
A Novel In Vitro Culture Model System to Study Merkel Cell Polyomavirus–Associated MCC Using Three-Dimensional Organotypic Raft Equivalents of Human Skin



Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture Conditions
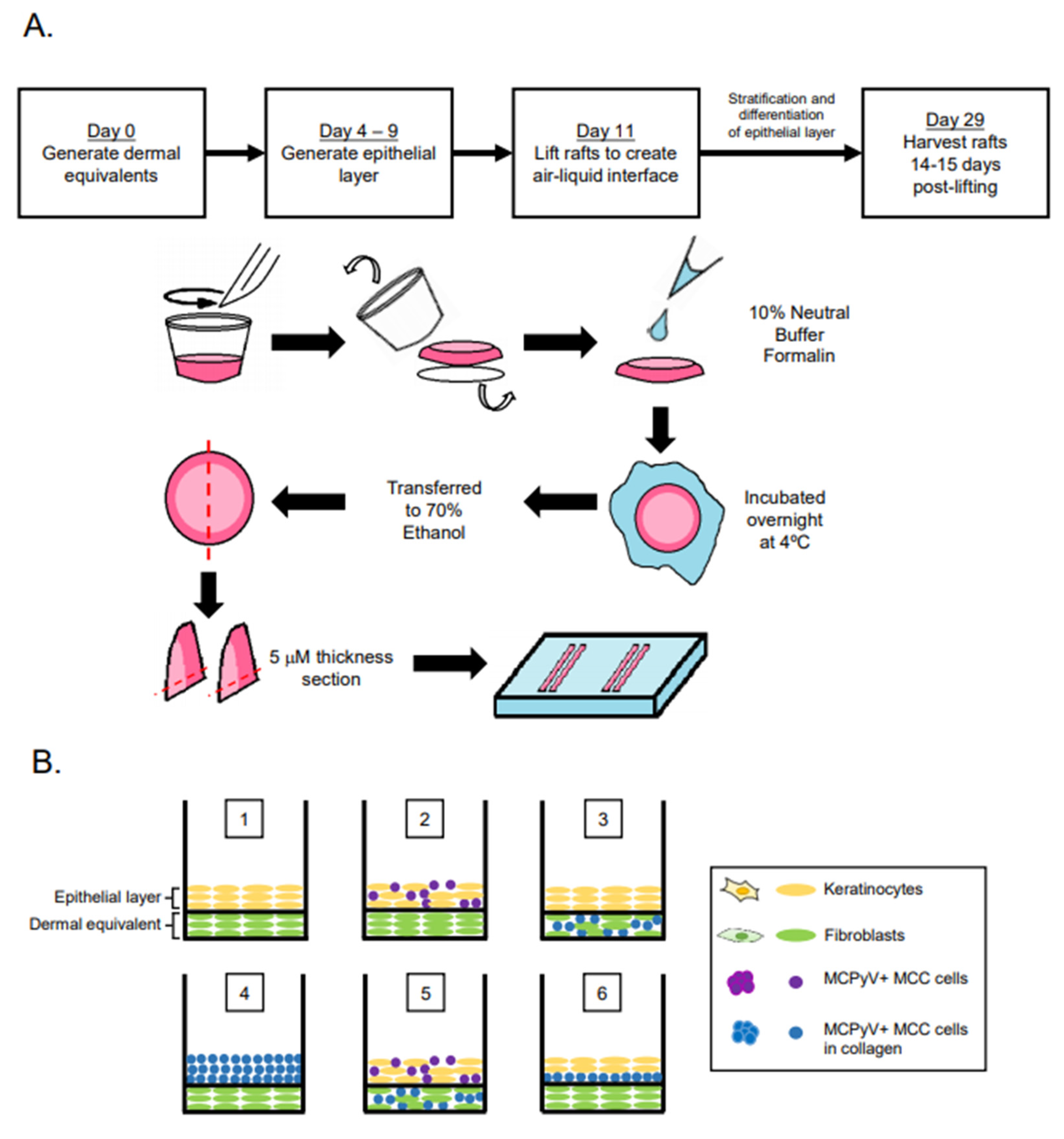
2.2. Organotypic Raft Culture
2.3. Immunofluorescence and Immunohistochemistry
2.4. Antibodies and Reagents
2.5. Immunoblotting
3. Results
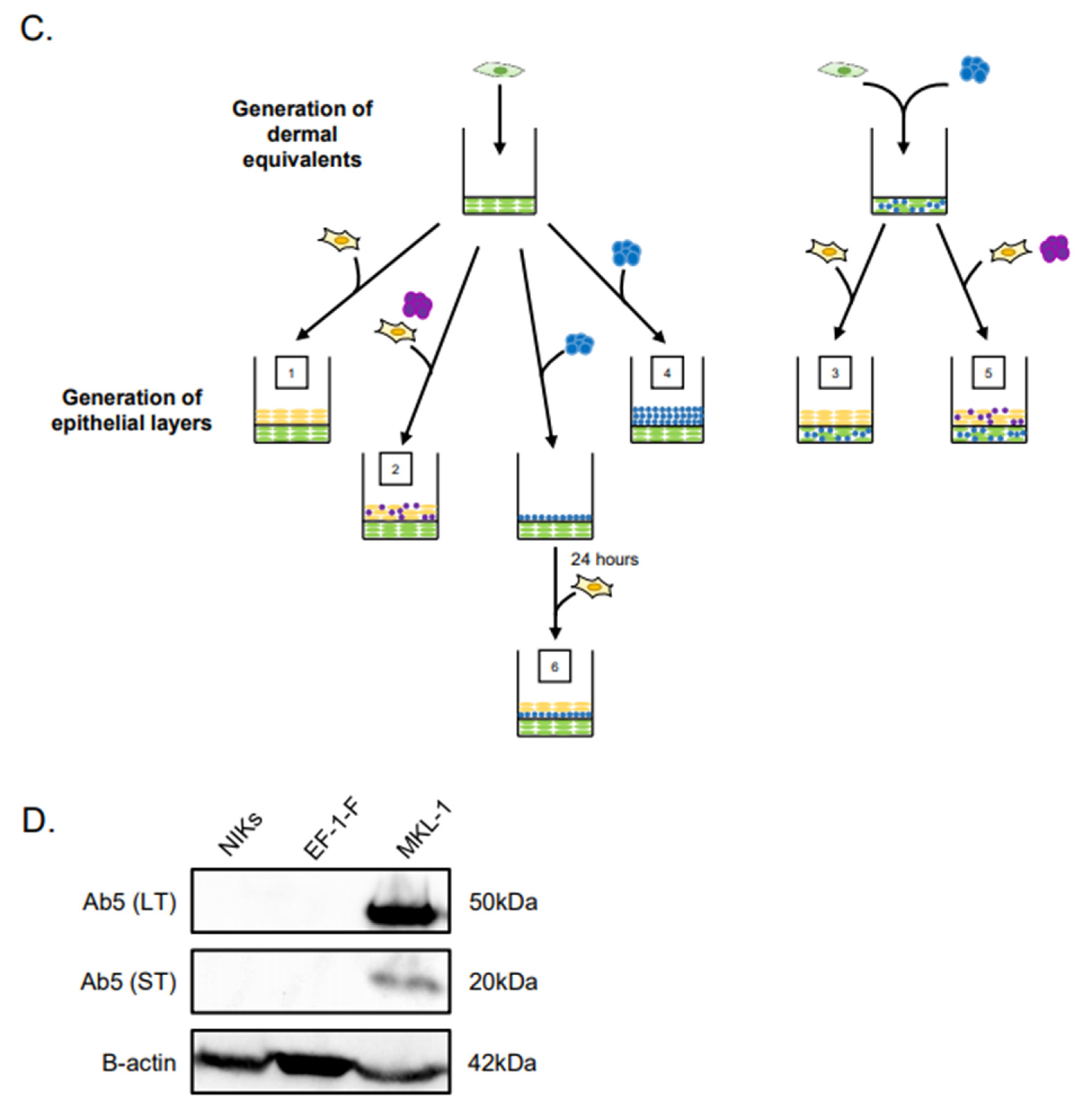
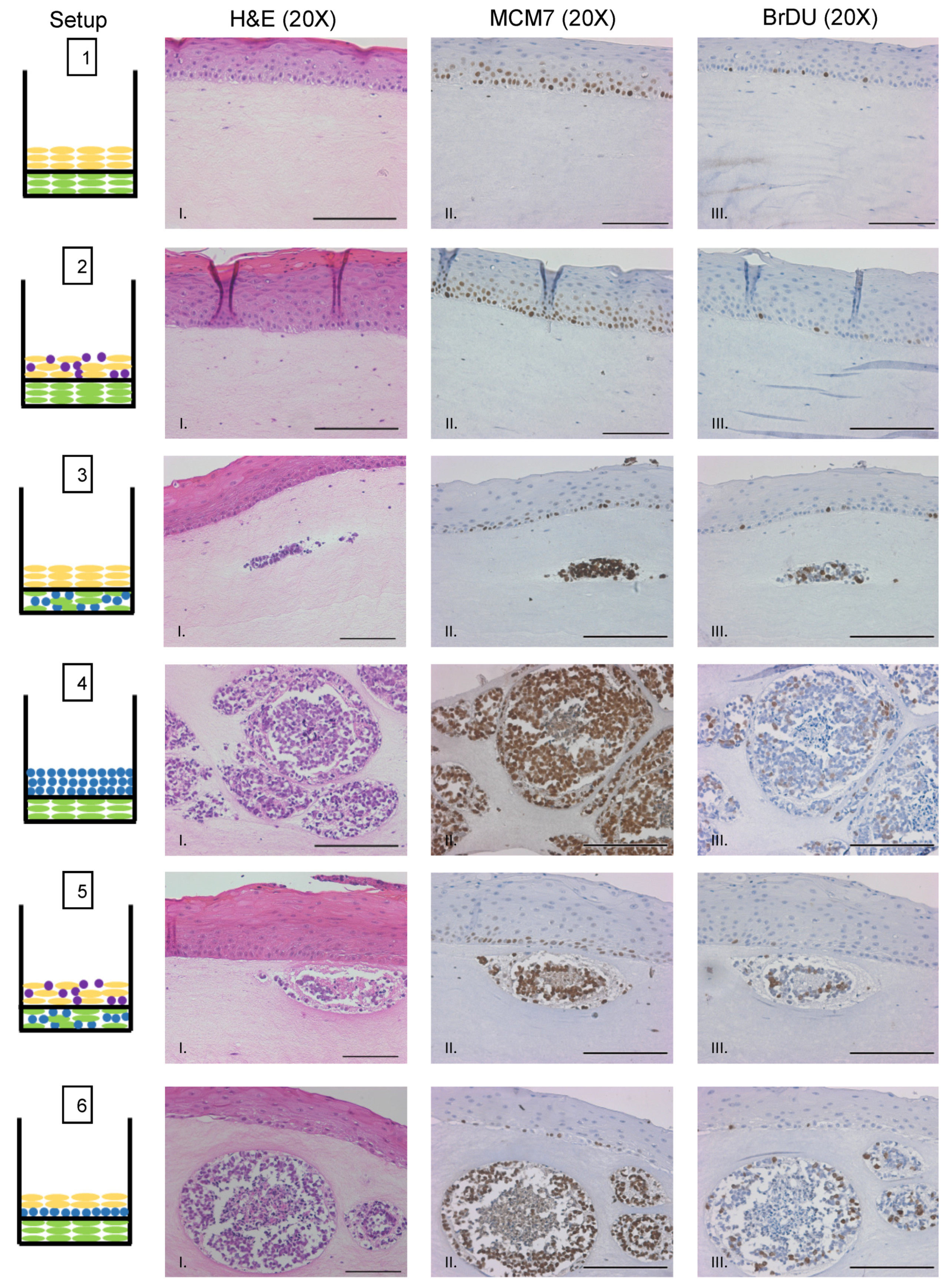
3.1. Experimental Design of MCC Culture in Organotypic Rafts
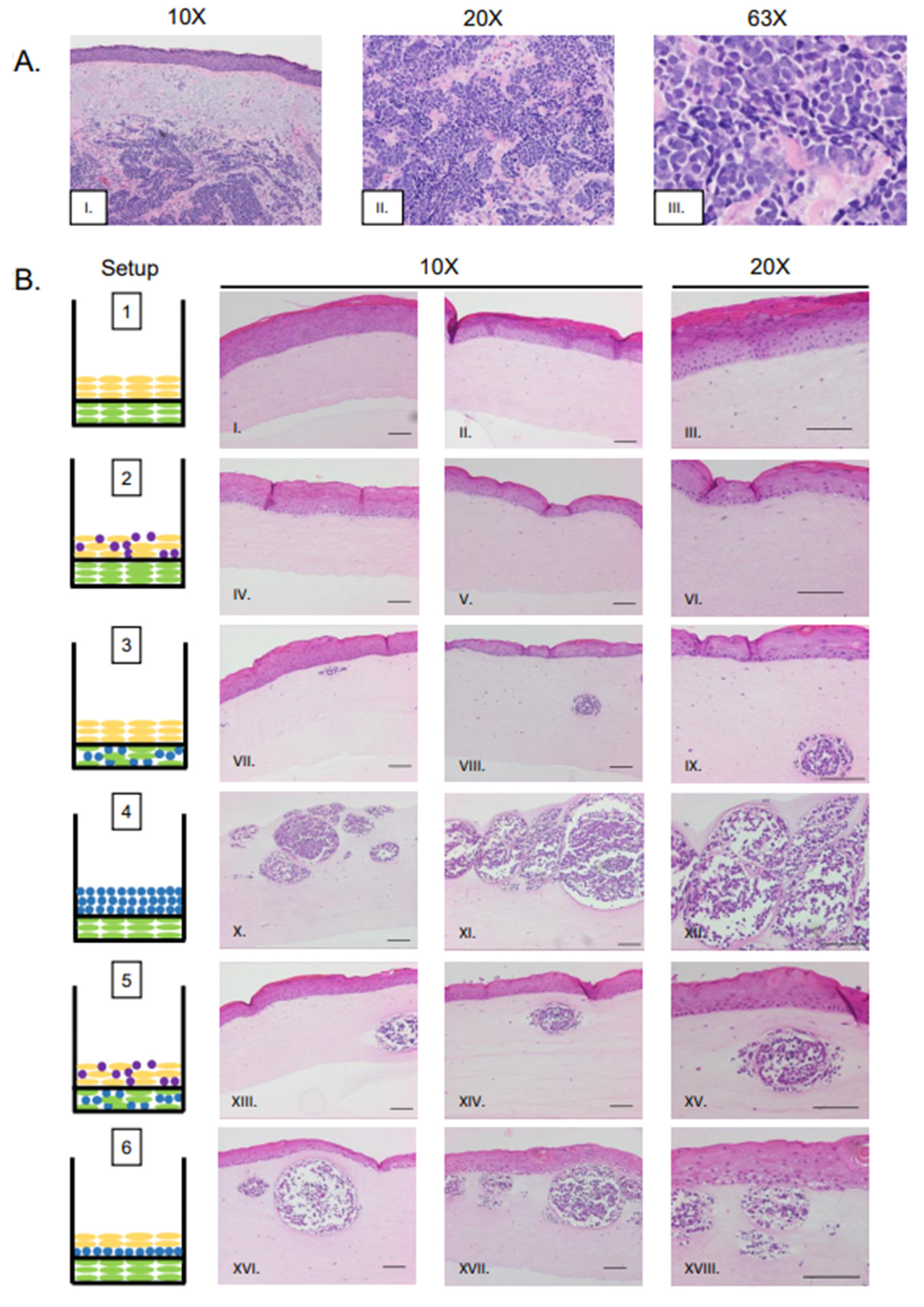
3.2. MCPyV+ MCC-Like Lesions Formed in Dermal Layer of Organotypic Raft Cultures but Not in the Epithelial Layer
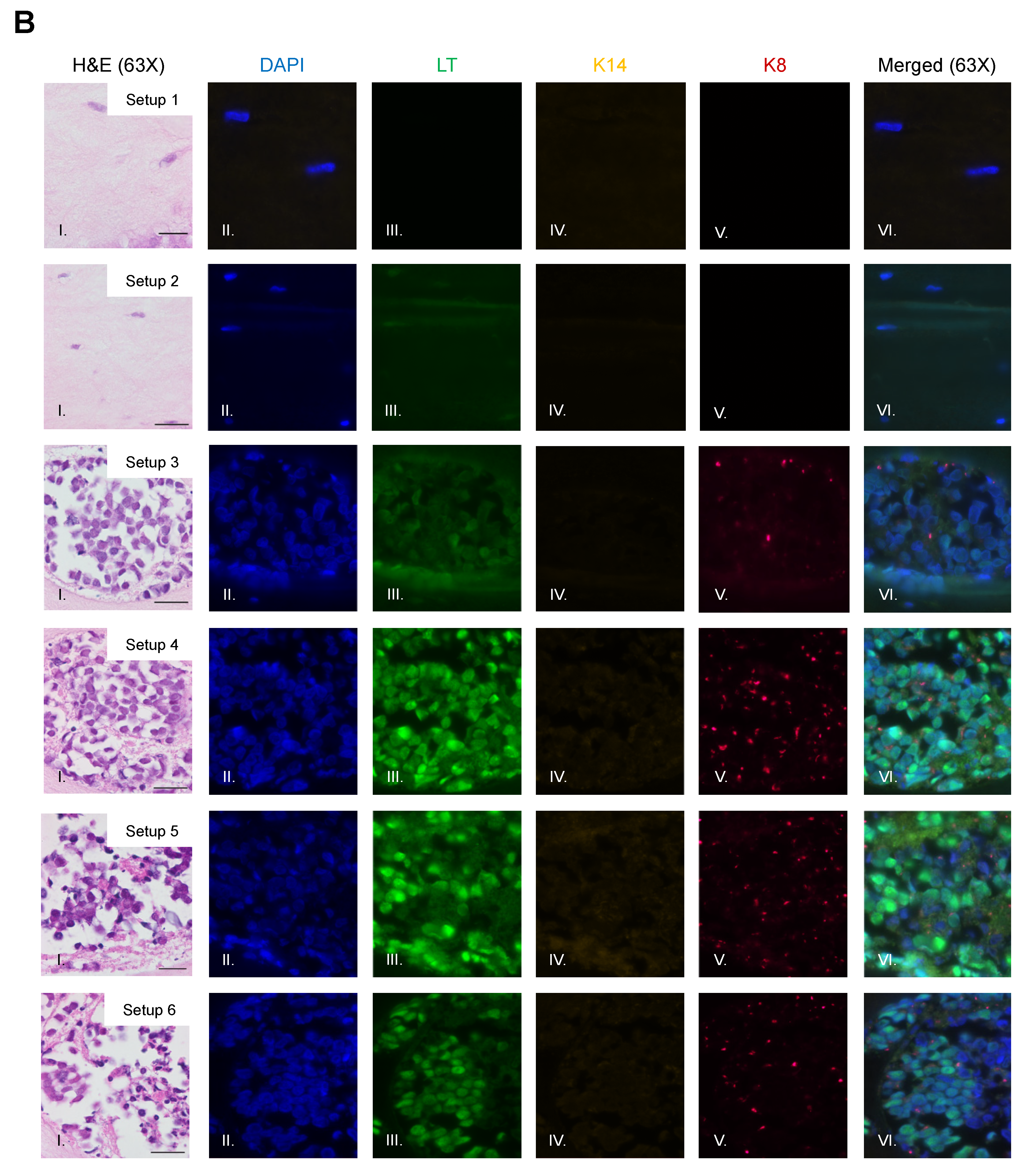
3.3. Identification of MCPyV+ MCC Cells, Epithelial Cells, and Fibroblasts within Raft Structures
3.4. MCPyV+ MCC Cells Retain LT Function and Proliferative Capacity in Organotypic Raft Cultures
4. Conclusions
5. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal Integration of a Polyomavirus in Human Merkel Cell Carcinoma. Science 2008, 319, 1096. [Google Scholar] [CrossRef]
- Toker, C. Trabecular Carcinoma of the Skin. Arch. Dermatol. 1972, 105, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.C.; Stang, A.; DeCaprio, J.A.; Cerroni, L.; Lebbe, C.; Veness, M.; Nghiem, P. Merkel cell carcinoma. Nat. Rev. Dis. Prim. 2017, 3, 17077. [Google Scholar] [CrossRef] [PubMed]
- Cook, L. Polyomaviruses. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef]
- DeCaprio, J.A.; Garcea, R.L. A cornucopia of human polyomaviruses. Nat. Rev. Microbiol. 2013, 11, 264–276. [Google Scholar] [CrossRef]
- Schowalter, R.M.; Pastrana, D.V.; Pumphrey, K.A.; Moyer, A.L.; Buck, C.B. Merkel Cell Polyomavirus and Two Previously Unknown Polyomaviruses are Chronically Shed from Human Skin. Cell Host Microbe 2010, 7, 509–515. [Google Scholar] [CrossRef]
- Tolstov, Y.L.; Knauer, A.; Chen, J.G.; Kensler, T.W.; Kingsley, L.A.; Moore, P.S.; Chang, Y. Asymptomatic primary Merkel cell polyomavirus infection among adults. Emerg. Infect. Dis. 2011, 17, 1371–1380. [Google Scholar] [CrossRef]
- Liu, W.; Yang, R.; Payne, A.S.; Schowalter, R.M.; Spurgeon, M.E.; Lambert, P.F.; Xu, X.; Buck, C.B.; You, J. Identifying the Target Cells and Mechanisms of Merkel Cell Polyomavirus Infection. Cell Host Microbe 2016, 19, 775–787. [Google Scholar] [CrossRef]
- Neumann, F.; Czech-Sioli, M.; Grundhoff, A.; Fischer, N. In Vitro Replication Assay for Merkel Cell Polyomavirus (MCPyV). Curr. Protoc. Microbiol. 2015, 38, 14F.12.11–14F.12.19. [Google Scholar] [CrossRef]
- Zur Hausen, A.; Rennspiess, D.; Winnepenninckx, V.; Speel, E.J.; Kurz, A.K. Early B-cell differentiation in Merkel cell carcinomas: Clues to cellular ancestry. Cancer Res. 2013, 73, 4982–4987. [Google Scholar] [CrossRef]
