
Site-Specific O-Glycosylation Analysis of SARS-CoV-2 Spike Protein Produced in Insect and Human Cells

 , , , , , and
, , , , , and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
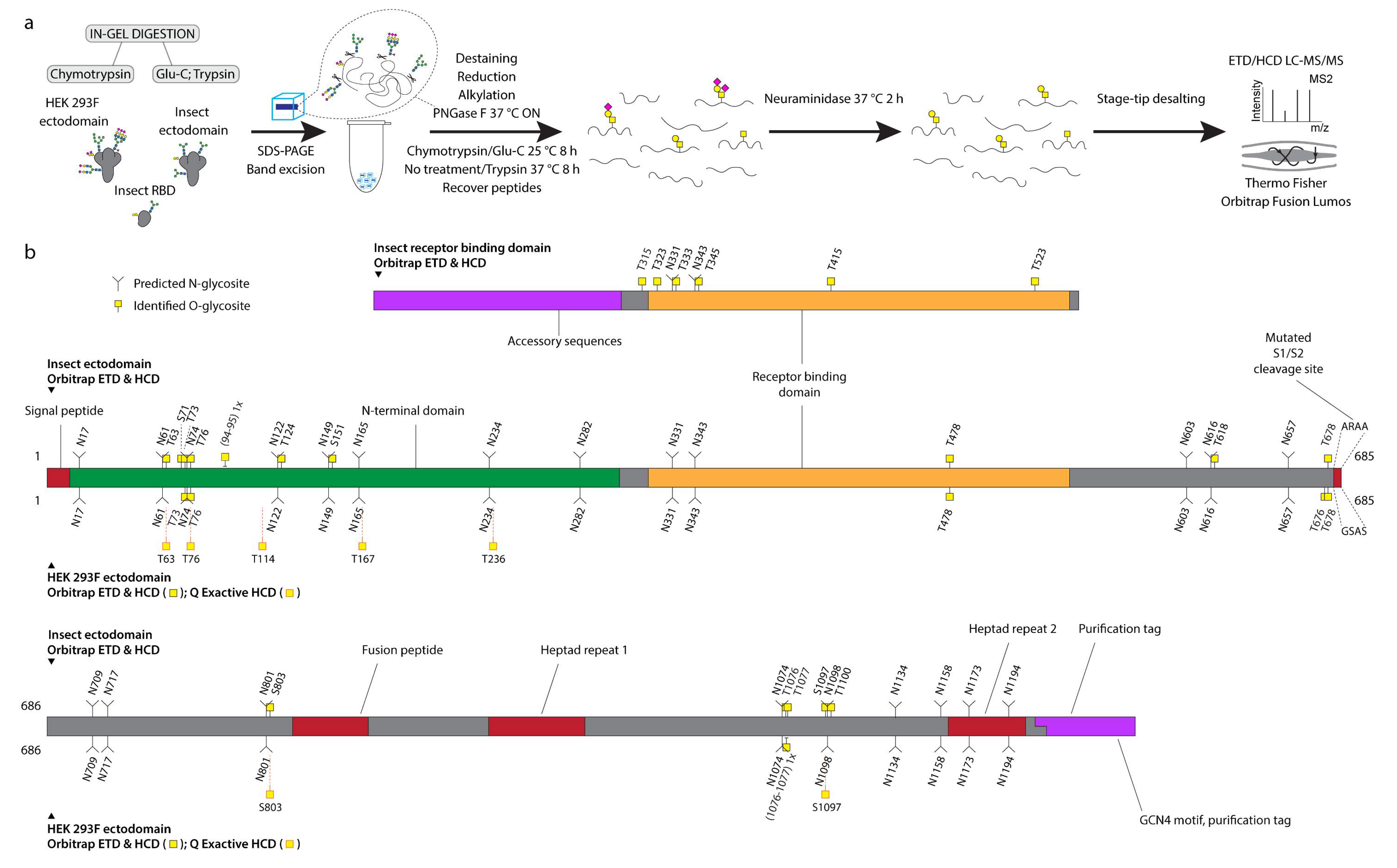
2.1. Protein Expression and Purification in S2 Cells
2.2. Protein Expression and Purification in HEK 293F Cells
2.3. In-Gel Digestion
2.4. Mass Spectrometry Analysis with Orbitrap Fusion Lumos Mass Spectrometer
2.5. In-Solution Digestion and Mass Spectrometry Analysis with Q Exactive HF-X Mass Spectrometer
2.6. Molecular Modelling
3. Results
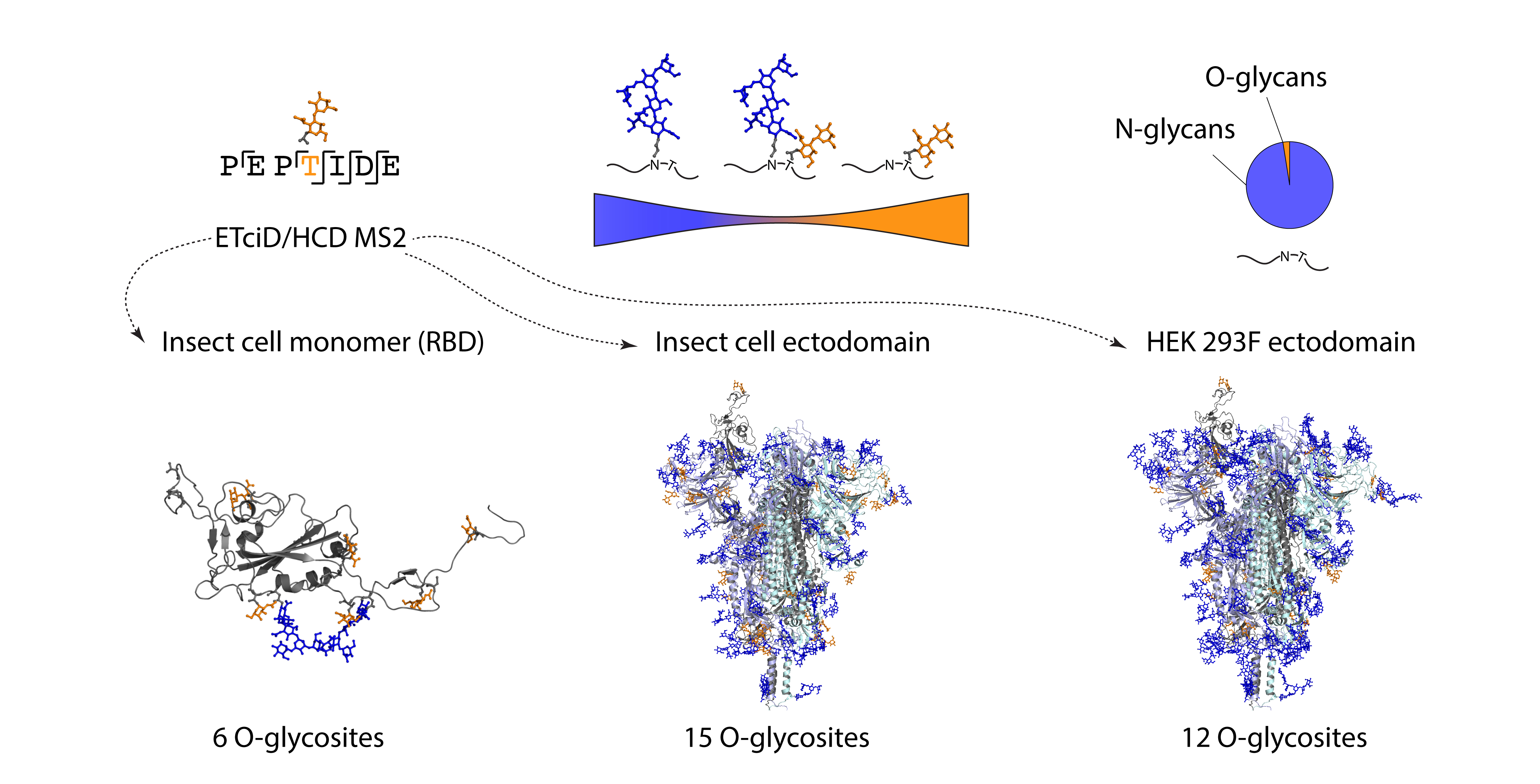
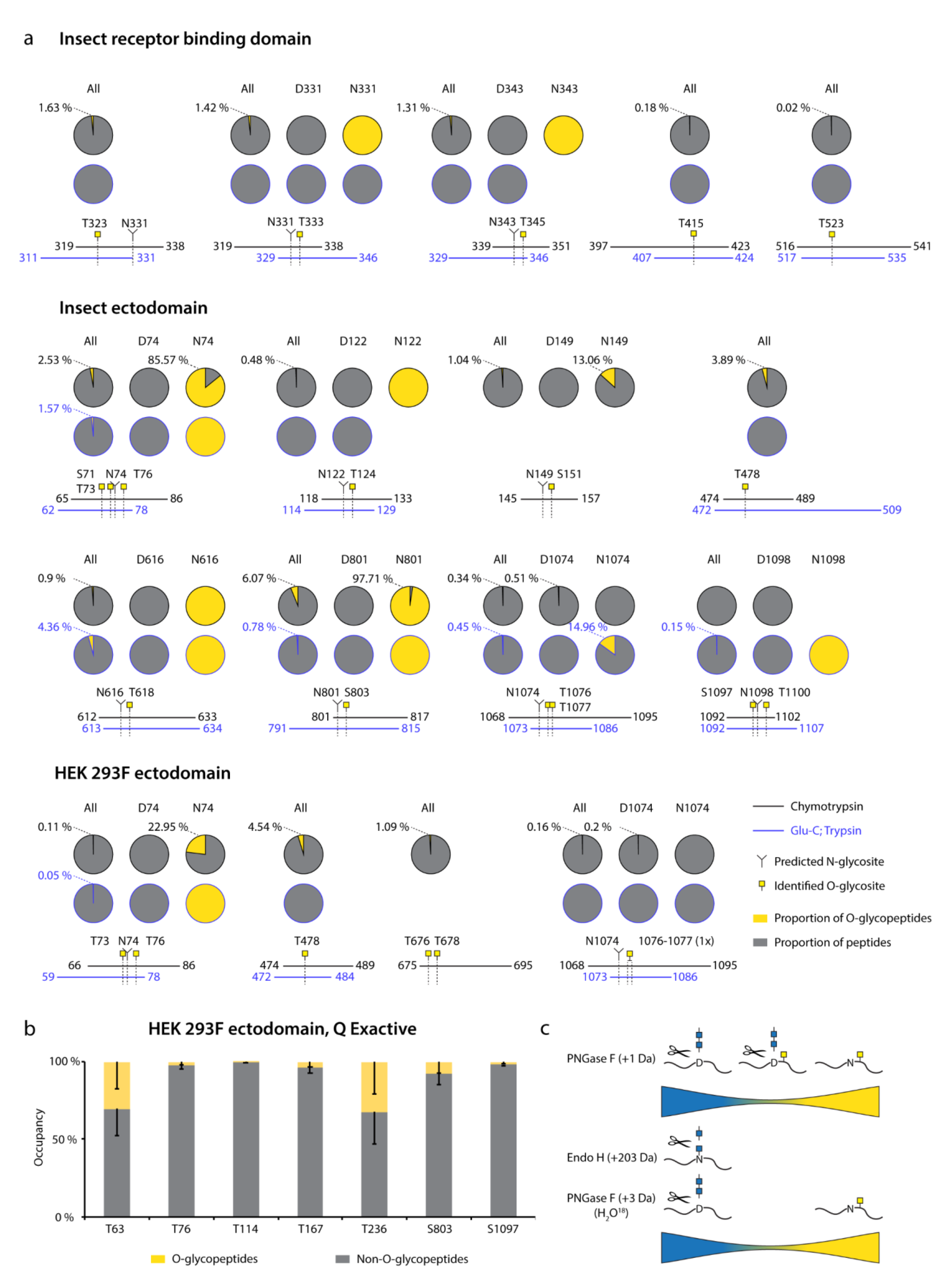
3.1. Recombinant SARS-CoV-2 Spike Proteins Are Modified with Multiple O-Glycosylation Sites
3.2. Differences in O-Glycosylation Patterns between Multimeric and Monomeric S
3.3. Majority of O-Glycosites Are Located in Vicinity of N-Glycosites
3.4. O-Glycosylation in the Context of S Protein Structural Features
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Cheng, Y.W.; Chao, T.L.; Li, C.L.; Chiu, M.F.; Kao, H.C.; Wang, S.H.; Pang, Y.H.; Lin, C.H.; Tsai, Y.M.; Lee, W.H.; et al. Furin Inhibitors Block SARS-CoV-2 Spike Protein Cleavage to Suppress Virus Production and Cytopathic Effects. Cell Rep. 2020, 33, 108254. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292. [Google Scholar] [CrossRef]
- Wrobel, A.G.; Benton, D.J.; Xu, P.; Roustan, C.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. SARS-CoV-2 and bat RaTG13 spike glycoprotein structures inform on virus evolution and furin-cleavage effects. Nat. Struct. Mol. Biol 2020, 27, 763–767. [Google Scholar] [CrossRef]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Marc, G.P.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef]
