
Detailed Molecular Interactions between Respiratory Syncytial Virus Fusion Protein and the TLR4/MD-2 Complex In Silico

 , , , ,
, , , , 

Abstract
1. Introduction
2. Materials and Methods
2.1. Protein Preparation
2.2. Protein–Protein Docking and Optimal Docking Model Selection
2.3. Calculation of the Binding Affinity
ICspolar/polar − 0.22671 ICspolar/apolar + 0.18681 %NISapolar + 0.13810 %NIScharged − 15.9433
2.4. Validation of the Present Docking Simulation
3. Results
3.1. Determination of Suitable Structures among the Candidates
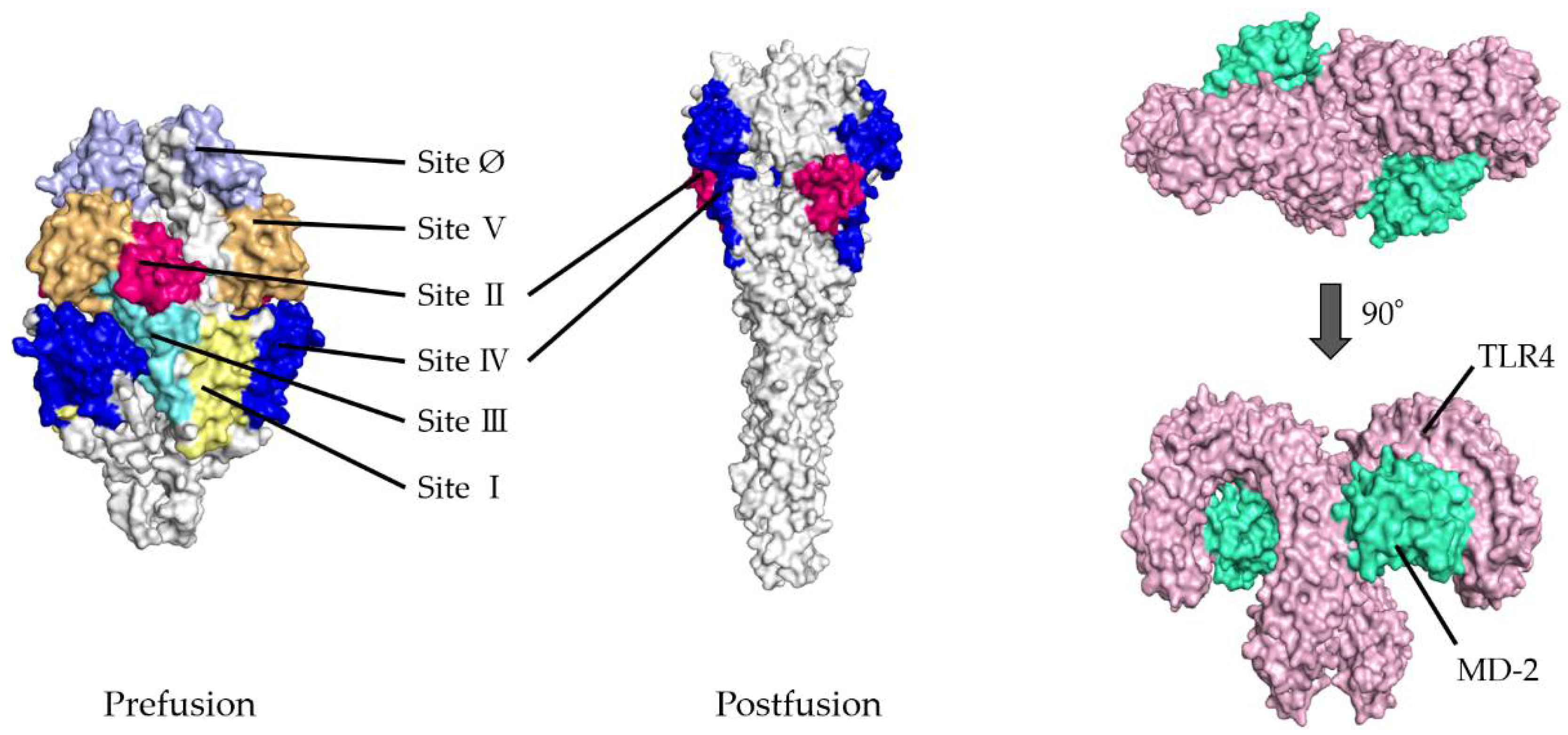
- To validate the docking simulation, the docking models generated by HDOCK were rescored using HawkDock and PPI-Affinity. As shown in Table 1(a,b), the top five ranked docking models for the prefusion protein were all models in which sites II and IV were designated as the binding site. Thus, the best-scored models in sites II and IV were determined as the optimal models in the present docking simulation, respectively. The rank of the selected model in site II was first, twenty-seventh, and nineth, and in site IV, second, eighth, and tenth, based on the HDOCK, HawkDock, and PPI-Affinity scores, respectively, in 120 docking models. Similarly, in postfusion proteins, the optimal models were ranked and selected from among the top 20 docking models based on the HDOCK, HawkDock, and PPI-Affinity scores-site II: first, third, and third, respectively; site IV: first, third, and third, respectively. To understand the 3D structures of the F proteins (prefusion/postfusion) and TLR4/MD-2 complex easily, the natural structures are illustrated in Figure 1.
3.2. Molecular Interactions between Prefusion Proteins and TLR4/MD-2
3.3. Molecular Interactions between Postfusion Proteins and TLR4/MD-2
3.4. Molecular Docking between TLR4/MD-2 and LPS, and RSV Prefusion Protein and Antibody CR9501
3.5. Comparison of Interacting Sites between the Present Docking Models and Prediction by ScanNet
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Peter, L.; Collins, R.A.K. Respiratory syncytial virus and metapneumovirus. In Fields Virology, 6th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 1086–1123. [Google Scholar]
- Leung, A.K.; Kellner, J.D.; Davies, H.D. Respiratory syncytial virus bronchiolitis. J. Natl. Med. Assoc. 2005, 97, 1708–1713. [Google Scholar] [PubMed]
- Yorita, K.L.; Holman, R.C.; Steiner, C.A.; Effler, P.V.; Miyamura, J.; Forbes, S.; Anderson, L.J.; Balaraman, V. Severe bronchiolitis and respiratory syncytial virus among young children in Hawaii. Pediatr. Infect. Dis. J. 2007, 26, 1081–1088. [Google Scholar] [CrossRef] [PubMed]
