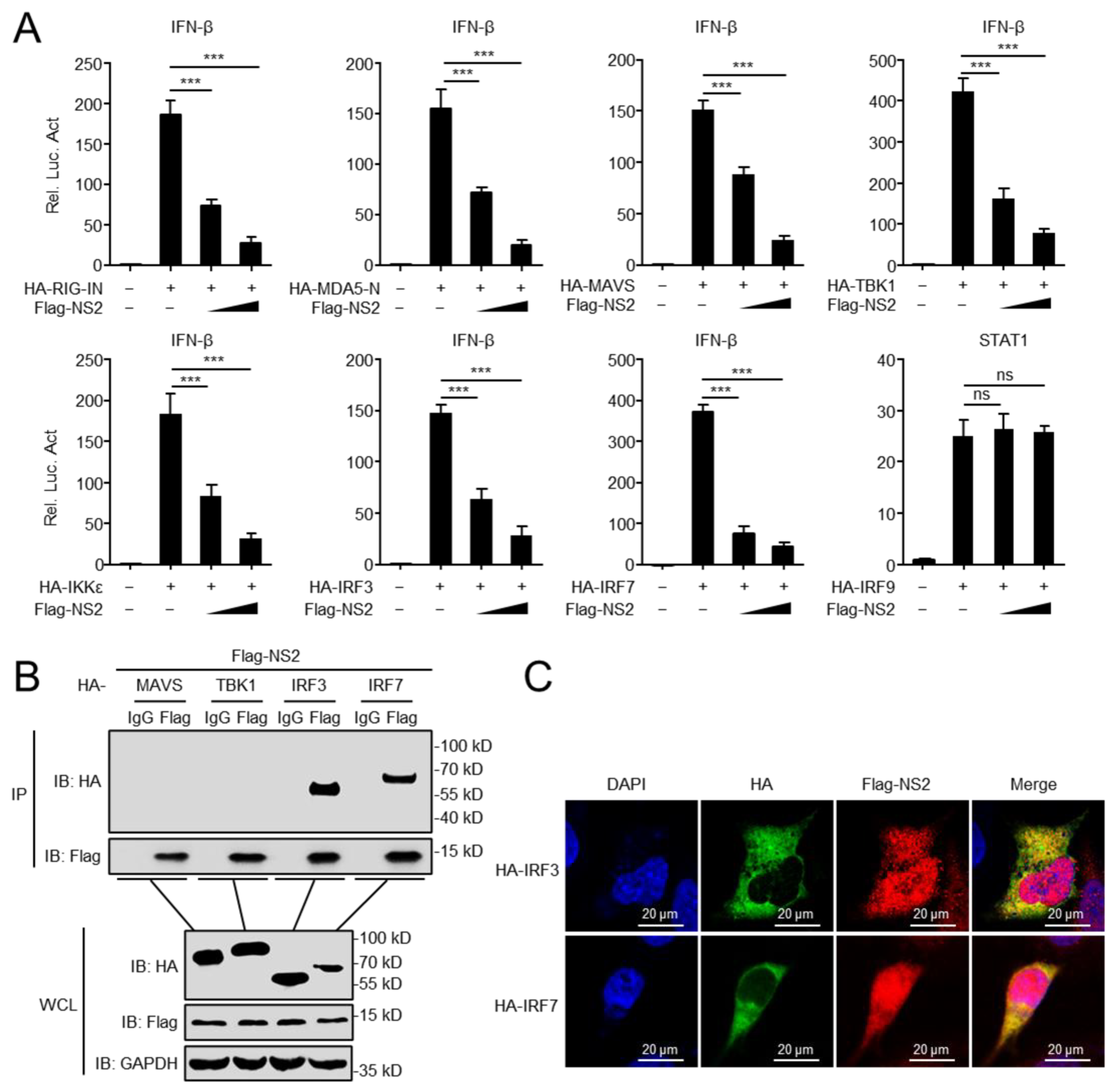
3.2. NS2 Interacts with IRF3 and IRF7
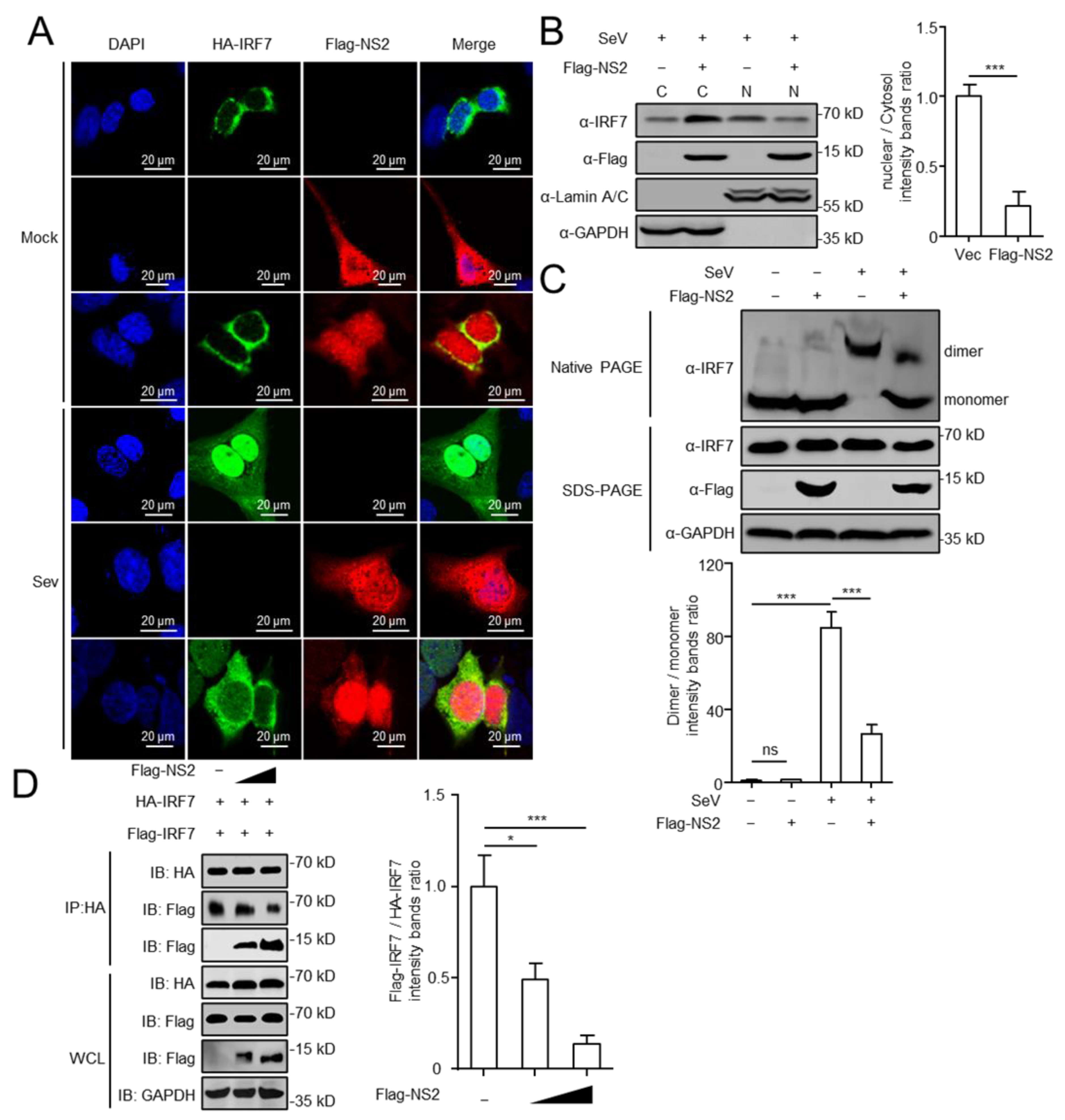
3.3. NS2 Targets IRF7 and Inhibits IFN-I Production
3.4. NS2 Blocks the Nuclear Translocation of IRF7
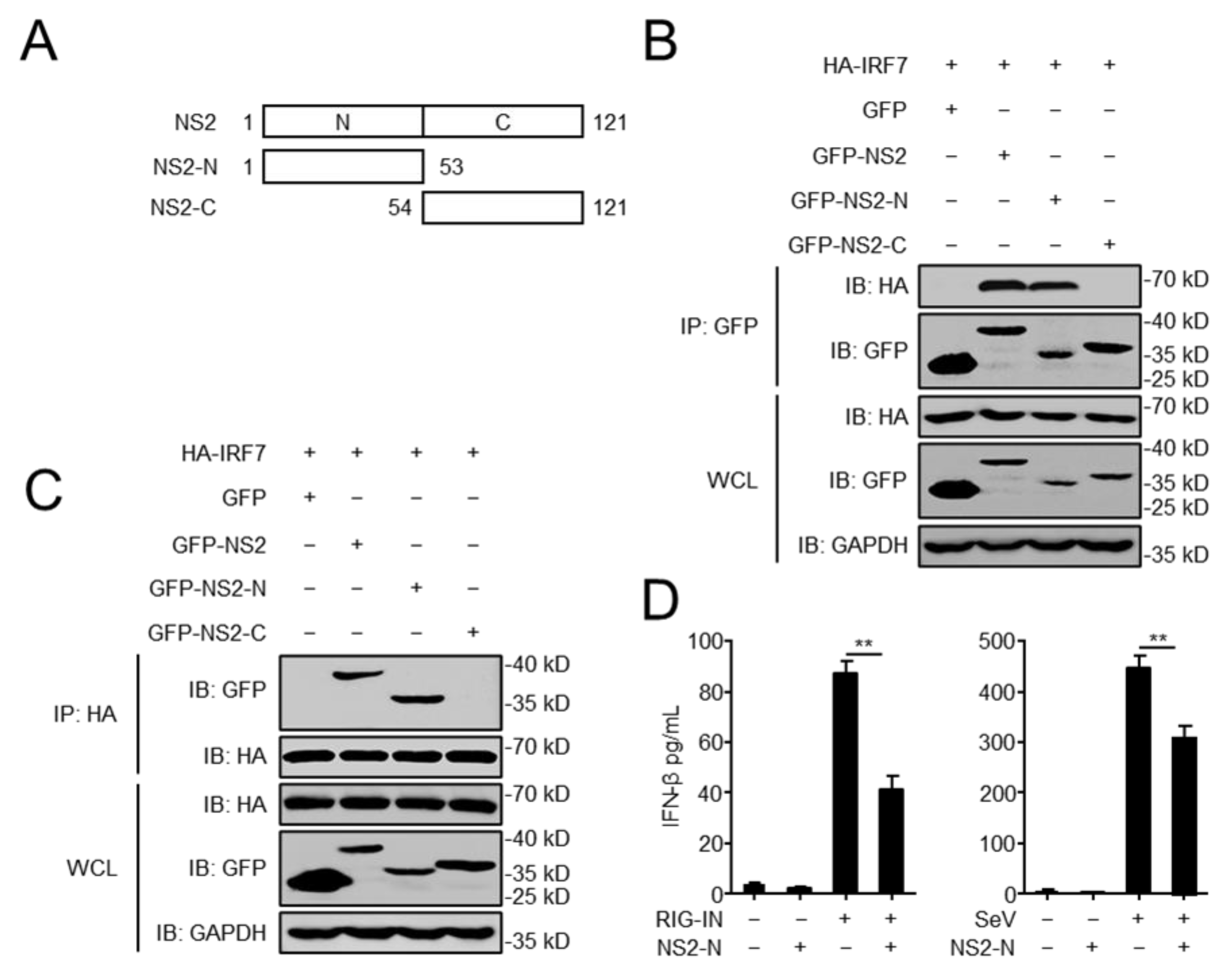
3.5. The N-Terminal Domain of NS2 Is Essential for the Interaction with IRF7
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lipsitch, M. Avian influenza: Ferret H7N9 flu model questioned. Nature 2013, 501, 33. [Google Scholar] [CrossRef] [PubMed]
- Medina, R.A.; Garcia-Sastre, A. Influenza A viruses: New research developments. Nat. Rev. Microbiol. 2011, 9, 590–603. [Google Scholar] [CrossRef]
- Zhang, Q.; Shi, J.; Deng, G.; Guo, J.; Zeng, X.; He, X.; Kong, H.; Gu, C.; Li, X.; Liu, J.; et al. H7N9 influenza viruses are transmissible in ferrets by respiratory droplet. Science 2013, 341, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Cui, P.; Zeng, X.; Li, X.; Li, Y.; Shi, J.; Zhao, C.; Qu, Z.; Wang, Y.; Guo, J.; Gu, W.; et al. Genetic and biological characteristics of the globally circulating H5N8 avian influenza viruses and the protective efficacy offered by the poultry vaccine currently used in China. Sci. China Life Sci. 2022, 65, 795–808. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Deng, G.; Zeng, X.; Cui, P.; Hou, Y.; Liu, Y.; Fang, J.; Pan, S.; Wang, D.; Chen, X.; et al. Genetic and biological properties of H7N9 avian influenza viruses detected after application of the H7N9 poultry vaccine in China. PLoS Pathog. 2021, 17, e1009561. [Google Scholar] [CrossRef]
- Zeng, X.-Y.; He, X.-W.; Meng, F.; Ma, Q.; Wang, Y.; Bao, H.-M.; Liu, Y.-J.; Deng, G.-H.; Shi, J.-Z.; Li, Y.-B.; et al. Protective efficacy of an H5/H7 trivalent inactivated vaccine (H5-Re13, H5-Re14, and H7-Re4 strains) in chickens, ducks, and geese against newly detected H5N1, H5N6, H5N8, and H7N9 viruses. J. Integr. Agric. 2022, 21, 2086–2094. [Google Scholar] [CrossRef]