- Voysey, M.; Clemens, S.A.C.; Madhi, S.A.; Weckx, L.Y.; Folegatti, P.M.; Aley, P.K.; Angus, B.; Baillie, V.L.; Barnabas, S.L.; Bhorat, Q.E.; et al. Safety and efficacy of the ChAdOx1 nCoV-19 vaccine (AZD1222) against SARS-CoV-2: An interim analysis of four randomised controlled trials in Brazil, South Africa, and the UK. Lancet 2021, 397, 99–111. [Google Scholar] [CrossRef]
- Zhang, Y.; Zeng, G.; Pan, H.; Li, C.; Hu, Y.; Chu, K.; Han, W.; Chen, Z.; Tang, R.; Yin, W.; et al. Safety, tolerability, and immunogenicity of an inactivated SARS-CoV-2 vaccine in healthy adults aged 2021, 18-59 years: A randomised, double-blind, placebo-controlled, phase 1/2 clinical trial. Lancet Infect. Dis. 2021, 21, 181–192. [Google Scholar] [CrossRef]
- Prüβ, B.M. Current State of the First COVID-19 Vaccines. Vaccines 2021, 9, 30. [Google Scholar] [CrossRef]
- Bagdonaite, I.; Vakhrushev, S.Y.; Joshi, H.J.; Wandall, H.H. Viral glycoproteomes: Technologies for characterization and outlook for vaccine design. FEBS Lett. 2018, 592, 3898–3920. [Google Scholar] [CrossRef]
- Bagdonaite, I.; Wandall, H.H. Global aspects of viral glycosylation. Glycobiology 2018, 28, 443–467. [Google Scholar] [CrossRef]
- Bangaru, S.; Ozorowski, G.; Turner, H.L.; Antanasijevic, A.; Huang, D.; Wang, X.; Torres, J.L.; Diedrich, J.K.; Tian, J.H.; Portnoff, A.D.; et al. Structural analysis of full-length SARS-CoV-2 spike protein from an advanced vaccine candidate. Science 2020, 370, 1089–1094. [Google Scholar] [CrossRef]