- Falsey, A.R.; Hennessey, P.A.; Formica, M.A.; Cox, C.; Walsh, E.E. Respiratory syncytial virus infection in elderly and high-risk adults. N. Engl. J. Med. 2005, 352, 1749–1759. [Google Scholar] [CrossRef] [PubMed]
- Weiss, R.A.; McMichael, A.J. Social and environmental risk factors in the emergence of infectious diseases. Nat. Med. 2004, 10 (Suppl. 12), S70–S76. [Google Scholar] [CrossRef]
- Lee, N.; Lui, G.C.; Wong, K.T.; Li, T.C.; Tse, E.C.; Chan, J.Y.; Yu, J.; Wong, S.S.; Choi, K.W.; Wong, R.Y.; et al. High morbidity and mortality in adults hospitalized for respiratory syncytial virus infections. Clin. Infect. Dis. 2013, 57, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Matias, G.; Taylor, R.; Haguinet, F.; Schuck-Paim, C.; Lustig, R.; Shinde, V. Estimates of mortality attributable to influenza and RSV in the United States during 1997–2009 by influenza type or subtype, age, cause of death, and risk status. Influenza Other Respir. Viruses 2014, 8, 507–515. [Google Scholar] [CrossRef]
- Roberts, A.R.G.D.P. Paramyxoviridae: The viruses and their replication. In Fields Virology, 6th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 957–995. [Google Scholar]
- Collins, P.L.; Fearns, R.; Graham, B.S. Respiratory syncytial virus: Virology, reverse genetics, and pathogenesis of disease. Curr. Top. Microbiol. Immunol. 2013, 372, 3–38. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.A.; Settembre, E.C.; Shaw, C.A.; Dey, A.K.; Rappuoli, R.; Mandl, C.W.; Dormitzer, P.R.; Carfi, A. Structural basis for immunization with postfusion respiratory syncytial virus fusion F glycoprotein (RSV F) to elicit high neutralizing antibody titers. Proc. Natl. Acad. Sci. USA 2011, 108, 9619–9624. [Google Scholar] [CrossRef] [PubMed]
- McLellan, J.S.; Chen, M.; Leung, S.; Graepel, K.W.; Du, X.; Yang, Y.; Zhou, T.; Baxa, U.; Yasuda, E.; Beaumont, T.; et al. Structure of RSV fusion glycoprotein trimer bound to a prefusion-specific neutralizing antibody. Science 2013, 340, 1113–1117. [Google Scholar] [CrossRef] [PubMed]
- Killikelly, A.M.; Kanekiyo, M.; Graham, B.S. Pre-fusion F is absent on the surface of formalin-inactivated respiratory syncytial virus. Sci. Rep. 2016, 6, 34108. [Google Scholar] [CrossRef] [PubMed]
- Beeler, J.A.; van Wyke Coelingh, K. Neutralization epitopes of the F glycoprotein of respiratory syncytial virus: Effect of mutation upon fusion function. J. Virol. 1989, 63, 2941–2950. [Google Scholar] [CrossRef]
- Gilman, M.S.; Castellanos, C.A.; Chen, M.; Ngwuta, J.O.; Goodwin, E.; Moin, S.M.; Mas, V.; Melero, J.A.; Wright, P.F.; Graham, B.S.; et al. Rapid profiling of RSV antibody repertoires from the memory B cells of naturally infected adult donors. Sci. Immunol. 2016, 1, eaaj1879. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Hosoya, M. Neutralizing epitopes of RSV and palivizumab resistance in Japan. Fukushima J. Med. Sci. 2017, 63, 127–134. [Google Scholar] [CrossRef]