- Cui, Y.; Li, Y.; Li, M.; Zhao, L.; Wang, D.; Tian, J.; Bai, X.; Ci, Y.; Wu, S.; Wang, F.; et al. Evolution and extensive reassortment of H5 influenza viruses isolated from wild birds in China over the past decade. Emerg. Microbes Infect. 2020, 9, 1793–1803. [Google Scholar] [CrossRef]
- Eisfeld, A.J.; Neumann, G.; Kawaoka, Y. At the centre: Influenza A virus ribonucleoproteins. Nat. Rev. Microbiol. 2015, 13, 28–41. [Google Scholar] [CrossRef]
- Lakdawala, S.S.; Wu, Y.; Wawrzusin, P.; Kabat, J.; Broadbent, A.J.; Lamirande, E.W.; Fodor, E.; Altan-Bonnet, N.; Shroff, H.; Subbarao, K. Influenza a virus assembly intermediates fuse in the cytoplasm. PLoS Pathog. 2014, 10, e1003971. [Google Scholar] [CrossRef]
- Wang, J.; Zeng, Y.; Xu, S.; Yang, J.; Wang, W.; Zhong, B.; Ge, J.; Yin, L.; Bu, Z.; Shu, H.B.; et al. A naturally occurring deletion in the effector domain of H5N1 swine influenza virus nonstructural protein 1 regulates viral fitness and host innate immunity. J. Virol. 2018, 92, e00149-18. [Google Scholar] [CrossRef]
- Hutchinson, E.C.; Charles, P.D.; Hester, S.S.; Thomas, B.; Trudgian, D.; Martinez-Alonso, M.; Fodor, E. Conserved and host-specific features of influenza virion architecture. Nat. Commun. 2014, 5, 4816. [Google Scholar] [CrossRef] [PubMed]
- Inglis, S.C.; Barrett, T.; Brown, C.M.; Almond, J.W. The smallest genome RNA segment of influenza virus contains two genes that may overlap. Proc. Natl. Acad. Sci. USA 1979, 76, 3790–3794. [Google Scholar] [CrossRef] [PubMed]
- Lamb, R.A.; Choppin, P.W. Segment 8 of the influenza virus genome is unique in coding for two polypeptides. Proc. Natl. Acad. Sci. USA 1979, 76, 4908–4912. [Google Scholar] [CrossRef]
- Bullido, R.; Gomez-Puertas, P.; Saiz, M.J.; Portela, A. Influenza A virus NEP (NS2 protein) downregulates RNA synthesis of model template RNAs. J. Virol. 2001, 75, 4912–4917. [Google Scholar] [CrossRef] [PubMed]
- Iwatsuki-Horimoto, K.; Horimoto, T.; Fujii, Y.; Kawaoka, Y. Generation of influenza A virus NS2 (NEP) mutants with an altered nuclear export signal sequence. J. Virol. 2004, 78, 10149–10155. [Google Scholar] [CrossRef] [PubMed]