- Schowalter, R.M.; Reinhold, W.C.; Buck, C.B. Entry tropism of BK and Merkel cell polyomaviruses in cell culture. PLoS ONE 2012, 7, e42181. [Google Scholar] [CrossRef] [PubMed]
- Neumann, F.; Borchert, S.; Schmidt, C.; Reimer, R.; Hohenberg, H.; Fischer, N.; Grundhoff, A. Replication, Gene Expression and Particle Production by a Consensus Merkel Cell Polyomavirus (MCPyV) Genome. PLoS ONE 2011, 6, e29112. [Google Scholar] [CrossRef] [PubMed]
- Kwun, H.J.; Chang, Y.; Moore, P.S. Protein-mediated viral latency is a novel mechanism for Merkel cell polyomavirus persistence. Proc. Natl. Acad. Sci. USA 2017, 114, E4040–E4047. [Google Scholar] [CrossRef] [PubMed]
- Moens, U.; Rasheed, K.; Abdulsalam, I.; Sveinbjørnsson, B. The Role of Merkel Cell Polyomavirus and Other Human Polyomaviruses in Emerging Hallmarks of Cancer. Viruses 2015, 7, 1871. [Google Scholar] [CrossRef] [PubMed]
- Schrama, D.; Sarosi, E.M.; Adam, C.; Ritter, C.; Kaemmerer, U.; Klopocki, E.; König, E.M.; Utikal, J.; Becker, J.C.; Houben, R. Characterization of six Merkel cell polyomavirus-positive Merkel cell carcinoma cell lines: Integration pattern suggest that large T antigen truncating events occur before or during integration. Int. J. Cancer 2019, 145, 1020–1032. [Google Scholar] [CrossRef]
- Shuda, M.; Feng, H.; Kwun, H.J.; Rosen, S.T.; Gjoerup, O.; Moore, P.S.; Chang, Y. T antigen mutations are a human tumor-specific signature for Merkel cell polyomavirus. Proc. Natl. Acad. Sci. USA 2008, 105, 16272–16277. [Google Scholar] [CrossRef]
- Hesbacher, S.; Pfitzer, L.; Wiedorfer, K.; Angermeyer, S.; Borst, A.; Haferkamp, S.; Scholz, C.J.; Wobser, M.; Schrama, D.; Houben, R. RB1 is the crucial target of the Merkel cell polyomavirus Large T antigen in Merkel cell carcinoma cells. Oncotarget 2016, 7, 32956–32968. [Google Scholar] [CrossRef]
- Pilon, A.A.; Desjardins, P.; Hassell, J.A.; Mes-Masson, A.M. Functional implications of mutations within polyomavirus large T antigen Rb-binding domain: Effects on pRb and p107 binding in vitro and immortalization activity in vivo. J. Virol. 1996, 70, 4457. [Google Scholar] [CrossRef]
- Cheng, J.; Rozenblatt-Rosen, O.; Paulson, K.G.; Nghiem, P.; DeCaprio, J.A. Merkel cell polyomavirus large T antigen has growth-promoting and inhibitory activities. J. Virol. 2013, 87, 6118–6126. [Google Scholar] [CrossRef]
- Fan, K.; Gravemeyer, J.; Ritter, C.; Rasheed, K.; Gambichler, T.; Moens, U.; Shuda, M.; Schrama, D.; Becker, J.C. MCPyV Large T Antigen-Induced Atonal Homolog 1 is a Lineage-Dependency Oncogene in Merkel Cell Carcinoma. J. Investig. Dermatol. 2020, 140, 56–65.e53. [Google Scholar] [CrossRef]