- Oda, K.; Kitano, H. A comprehensive map of the toll-like receptor signaling network. Mol. Syst. Biol. 2006, 2, 2006-0015. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, K.; Takeuchi, O.; Kawai, T.; Sanjo, H.; Ogawa, T.; Takeda, Y.; Takeda, K.; Akira, S. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: Evidence for TLR4 as the Lps gene product. J. Immunol. 1999, 162, 3749–3752. [Google Scholar]
- Kurt-Jones, E.A.; Popova, L.; Kwinn, L.; Haynes, L.M.; Jones, L.P.; Tripp, R.A.; Walsh, E.E.; Freeman, M.W.; Golenbock, D.T.; Anderson, L.J.; et al. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat. Immunol. 2000, 1, 398–401. [Google Scholar] [CrossRef] [PubMed]
- Viriyakosol, S.; Tobias, P.S.; Kitchens, R.L.; Kirkland, T.N. MD-2 binds to bacterial lipopolysaccharide. J. Biol. Chem. 2001, 276, 38044–38051. [Google Scholar] [CrossRef] [PubMed]
- Shimazu, R.; Akashi, S.; Ogata, H.; Nagai, Y.; Fukudome, K.; Miyake, K.; Kimoto, M. MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J. Exp. Med. 1999, 189, 1777–1782. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Adachi, O.; Ogawa, T.; Takeda, K.; Akira, S. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity 1999, 11, 115–122. [Google Scholar] [CrossRef]
- Jiang, Z.; Georgel, P.; Du, X.; Shamel, L.; Sovath, S.; Mudd, S.; Huber, M.; Kalis, C.; Keck, S.; Galanos, C.; et al. CD14 is required for MyD88-independent LPS signaling. Nat. Immunol. 2005, 6, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Tabor, D.E.; Fernandes, F.; Langedijk, A.C.; Wilkins, D.; Lebbink, R.J.; Tovchigrechko, A.; Ruzin, A.; Kragten-Tabatabaie, L.; Jin, H.; Esser, M.T.; et al. Global Molecular Epidemiology of Respiratory Syncytial Virus from the 2017–2018 INFORM-RSV Study. J. Clin. Microbiol. 2020, 59, e01828-20. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Zhang, D.; Zhou, P.; Li, B.; Huang, S.Y. HDOCK: A web server for protein-protein and protein-DNA/RNA docking based on a hybrid strategy. Nucleic Acids Res. 2017, 45, W365–W373. [Google Scholar] [CrossRef] [PubMed]
- Weng, G.; Wang, E.; Wang, Z.; Liu, H.; Zhu, F.; Li, D.; Hou, T. HawkDock: A web server to predict and analyze the protein-protein complex based on computational docking and MM/GBSA. Nucleic Acids Res. 2019, 47, W322–W330. [Google Scholar] [CrossRef] [PubMed]