- Paterson, D.; Fodor, E. Emerging roles for the influenza A virus nuclear export protein (NEP). PLoS Pathog. 2012, 8, e1003019. [Google Scholar] [CrossRef]
- Gong, W.; He, X.; Huang, K.; Zhang, Y.; Li, C.; Yang, Y.; Zou, Z.; Jin, M. Interaction of NEP with G protein pathway suppressor 2 facilitates influenza A virus replication by weakening the inhibition of GPS2 to RNA synthesis and ribonucleoprotein assembly. J. Virol. 2021, 95, e00008-21. [Google Scholar] [CrossRef]
- Gorai, T.; Goto, H.; Noda, T.; Watanabe, T.; Kozuka-Hata, H.; Oyama, M.; Takano, R.; Neumann, G.; Watanabe, S.; Kawaoka, Y. F1Fo-ATPase, F-type proton-translocating ATPase, at the plasma membrane is critical for efficient influenza virus budding. Proc. Natl. Acad. Sci. USA 2012, 109, 4615–4620. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. Innate immune recognition of viral infection. Nat. Immunol. 2006, 7, 131–137. [Google Scholar] [CrossRef]
- Thompson, M.R.; Kaminski, J.J.; Kurt-Jones, E.A.; Fitzgerald, K.A. Pattern recognition receptors and the innate immune response to viral infection. Viruses 2011, 3, 920–940. [Google Scholar] [CrossRef]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef]
- Xu, L.G.; Wang, Y.Y.; Han, K.J.; Li, L.Y.; Zhai, Z.; Shu, H.B. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol. Cell 2005, 19, 727–740. [Google Scholar] [CrossRef]
- Chen, Y.; Lei, X.; Jiang, Z.; Fitzgerald, K.A. Cellular nucleic acid-binding protein is essential for type I interferon-mediated immunity to RNA virus infection. Proc. Natl. Acad. Sci. USA 2021, 118, e2100383118. [Google Scholar] [CrossRef] [PubMed]
- Negishi, H.; Taniguchi, T.; Yanai, H. The interferon (IFN) class of cytokines and the IFN regulatory factor (IRF) transcription factor family. Cold Spring Harb. Perspect. Biol. 2018, 10, a028423. [Google Scholar] [CrossRef] [PubMed]