- Kervarrec, T.; Samimi, M.; Hesbacher, S.; Berthon, P.; Wobser, M.; Sallot, A.; Sarma, B.; Schweinitzer, S.; Gandon, T.; Destrieux, C.; et al. Merkel Cell Polyomavirus T Antigens Induce Merkel Cell-Like Differentiation in GLI1-Expressing Epithelial Cells. Cancers 2020, 12, 1989. [Google Scholar] [CrossRef] [PubMed]
- Kwun, H.J.; Shuda, M.; Feng, H.; Camacho, C.J.; Moore, P.S.; Chang, Y. Merkel Cell Polyomavirus Small T Antigen Controls Viral Replication and Oncoprotein Expression by Targeting the Cellular Ubiquitin Ligase SCFFbw7. Cell Host Microbe 2013, 14, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Shuda, M.; Guastafierro, A.; Geng, X.; Shuda, Y.; Ostrowski, S.M.; Lukianov, S.; Jenkins, F.J.; Honda, K.; Maricich, S.M.; Moore, P.S.; et al. Merkel Cell Polyomavirus Small T Antigen Induces Cancer and Embryonic Merkel Cell Proliferation in a Transgenic Mouse Model. PLoS ONE 2015, 10, e0142329. [Google Scholar] [CrossRef] [PubMed]
- Shuda, M.; Kwun, H.J.; Feng, H.; Chang, Y.; Moore, P.S. Human Merkel cell polyomavirus small T antigen is an oncoprotein targeting the 4E-BP1 translation regulator. J. Clin. Investig. 2011, 121, 3623–3634. [Google Scholar] [CrossRef]
- Spurgeon, M.E.; Cheng, J.; Bronson, R.T.; Lambert, P.F.; DeCaprio, J.A. Tumorigenic activity of merkel cell polyomavirus T antigens expressed in the stratified epithelium of mice. Cancer Res. 2015, 75, 1068–1079. [Google Scholar] [CrossRef]
- Verhaegen, M.E.; Mangelberger, D.; Harms, P.W.; Eberl, M.; Wilbert, D.M.; Meireles, J.; Bichakjian, C.K.; Saunders, T.L.; Wong, S.Y.; Dlugosz, A.A. Merkel Cell Polyomavirus Small T Antigen Initiates Merkel Cell Carcinoma-like Tumor Development in Mice. Cancer Res. 2017, 77, 3151–3157. [Google Scholar] [CrossRef]
- Houben, R.; Shuda, M.; Weinkam, R.; Schrama, D.; Feng, H.; Chang, Y.; Moore, P.S.; Becker, J.C. Merkel Cell Polyomavirus-Infected Merkel Cell Carcinoma Cells Require Expression of Viral T Antigens. J. Virol. 2010, 84, 7064. [Google Scholar] [CrossRef]
- Harold, A.; Amako, Y.; Hachisuka, J.; Bai, Y.; Li, M.Y.; Kubat, L.; Gravemeyer, J.; Franks, J.; Gibbs, J.R.; Park, H.J.; et al. Conversion of Sox2-dependent Merkel cell carcinoma to a differentiated neuron-like phenotype by T antigen inhibition. Proc. Natl. Acad. Sci. USA 2019, 116, 20104–20114. [Google Scholar] [CrossRef]
- Guastafierro, A.; Feng, H.; Thant, M.; Kirkwood, J.M.; Chang, Y.; Moore, P.S.; Shuda, M. Characterization of an early passage Merkel cell polyomavirus-positive Merkel cell carcinoma cell line, MS-1, and its growth in NOD scid gamma mice. J. Virol. Methods 2013, 187, 6–14. [Google Scholar] [CrossRef]
- Martin, E.M.; Gould, V.E.; Hoog, A.; Rosen, S.T.; Radosevich, J.A.; Deftos, L.J. Parathyroid hormone-related protein, chromogranin A, and calcitonin gene products in the neuroendocrine skin carcinoma cell lines MKL1 and MKL2. Bone Miner. 1991, 14, 113–120. [Google Scholar] [CrossRef]