- Romero-Molina, S.; Ruiz-Blanco, Y.B.; Mieres-Perez, J.; Harms, M.; Munch, J.; Ehrmann, M.; Sanchez-Garcia, E. PPI-Affinity: A Web Tool for the Prediction and Optimization of Protein-Peptide and Protein-Protein Binding Affinity. J. Proteome Res. 2022, 21, 1829–1841. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Jablonska, J.; Pravda, L.; Varekova, R.S.; Thornton, J.M. PDBsum: Structural summaries of PDB entries. Protein Sci. 2018, 27, 129–134. [Google Scholar] [CrossRef]
- Vangone, A.; Bonvin, A.M. Contacts-based prediction of binding affinity in protein-protein complexes. eLife 2015, 4, e07454. [Google Scholar] [CrossRef]
- Kastritis, P.L.; Rodrigues, J.P.; Folkers, G.E.; Boelens, R.; Bonvin, A.M. Proteins feel more than they see: Fine-tuning of binding affinity by properties of the non-interacting surface. J. Mol. Biol. 2014, 426, 2632–2652. [Google Scholar] [CrossRef]
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef]
- Tubiana, J.; Schneidman-Duhovny, D.; Wolfson, H.J. ScanNet: An interpretable geometric deep learning model for structure-based protein binding site prediction. Nat. Methods 2022, 19, 730–739. [Google Scholar] [CrossRef]
- Battles, M.B.; McLellan, J.S. Respiratory syncytial virus entry and how to block it. Nat. Rev. Microbiol. 2019, 17, 233–245. [Google Scholar] [CrossRef]
- Park, B.S.; Song, D.H.; Kim, H.M.; Choi, B.S.; Lee, H.; Lee, J.O. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature 2009, 458, 1191–1195. [Google Scholar] [CrossRef] [PubMed]
- Alberts, B. Molecular Biology of the Cell, 5th ed.; Garland Science: New York City, NY, USA, 2008; pp. 617–628. [Google Scholar]
- Hernandez, J.M.; Podbilewicz, B. The hallmarks of cell-cell fusion. Development 2017, 144, 4481–4495. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.Y.; Zhang, H.X.; Mezei, M.; Cui, M. Molecular docking: A powerful approach for structure-based drug discovery. Curr. Comput. Aided Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Graves, A.P.; Shivakumar, D.M.; Boyce, S.E.; Jacobson, M.P.; Case, D.A.; Shoichet, B.K. Rescoring docking hit lists for model cavity sites: Predictions and experimental testing. J. Mol. Biol. 2008, 377, 914–934. [Google Scholar] [CrossRef]
- Li, J.; Fu, A.; Zhang, L. An Overview of Scoring Functions Used for Protein-Ligand Interactions in Molecular Docking. Interdiscip. Sci. 2019, 11, 320–328. [Google Scholar] [CrossRef]
- Huang, S.Y.; Zou, X. An iterative knowledge-based scoring function for protein-protein recognition. Proteins 2008, 72, 557–579. [Google Scholar] [CrossRef]
- Gilman, M.S.A.; Furmanova-Hollenstein, P.; Pascual, G.; van ‘t Wout, A.B.; Langedijk, J.P.M.; McLellan, J.S. Transient opening of trimeric prefusion RSV F proteins. Nat. Commun. 2019, 10, 2105. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (a) | ||||
| Ranking | HDOCK | HawkDock | PPI-Affinity | Antigenic Sites |
| 1st | 2 | 8 | 10 | Site IV |
| 2nd | 12 | 15 | 7 | |