- Kong, H.; Burke, D.F.; da Silva Lopes, T.J.; Takada, K.; Imai, M.; Zhong, G.; Hatta, M.; Fan, S.; Chiba, S.; Smith, D.; et al. Plasticity of the Influenza Virus H5 HA Protein. Mbio 2021, 12, e03324-20. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liang, L.; Jiang, L.; Wang, Q.; Wen, X.; Zhao, Y.; Cui, P.; Zhang, Y.; Wang, G.; Li, Q.; et al. Viral RNA-binding ability conferred by SUMOylation at PB1 K612 of influenza A virus is essential for viral pathogenesis and transmission. PLoS Pathog. 2021, 17, e1009336. [Google Scholar] [CrossRef]
- Wang, G.; Zhao, Y.; Zhou, Y.; Jiang, L.; Liang, L.; Kong, F.; Yan, Y.; Wang, X.; Wang, Y.; Wen, X.; et al. PIAS1-mediated SUMOylation of influenza A virus PB2 restricts viral replication and virulence. PLoS Pathog. 2022, 18, e1010446. [Google Scholar] [CrossRef]
- Wang, X.; Jiang, L.; Wang, G.; Shi, W.; Hu, Y.; Wang, B.; Zeng, X.; Tian, G.; Deng, G.; Shi, J.; et al. Influenza A virus use of BinCARD1 to facilitate the binding of viral NP to importin α7 is counteracted by TBK1-p62 axis-mediated autophagy. Cell. Mol. Immunol. 2022, 19, 1168–1184. [Google Scholar] [CrossRef]
- Krug, R.M. Functions of the influenza A virus NS1 protein in antiviral defense. Curr. Opin. Virol. 2015, 12, 1–6. [Google Scholar] [CrossRef]
- Zhu, Q.; Yang, H.; Chen, W.; Cao, W.; Zhong, G.; Jiao, P.; Deng, G.; Yu, K.; Yang, C.; Bu, Z.; et al. A naturally occurring deletion in its NS gene contributes to the attenuation of an H5N1 swine influenza virus in chickens. J. Virol. 2008, 82, 220–228. [Google Scholar] [CrossRef]
- Liu, X.; Yang, C.; Hu, Y.; Lei, E.; Lin, X.; Zhao, L.; Zou, Z.; Zhang, A.; Zhou, H.; Chen, H.; et al. HIST1H1C regulates interferon-beta and inhibits influenza virus replication by interacting with IRF3. Front. Immunol. 2017, 8, 350. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Xu, S.; Wei, Y.; Zhang, X.; Wang, Q.; Jia, Y.; Wang, W.; Han, L.; Chen, Z.; Wang, Z.; et al. The PB1 protein of influenza A virus inhibits the innate immune response by targeting MAVS for NBR1-mediated selective autophagic degradation. PLoS Pathog. 2021, 17, e1009300. [Google Scholar] [CrossRef] [PubMed]