- Rosen, S.T.; Gould, V.E.; Salwen, H.R.; Herst, C.V.; Le Beau, M.M.; Lee, I.; Bauer, K.; Marder, R.J.; Andersen, R.; Kies, M.S.; et al. Establishment and characterization of a neuroendocrine skin carcinoma cell line. Lab. Investig. 1987, 56, 302–312. [Google Scholar] [PubMed]
- Velásquez, C.; Amako, Y.; Harold, A.; Toptan, T.; Chang, Y.; Shuda, M. Characterization of a Merkel Cell Polyomavirus-Positive Merkel Cell Carcinoma Cell Line CVG-1. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Becker, M.; Dominguez, M.; Greune, L.; Soria-Martinez, L.; Pfleiderer, M.M.; Schowalter, R.; Buck, C.B.; Blaum, B.S.; Schmidt, M.A.; Schelhaas, M. Infectious Entry of Merkel Cell Polyomavirus. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed]
- Koljonen, V. Merkel cell carcinoma. World J. Surg. Oncol. 2006, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Swann, M.H.; Yoon, J. Merkel Cell Carcinoma. Semin. Oncol. 2007, 34, 51–56. [Google Scholar] [CrossRef]
- Asselineau, D.; Prunieras, M. Reconstruction of ‘simplified’ skin: Control of fabrication. Br. J. Dermatol. 1984, 111, 219–222. [Google Scholar] [CrossRef]
- Aasen, T.; Hodgins, M.B.; Edward, M.; Graham, S.V. The relationship between connexins, gap junctions, tissue architecture and tumour invasion, as studied in a novel in vitro model of HPV-16-associated cervical cancer progression. Oncogene 2003, 22, 7969–7980. [Google Scholar] [CrossRef]
- Allen-Hoffmann, B.L.; Schlosser, S.J.; Ivarie, C.A.R.; Meisner, L.F.; O’Connor, S.L.; Sattler, C.A. Normal Growth and Differentiation in a Spontaneously Immortalized Near-Diploid Human Keratinocyte Cell Line, NIKS. J. Investig. Dermatol. 2000, 114, 444–455. [Google Scholar] [CrossRef]
- Flores, E.R.; Allen-Hoffmann, B.L.; Lee, D.; Sattler, C.A.; Lambert, P.F. Establishment of the human papillomavirus type 16 (HPV-16) life cycle in an immortalized human foreskin keratinocyte cell line. Virology 1999, 262, 344–354. [Google Scholar] [CrossRef][Green Version]
- Lambert, P.F.; Ozbun, M.A.; Collins, A.; Holmgren, S.; Lee, D.; Nakahara, T. Using an immortalized cell line to study the HPV life cycle in organotypic “raft” cultures. Methods Mol. Med. 2005, 119, 141–155. [Google Scholar] [CrossRef]
- Lee, D.; Norby, K.; Hayes, M.; Chiu, Y.F.; Sugden, B.; Lambert, P.F. Using Organotypic Epithelial Tissue Culture to Study the Human Papillomavirus Life Cycle. Curr. Protoc. Microbiol. 2016, 41, 14b–18. [Google Scholar] [CrossRef] [PubMed]
- Meyers, C. Organotypic (raft) epithelial tissue culture system for the differentiation-dependent replication of papillomavirus. Methods Cell Sci. 1996, 18, 201–210. [Google Scholar] [CrossRef]