| 3rd | 1 | 27 | 9 | Site II |
| 3rd | 15 | 12 | 10 | |
| 5th | 25 | 3 | 11 | |
| (b) | ||||
| Ranking | HDOCK | HawkDock | PPI-Affinity | Antigenic Sites |
| 1st | 1 | 3 | 3 | Site II |
| 2nd | 2 | 4 | 2 | |
| 3rd | 5 | 2 | 3 | |
| 4th | 4 | 6 | 4 | |
| 4th | 6 | 7 | 1 | |
| 1st | 1 | 3 | 3 | Site IV |
| 2nd | 6 | 5 | 1 | |
| 3rd | 2 | 9 | 2 | |
| 4th | 5 | 7 | 3 | |
| 5th | 11 | 2 | 3 | |
| Interaction Type | Fusion Protein | TLR4 | Distance (Å) |
|---|---|---|---|
| Hydrogen bonds | Lys272 | Lys541 | 2.81 |
| Asn276 | Arg496 | 2.80 | |
| Asn276 | Asn517 | 3.06 | |
| Asn276 | Ser520 | 2.98 |
| Fusion Protein | Prefusion | Postfusion | ||
|---|---|---|---|---|
| Antigenic Sites | Site II | Site IV | Site II | Site IV |
| ICs charged/charged (no.) | 14 | 7 | 1 | 14 |
| ICs charged/apolar (no.) | 28 | 29 | 14 | 40 |
| ICs polar/polar (no.) | 33 | 13 | 14 | 21 |
| ICs polar/apolar (no.) | 20 | 25 | 46 | 30 |
| %NIS apolar (%) | 34.42 | 33.91 | 34.48 | 34.38 |
| %NIS charged (%) | 24.39 | 24.68 | 23.69 | 23.52 |
| Binding Affinity (kcal/mol) | −8.3 | −12.9 | −15.4 | −14.3 |
| Interaction Type | Fusion Protein | TLR4 | Distance (Å) |
|---|---|---|---|
| Hydrogen bonds | Lys465 | Pro78 | 3.13 |
| Lys465 | Glu79 | 2.51 | |
| Salt bridges | Lys465 | Glu79 | 2.51 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akagawa, M.; Shirai, T.; Sada, M.; Nagasawa, N.; Kondo, M.; Takeda, M.; Nagasawa, K.; Kimura, R.; Okayama, K.; Hayashi, Y.; et al. Detailed Molecular Interactions between Respiratory Syncytial Virus Fusion Protein and the TLR4/MD-2 Complex In Silico. Viruses 2022, 14, 2382. https://doi.org/10.3390/v14112382
Akagawa M, Shirai T, Sada M, Nagasawa N, Kondo M, Takeda M, Nagasawa K, Kimura R, Okayama K, Hayashi Y, et al. Detailed Molecular Interactions between Respiratory Syncytial Virus Fusion Protein and the TLR4/MD-2 Complex In Silico. Viruses. 2022; 14(11):2382. https://doi.org/10.3390/v14112382
Chicago/Turabian StyleAkagawa, Mao, Tatsuya Shirai, Mitsuru Sada, Norika Nagasawa, Mayumi Kondo, Makoto Takeda, Koo Nagasawa, Ryusuke Kimura, Kaori Okayama, Yuriko Hayashi, and et al. 2022. "Detailed Molecular Interactions between Respiratory Syncytial Virus Fusion Protein and the TLR4/MD-2 Complex In Silico" Viruses 14, no. 11: 2382. https://doi.org/10.3390/v14112382
APA StyleAkagawa, M., Shirai, T., Sada, M., Nagasawa, N., Kondo, M., Takeda, M., Nagasawa, K., Kimura, R., Okayama, K., Hayashi, Y., Sugai, T., Tsugawa, T., Ishii, H., Kawashima, H., Katayama, K., Ryo, A., & Kimura, H. (2022). Detailed Molecular Interactions between Respiratory Syncytial Virus Fusion Protein and the TLR4/MD-2 Complex In Silico. Viruses, 14(11), 2382. https://doi.org/10.3390/v14112382