- Sanda, M.; Morrison, L.; Goldman, R. N- and O-Glycosylation of the SARS-CoV-2 Spike Protein. Anal. Chem. 2021, 93, 2003–2009. [Google Scholar] [CrossRef]
- Shajahan, A.; Supekar, N.T.; Gleinich, A.S.; Azadi, P. Deducing the N- and O-glycosylation profile of the spike protein of novel coronavirus SARS-CoV-2. Glycobiology 2020, 30, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Allen, J.D.; Wrapp, D.; McLellan, J.S.; Crispin, M. Site-specific glycan analysis of the SARS-CoV-2 spike. Science 2020, 369, 330–333. [Google Scholar] [PubMed]
- Zhao, P.; Praissman, J.L.; Grant, O.C.; Cai, Y.; Xiao, T.; Rosenbalm, K.E.; Aoki, K.; Kellman, B.P.; Bridger, R.; Barouch, D.H.; et al. Virus-Receptor Interactions of Glycosylated SARS-CoV-2 Spike and Human ACE2 Receptor. Cell Host Microbe 2020, 28, 586–601. [Google Scholar] [CrossRef] [PubMed]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [PubMed]
- Bennett, E.P.; Mandel, U.; Clausen, H.; Gerken, T.A.; Fritz, T.A.; Tabak, L.A. Control of mucin-type O-glycosylation: A classification of the polypeptide GalNAc-transferase gene family. Glycobiology 2012, 22, 736–756. [Google Scholar] [CrossRef]
- Tran, D.T.; Ten Hagen, K.G. Mucin-type O-glycosylation during development. J. Biol. Chem. 2013, 288, 6921–6929. [Google Scholar] [CrossRef] [PubMed]
- Fujitani, N.; Furukawa, J.; Araki, K.; Fujioka, T.; Takegawa, Y.; Piao, J.; Nishioka, T.; Tamura, T.; Nikaido, T.; Ito, M.; et al. Total cellular glycomics allows characterizing cells and streamlining the discovery process for cellular biomarkers. Proc. Natl. Acad. Sci. USA 2013, 110, 2105–2110. [Google Scholar] [CrossRef]