- Brunn, D.; Turkowski, K.; Gunther, S.; Weigert, A.; Muley, T.; Kriegsmann, M.; Winter, H.; Dammann, R.H.; Stathopoulos, G.T.; Thomas, M.; et al. Interferon Regulatory Factor 9 Promotes Lung Cancer Progression via Regulation of Versican. Cancers 2021, 13, 208. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lei, C.Q.; Ji, Y.; Zhou, H.; Ren, Y.; Peng, Q.; Zeng, Y.; Jia, Y.; Ge, J.; Zhong, B.; et al. Duck tembusu virus nonstructural protein 1 antagonizes IFN-beta signaling pathways by targeting VISA. J. Virol. 2016, 197, 4704–4713. [Google Scholar] [CrossRef]
- Neumann, G.; Hughes, M.T.; Kawaoka, Y. Influenza A virus NS2 protein mediates vRNP nuclear export through NES-independent interaction with hCRM1. EMBO J. 2000, 19, 6751–6758. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, R.E.; Talon, J.; Palese, P. The influenza virus NEP (NS2 protein) mediates the nuclear export of viral ribonucleoproteins. EMBO J. 1998, 17, 288–296. [Google Scholar] [CrossRef]
- Brunotte, L.; Flies, J.; Bolte, H.; Reuther, P.; Vreede, F.; Schwemmle, M. The nuclear export protein of H5N1 influenza A viruses recruits Matrix 1 (M1) protein to the viral ribonucleoprotein to mediate nuclear export. J. Biol. Chem. 2014, 289, 20067–20077. [Google Scholar] [CrossRef]
- Ning, S.; Pagano, J.S.; Barber, G.N. IRF7: Activation, regulation, modification and function. Genes Immun. 2011, 12, 399–414. [Google Scholar] [CrossRef]
- Akarsu, H.; Burmeister, W.P.; Petosa, C.; Petit, I.; Muller, C.W.; Ruigrok, R.W.; Baudin, F. Crystal structure of the M1 protein-binding domain of the influenza A virus nuclear export protein (NEP/NS2). EMBO J. 2003, 22, 4646–4655. [Google Scholar] [CrossRef]
- Shimizu, T.; Takizawa, N.; Watanabe, K.; Nagata, K.; Kobayashi, N. Crucial role of the influenza virus NS2 (NEP) C-terminal domain in M1 binding and nuclear export of vRNP. FEBS Lett. 2011, 585, 41–46. [Google Scholar] [CrossRef]