- Meyers, C.; Frattini, M.G.; Hudson, J.B.; Laimins, L.A. Biosynthesis of human papillomavirus from a continuous cell line upon epithelial differentiation. Science 1992, 257, 971–973. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, T.; Peh, W.L.; Doorbar, J.; Lee, D.; Lambert, P.F. Human Papillomavirus Type 16 E1^E4 Contributes to Multiple Facets of the Papillomavirus Life Cycle. J. Virol. 2005, 79, 13150. [Google Scholar] [CrossRef] [PubMed]
- Hukkanen, V.; Mikola, H.; Nykänen, M.; Syrjänen, S. Herpes simplex virus type 1 infection has two separate modes of spread in three-dimensional keratinocyte culture. J. Gen. Virol. 1999, 80, 2149–2155. [Google Scholar] [CrossRef][Green Version]
- Visalli, R.J.; Courtney, R.J.; Meyers, C. Infection and Replication of Herpes Simplex Virus Type 1 in an Organotypic Epithelial Culture System. Virology 1997, 230, 236–243. [Google Scholar] [CrossRef]
- Meyers, C.; Mane, M.; Kokorina, N.; Alam, S.; Hermonat, P.L. Ubiquitous Human Adeno-Associated Virus Type 2 Autonomously Replicates in Differentiating Keratinocytes of a Normal Skin Model. Virology 2000, 272, 338–346. [Google Scholar] [CrossRef]
- Andrei, G.; van den Oord, J.; Fiten, P.; Opdenakker, G.; De Wolf-Peeters, C.; De Clercq, E.; Snoeck, R. Organotypic Epithelial Raft Cultures as a Model for Evaluating Compounds against Alphaherpesviruses. Antimicrob. Agents Chemother. 2005, 49, 4671. [Google Scholar] [CrossRef]
- Makielski, K.R.; Lee, D.; Lorenz, L.D.; Nawandar, D.M.; Chiu, Y.F.; Kenney, S.C.; Lambert, P.F. Human papillomavirus promotes Epstein-Barr virus maintenance and lytic reactivation in immortalized oral keratinocytes. Virology 2016, 495, 52–62. [Google Scholar] [CrossRef]
- Nawandar, D.M.; Ohashi, M.; Djavadian, R.; Barlow, E.; Makielski, K.; Ali, A.; Lee, D.; Lambert, P.F.; Johannsen, E.; Kenney, S.C. Differentiation-Dependent LMP1 Expression Is Required for Efficient Lytic Epstein-Barr Virus Reactivation in Epithelial Cells. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Nawandar, D.M.; Wang, A.; Makielski, K.; Lee, D.; Ma, S.; Barlow, E.; Reusch, J.; Jiang, R.; Wille, C.K.; Greenspan, D.; et al. Differentiation-Dependent KLF4 Expression Promotes Lytic Epstein-Barr Virus Infection in Epithelial Cells. PLoS Pathog. 2015, 11, e1005195. [Google Scholar] [CrossRef] [PubMed]
- Temple, R.M.; Zhu, J.; Budgeon, L.; Christensen, N.D.; Meyers, C.; Sample, C.E. Efficient replication of Epstein–Barr virus in stratified epithelium in vitro. Proc. Natl. Acad. Sci. USA 2014, 111, 16544. [Google Scholar] [CrossRef] [PubMed]
- Harms, P.W. Update on Merkel Cell Carcinoma. Clin. Lab. Med. 2017, 37, 485–501. [Google Scholar] [CrossRef] [PubMed]
- Harms, P.W.; Harms, K.L.; Moore, P.S.; De Caprio, J.A.; Nghiem, P.; Wong, M.K.K.; Brownell, I.; International Workshop on Merkel Cell Carcinoma Research Working Group. The biology and treatment of Merkel cell carcinoma: Current understanding and research priorities. Nat. Rev. Clin. Oncol. 2018, 15, 763–776. [Google Scholar] [CrossRef]