- Aoki, K.; Porterfield, M.; Lee, S.S.; Dong, B.; Nguyen, K.; McGlamry, K.H.; Tiemeyer, M. The diversity of O-linked glycans expressed during Drosophila melanogaster development reflects stage- and tissue-specific requirements for cell signaling. J. Biol. Chem 2008, 283, 30385–30400. [Google Scholar] [CrossRef]
- Müller, R.; Hülsmeier, A.J.; Altmann, F.; Ten Hagen, K.; Tiemeyer, M.; Hennet, T. Characterization of mucin-type core-1 beta1-3 galactosyltransferase homologous enzymes in Drosophila melanogaster. FEBS J. 2005, 272, 4295–4305. [Google Scholar] [CrossRef] [PubMed]
- Levery, S.B.; Steentoft, C.; Halim, A.; Narimatsu, Y.; Clausen, H.; Vakhrushev, S.Y. Advances in mass spectrometry driven O-glycoproteomics. Biochim. Biophys. Acta 2015, 1850, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Riley, N.M.; Malaker, S.A.; Driessen, M.D.; Bertozzi, C.R. Optimal Dissociation Methods Differ for N- and O-Glycopeptides. J. Proteome Res. 2020, 19, 3286–3301. [Google Scholar] [CrossRef]
- Bagdonaite, I.; Norden, R.; Joshi, H.J.; Dabelsteen, S.; Nystrom, K.; Vakhrushev, S.Y.; Olofsson, S.; Wandall, H.H. A strategy for O-glycoproteomics of enveloped viruses—the O-glycoproteome of herpes simplex virus type 1. PLoS Pathog. 2015, 11, e1004784. [Google Scholar] [CrossRef] [PubMed]
- Bagdonaite, I.; Norden, R.; Joshi, H.J.; King, S.L.; Vakhrushev, S.Y.; Olofsson, S.; Wandall, H.H. Global Mapping of O-Glycosylation of Varicella Zoster Virus, Human Cytomegalovirus, and Epstein-Barr Virus. J. Biol. Chem. 2016, 291, 12014–12028. [Google Scholar] [CrossRef]