- Manz, B.; Brunotte, L.; Reuther, P.; Schwemmle, M. Adaptive mutations in NEP compensate for defective H5N1 RNA replication in cultured human cells. Nat. Commun. 2012, 3, 802. [Google Scholar] [CrossRef] [PubMed]
- Janeway, C.A., Jr.; Medzhitov, R. Innate immune recognition. Annu. Rev. Immunol. 2002, 20, 197–216. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Ketkar, H.; Geng, T.; Lo, E.; Wang, L.; Xi, J.; Sun, Q.; Zhu, Z.; Cui, Y.; Yang, L.; et al. Zika virus non-structural protein 4A blocks the RLR-MAVS signaling. Front. Microbiol. 2018, 9, 1350. [Google Scholar] [CrossRef] [PubMed]
- Qian, S.; Fan, W.; Liu, T.; Wu, M.; Zhang, H.; Cui, X.; Zhou, Y.; Hu, J.; Wei, S.; Chen, H.; et al. Seneca valley virus suppresses host type I interferon production by targeting adaptor proteins MAVS, TRIF, and TANK for cleavage. J. Virol. 2017, 91, e00823-17. [Google Scholar] [CrossRef]
- Shi, C.S.; Qi, H.Y.; Boularan, C.; Huang, N.N.; Abu-Asab, M.; Shelhamer, J.H.; Kehrl, J.H. SARS-coronavirus open reading frame-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J. Virol. 2014, 193, 3080–3089. [Google Scholar] [CrossRef]
- Sun, N.; Jiang, L.; Ye, M.; Wang, Y.; Wang, G.; Wan, X.; Zhao, Y.; Wen, X.; Liang, L.; Ma, S.; et al. TRIM35 mediates protection against influenza infection by activating TRAF3 and degrading viral PB2. Protein Cell 2020, 11, 894–914. [Google Scholar] [CrossRef]
- Gack, M.U.; Albrecht, R.A.; Urano, T.; Inn, K.S.; Huang, I.C.; Carnero, E.; Farzan, M.; Inoue, S.; Jung, J.U.; Garcia-Sastre, A. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe 2009, 5, 439–449. [Google Scholar] [CrossRef]
- Graef, K.M.; Vreede, F.T.; Lau, Y.F.; McCall, A.W.; Carr, S.M.; Subbarao, K.; Fodor, E. The PB2 subunit of the influenza virus RNA polymerase affects virulence by interacting with the mitochondrial antiviral signaling protein and inhibiting expression of beta interferon. J. Virol. 2010, 84, 8433–8445. [Google Scholar] [CrossRef]