- Smith, P.D.; Patterson, J.W. Merkel cell carcinoma (neuroendocrine carcinoma of the skin). Am. J. Clin. Pathol. 2001, 115, S68–S78. [Google Scholar] [CrossRef]
- Maricich, S.M.; Wellnitz, S.A.; Nelson, A.M.; Lesniak, D.R.; Gerling, G.J.; Lumpkin, E.A.; Zoghbi, H.Y. Merkel Cells Are Essential for Light-Touch Responses. Science 2009, 324, 1580. [Google Scholar] [CrossRef]
- Moll, I.; Zieger, W.; Schmelz, M. Proliferative merkel cells were not detected in human skin. Arch. Dermatol. Res. 1996, 288, 184–187. [Google Scholar] [CrossRef]
- Morrison, K.M.; Miesegaes, G.R.; Lumpkin, E.A.; Maricich, S.M. Mammalian Merkel cells are descended from the epidermal lineage. Dev. Biol. 2009, 336, 76–83. [Google Scholar] [CrossRef]
- Brown, H.A.; Sawyer, D.M.; Woo, T. Intraepidermal Merkel cell carcinoma with no dermal involvement. Am. J. Dermatopathol. 2000, 22, 65–69. [Google Scholar] [CrossRef]
- Jour, G.A.-O.; Aung, P.P.; Rozas-Muñoz, E.; Curry, J.L.; Prieto, V.; Ivan, D. Intraepidermal Merkel cell carcinoma: A case series of a rare entity with clinical follow up. J. Cutan. Pathol. 2017, 44, 684–691. [Google Scholar] [CrossRef]
- Ostrowski, S.M.; Wright, M.C.; Bolock, A.M.; Geng, X.; Maricich, S.M. Ectopic Atoh1 expression drives Merkel cell production in embryonic, postnatal and adult mouse epidermis. Development 2015, 142, 2533–2544. [Google Scholar] [CrossRef] [PubMed]
- Wright, M.C.; Reed-Geaghan, E.G.; Bolock, A.M.; Fujiyama, T.; Hoshino, M.; Maricich, S.M. Unipotent, Atoh1+ progenitors maintain the Merkel cell population in embryonic and adult mice. J. Cell Biol. 2015, 208, 367–379. [Google Scholar] [CrossRef]
- Moll, R.; Osborn, M.; Hartschuh, W.; Moll, I.; Mahrle, G.; Weber, K. Variability of expression and arrangement of cytokeratin and neurofilaments in cutaneous neuroendocrine carcinomas (Merkel cell tumors): Immunocytochemical and biochemical analysis of twelve cases. Ultrastruct. Pathol. 1986, 10, 473–495. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, U.; Müller, U.; Metz, K.A.; Leder, L.D. Cytokeratin and neurofilament protein staining in Merkel cell carcinoma of the small cell type and small cell carcinoma of the lung. Am. J. Dermatopathol. 1998, 20, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.B.; Cohen, I.; Kumar, V.; Xu, Z.; Bar, C.; Dauber-Decker, K.L.; Tsai, P.C.; Marangoni, P.; Klein, O.D.; Hsu, Y.C.; et al. FGF signalling controls the specification of hair placode-derived SOX9 positive progenitors to Merkel cells. Nat. Commun. 2018, 9, 2333. [Google Scholar] [CrossRef] [PubMed]