- Steentoft, C.; Vakhrushev, S.Y.; Joshi, H.J.; Kong, Y.; Vester-Christensen, M.B.; Schjoldager, K.T.; Lavrsen, K.; Dabelsteen, S.; Pedersen, N.B.; Marcos-Silva, L.; et al. Precision mapping of the human O-GalNAc glycoproteome through SimpleCell technology. EMBO J. 2013, 32, 1478–1488. [Google Scholar] [CrossRef]
- Steentoft, C.; Vakhrushev, S.Y.; Vester-Christensen, M.B.; Schjoldager, K.T.; Kong, Y.; Bennett, E.P.; Mandel, U.; Wandall, H.; Levery, S.B.; Clausen, H. Mining the O-glycoproteome using zinc-finger nuclease-glycoengineered SimpleCell lines. Nat. Methods 2011, 8, 977–982. [Google Scholar] [CrossRef] [PubMed]
- Vakhrushev, S.Y.; Steentoft, C.; Vester-Christensen, M.B.; Bennett, E.P.; Clausen, H.; Levery, S.B. Enhanced mass spectrometric mapping of the human GalNAc-type O-glycoproteome with SimpleCells. Mol. Cell Proteom. 2013, 12, 932–944. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Pauthner, M.; Andrabi, R.; Rantalainen, K.; Berndsen, Z.; Diedrich, J.K.; Menis, S.; Sok, D.; Bastidas, R.; Park, S.R.; et al. Differential processing of HIV envelope glycans on the virus and soluble recombinant trimer. Nat. Commun. 2018, 9, 3693. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.; Park, S.J.; Choi, Y.K.; Park, T.; Tanveer, M.; Cao, Y.; Kern, N.R.; Lee, J.; Yeom, M.S.; Croll, T.I.; et al. Developing a Fully Glycosylated Full-Length SARS-CoV-2 Spike Protein Model in a Viral Membrane. J. Phys. Chem. B 2020, 124, 7128–7137. [Google Scholar] [CrossRef] [PubMed]
- Frishman, D.; Argos, P. Knowledge-based protein secondary structure assignment. Proteins 1995, 23, 566–579. [Google Scholar] [CrossRef]