- Khaperskyy, D.A.; Schmaling, S.; Larkins-Ford, J.; McCormick, C.; Gaglia, M.M. Selective degradation of host RNA polymerase II transcripts by influenza A virus PA-X host shutoff protein. PLoS Pathog. 2016, 12, e1005427. [Google Scholar] [CrossRef]
- Wang, R.; Zhu, Y.; Ren, C.; Yang, S.; Tian, S.; Chen, H.; Jin, M.; Zhou, H. Influenza A virus protein PB1-F2 impairs innate immunity by inducing mitophagy. Autophagy 2021, 17, 496–511. [Google Scholar] [CrossRef]
- Wei, Y.; Zeng, Y.; Zhang, X.; Xu, S.; Wang, Z.; Du, Y.; Zhang, B.; Lei, C.Q.; Zhu, Q. The nucleoprotein of H7N9 influenza virus positively regulates TRAF3-mediated innate signaling and attenuates viral virulence in mice. J. Virol. 2020, 94, e01640-20. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Taniguchi, T. IRFs: Master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat. Rev. Immunol. 2006, 6, 644–658. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.W.; Kim, D.; Jung, J.U.; Lee, H.R. KSHV-encoded viral interferon regulatory factor 4 (vIRF4) interacts with IRF7 and inhibits interferon alpha production. Biochem. Biophys. Res. Commun. 2017, 486, 700–705. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Mamane, Y.; Hiscott, J. Multiple regulatory domains control IRF-7 activity in response to virus infection. J. Biol. Chem. 2000, 275, 34320–34327. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhao, J.; Ren, J.; Hall, K.H.; Moorman, J.P.; Yao, Z.Q.; Ning, S. Protein phosphatase 1 abrogates IRF7-mediated type I IFN response in antiviral immunity. Eur. J. Immunol. 2016, 46, 2409–2419. [Google Scholar] [CrossRef]
- Lin, R.; Noyce, R.S.; Collins, S.E.; Everett, R.D.; Mossman, K.L. The herpes simplex virus ICP0 RING finger domain inhibits IRF3- and IRF7-mediated activation of interferon-stimulated genes. J. Virol. 2004, 78, 1675–1684. [Google Scholar] [CrossRef]
- Wu, L.; Fossum, E.; Joo, C.H.; Inn, K.S.; Shin, Y.C.; Johannsen, E.; Hutt-Fletcher, L.M.; Hass, J.; Jung, J.U. Epstein-Barr virus LF2: An antagonist to type I interferon. J. Virol. 2009, 83, 1140–1146. [Google Scholar] [CrossRef]