- Perdigoto, C.N.; Bardot, E.S.; Valdes, V.J.; Santoriello, F.J.; Ezhkova, E. Embryonic maturation of epidermal Merkel cells is controlled by a redundant transcription factor network. Development 2014, 141, 4690. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Thoresen, D.T.; Miao, L.; Williams, J.S.; Wang, C.; Atit, R.P.; Wong, S.Y.; Brownell, I. A Cascade of Wnt, Eda, and Shh Signaling Is Essential for Touch Dome Merkel Cell Development. PLoS Genet. 2016, 12, e1006150. [Google Scholar] [CrossRef]
- Hiraiwa, A.; Fujita, M.; Nagasaka, T.; Adachi, A.; Ohashi, M.; Ishibashi, M. Immunolocalization of hCDC47 protein in normal and neoplastic human tissues and its relation to growth. Int. J. Cancer 1997, 74, 180–184. [Google Scholar] [CrossRef]
- Suzuki, S.; Adachi, A.; Hiraiwa, A.; Ohashi, M.; Ishibashi, M.; Kiyono, T. Cloning and characterization of human MCM7 promoter. Gene 1998, 216, 85–91. [Google Scholar] [CrossRef]
- Meier, F.; Nesbit, M.; Hsu, M.-Y.; Martin, B.; Van Belle, P.; Elder, D.E.; Schaumburg-Lever, G.; Garbe, C.; Walz, T.M.; Donatien, P.; et al. Human Melanoma Progression in Skin Reconstructs: Biological Significance of bFGF. Am. J. Pathol. 2000, 156, 193–200. [Google Scholar] [CrossRef]
- Eberle, J.; Hossini, A.M. Expression and function of bcl-2 proteins in melanoma. Curr. Genom. 2008, 9, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Moll, I.; Gillardon, F.; Waltering, S.; Schmelz, M.; Moll, R. Differences of bcl-2 protein expression between Merkel cells and Merkel cell carcinomas. J. Cutan. Pathol. 1996, 23, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Plettenberg, A.; Pammer, J.; Tschachler, E. Merkel cells and Merkel cell carcinoma express the BCL-2 proto-oncogene. Exp. Dermatol. 1996, 5, 102–107. [Google Scholar] [CrossRef]
- Spurgeon, M.E.; den Boon, J.A.; Horswill, M.; Barthakur, S.; Forouzan, O.; Rader, J.S.; Beebe, D.J.; Roopra, A.; Ahlquist, P.; Lambert, P.F. Human papillomavirus oncogenes reprogram the cervical cancer microenvironment independently of and synergistically with estrogen. Proc. Natl. Acad. Sci. USA 2017, 114, E9076–E9085. [Google Scholar] [CrossRef]
- Flores, E.R.; Allen-Hoffmann, B.L.; Lee, D.; Lambert, P.F. The Human Papillomavirus Type 16 E7 Oncogene Is Required for the Productive Stage of the Viral Life Cycle. J. Virol. 2000, 74, 6622. [Google Scholar] [CrossRef]
- Kaushik, S.; Pickup, M.W.; Weaver, V.M. From transformation to metastasis: Deconstructing the extracellular matrix in breast cancer. Cancer Metastasis Rev. 2016, 35, 655–667. [Google Scholar] [CrossRef]
- Infanger, D.W.; Lynch, M.E.; Fischbach, C. Engineered Culture Models for Studies of Tumor-Microenvironment Interactions. Ann. Rev. Biomed. Eng. 2013, 15, 29–53. [Google Scholar] [CrossRef]
- Kalli, M.; Stylianopoulos, T. Defining the Role of Solid Stress and Matrix Stiffness in Cancer Cell Proliferation and Metastasis. Front. Oncol. 2018, 8. [Google Scholar] [CrossRef]