- Krieger, E.; Vriend, G. YASARA View—Molecular graphics for all devices—From smartphones to workstations. Bioinformatics 2014, 30, 2981–2982. [Google Scholar] [CrossRef] [PubMed]
- Wilson, I.B. Glycosylation of proteins in plants and invertebrates. Curr. Opin. Struct. Biol. 2002, 12, 569–577. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bagdonaite, I.; Thompson, A.J.; Wang, X.; Søgaard, M.; Fougeroux, C.; Frank, M.; Diedrich, J.K.; Yates, J.R., III; Salanti, A.; Vakhrushev, S.Y.; et al. Site-Specific O-Glycosylation Analysis of SARS-CoV-2 Spike Protein Produced in Insect and Human Cells. Viruses 2021, 13, 551. https://doi.org/10.3390/v13040551
Bagdonaite I, Thompson AJ, Wang X, Søgaard M, Fougeroux C, Frank M, Diedrich JK, Yates JR III, Salanti A, Vakhrushev SY, et al. Site-Specific O-Glycosylation Analysis of SARS-CoV-2 Spike Protein Produced in Insect and Human Cells. Viruses. 2021; 13(4):551. https://doi.org/10.3390/v13040551
Chicago/Turabian StyleBagdonaite, Ieva, Andrew J. Thompson, Xiaoning Wang, Max Søgaard, Cyrielle Fougeroux, Martin Frank, Jolene K. Diedrich, John R. Yates, III, Ali Salanti, Sergey Y. Vakhrushev, and et al. 2021. "Site-Specific O-Glycosylation Analysis of SARS-CoV-2 Spike Protein Produced in Insect and Human Cells" Viruses 13, no. 4: 551. https://doi.org/10.3390/v13040551
APA StyleBagdonaite, I., Thompson, A. J., Wang, X., Søgaard, M., Fougeroux, C., Frank, M., Diedrich, J. K., Yates, J. R., III, Salanti, A., Vakhrushev, S. Y., Paulson, J. C., & Wandall, H. H. (2021). Site-Specific O-Glycosylation Analysis of SARS-CoV-2 Spike Protein Produced in Insect and Human Cells. Viruses, 13(4), 551. https://doi.org/10.3390/v13040551