- Zhu, F.X.; King, S.M.; Smith, E.J.; Levy, D.E.; Yuan, Y. A Kaposi’s sarcoma-associated herpesviral protein inhibits virus-mediated induction of type I interferon by blocking IRF-7 phosphorylation and nuclear accumulation. Proc. Natl. Acad. Sci. USA 2002, 99, 5573–5578. [Google Scholar] [CrossRef]
- Chen, S.; Yang, C.; Zhang, W.; Mahalingam, S.; Wang, M.; Cheng, A. Flaviviridae virus nonstructural proteins 5 and 5A mediate viral immune evasion and are promising targets in drug development. Pharmacol. Ther. 2018, 190, 1–14. [Google Scholar] [CrossRef]
- Chowdhury, J.B.; Kim, H.; Ray, R.; Ray, R.B. Hepatitis C virus NS5A protein modulates IRF-7-mediated interferon-alpha signaling. J. Interferon Cytokine Res. 2014, 34, 16–21. [Google Scholar] [CrossRef]
- Raychoudhuri, A.; Shrivastava, S.; Steele, R.; Dash, S.; Kanda, T.; Ray, R.; Ray, R.B. Hepatitis C virus infection impairs IRF-7 translocation and Alpha interferon synthesis in immortalized human hepatocytes. J. Virol. 2010, 84, 10991–10998. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, B.; Liu, M.; Huang, J.; Zeng, Q.; Zhu, Q.; Xu, S.; Chen, H. H1N1 Influenza A Virus Protein NS2 Inhibits Innate Immune Response by Targeting IRF7. Viruses 2022, 14, 2411. https://doi.org/10.3390/v14112411
Zhang B, Liu M, Huang J, Zeng Q, Zhu Q, Xu S, Chen H. H1N1 Influenza A Virus Protein NS2 Inhibits Innate Immune Response by Targeting IRF7. Viruses. 2022; 14(11):2411. https://doi.org/10.3390/v14112411
Chicago/Turabian StyleZhang, Bo, Minxuan Liu, Jiaxin Huang, Qiaoying Zeng, Qiyun Zhu, Shuai Xu, and Hualan Chen. 2022. "H1N1 Influenza A Virus Protein NS2 Inhibits Innate Immune Response by Targeting IRF7" Viruses 14, no. 11: 2411. https://doi.org/10.3390/v14112411
APA StyleZhang, B., Liu, M., Huang, J., Zeng, Q., Zhu, Q., Xu, S., & Chen, H. (2022). H1N1 Influenza A Virus Protein NS2 Inhibits Innate Immune Response by Targeting IRF7. Viruses, 14(11), 2411. https://doi.org/10.3390/v14112411