- Paulson, K.G.; Park, S.Y.; Vandeven, N.A.; Lachance, K.; Thomas, H.; Chapuis, A.G.; Harms, K.L.; Thompson, J.A.; Bhatia, S.; Stang, A.; et al. Merkel cell carcinoma: Current US incidence and projected increases based on changing demographics. J. Am. Acad. Dermatol. 2018, 78, 457–463.e452. [Google Scholar] [CrossRef]
- Liang, E.; Brower, J.V.; Rice, S.R.; Buehler, D.G.; Saha, S.; Kimple, R.J. Merkel Cell Carcinoma Analysis of Outcomes: A 30-Year Experience. PLoS ONE 2015, 10, e0129476. [Google Scholar] [CrossRef]
- Tello, T.L.; Coggshall, K.; Yom, S.S.; Yu, S.S. Merkel cell carcinoma: An update and review: Current and future therapy. J. Am. Acad. Dermatol. 2018, 78, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.S.; Byrne, P.J.; Jacobs, L.K.; Taube, J.M. Merkel cell carcinoma: Update and review. Semin. Cutan. Med. Surg. 2011, 30, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Harms, P.W.; Vats, P.; Verhaegen, M.E.; Robinson, D.R.; Wu, Y.-M.; Dhanasekaran, S.M.; Palanisamy, N.; Siddiqui, J.; Cao, X.; Su, F.; et al. The Distinctive Mutational Spectra of Polyomavirus-Negative Merkel Cell Carcinoma. Cancer Res. 2015, 75, 3720. [Google Scholar] [CrossRef] [PubMed]
- Starrett, G.J.; Thakuria, M.; Chen, T.; Marcelus, C.; Cheng, J.; Nomburg, J.; Thorner, A.R.; Slevin, M.K.; Powers, W.; Burns, R.T.; et al. Clinical and molecular characterization of virus-positive and virus-negative Merkel cell carcinoma. Genome Med. 2020, 12, 30. [Google Scholar] [CrossRef]
- Wong, S.Q.; Waldeck, K.; Vergara, I.A.; Schröder, J.; Madore, J.; Wilmott, J.S.; Colebatch, A.J.; De Paoli-Iseppi, R.; Li, J.; Lupat, R.; et al. UV-Associated Mutations Underlie the Etiology of MCV-Negative Merkel Cell Carcinomas. Cancer Res. 2015, 75, 5228. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MKL-1 | NIKS | EF-1-F | |
|---|---|---|---|
| LT antigen (CM2B4) | + | - | - |
| Cytokeratin 14 (K14) | − | + | − |
| Cytokeratin 10 (K10) * | − | + | − |
| Cytokeratin 8 (K8) | + | − | − |
| α-smooth muscle (SMA) | − | − | + |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loke, A.S.W.; Longley, B.J.; Lambert, P.F.; Spurgeon, M.E. A Novel In Vitro Culture Model System to Study Merkel Cell Polyomavirus–Associated MCC Using Three-Dimensional Organotypic Raft Equivalents of Human Skin. Viruses 2021, 13, 138. https://doi.org/10.3390/v13010138
Loke ASW, Longley BJ, Lambert PF, Spurgeon ME. A Novel In Vitro Culture Model System to Study Merkel Cell Polyomavirus–Associated MCC Using Three-Dimensional Organotypic Raft Equivalents of Human Skin. Viruses. 2021; 13(1):138. https://doi.org/10.3390/v13010138
Chicago/Turabian StyleLoke, Amanda S. W., B. Jack Longley, Paul F. Lambert, and Megan E. Spurgeon. 2021. "A Novel In Vitro Culture Model System to Study Merkel Cell Polyomavirus–Associated MCC Using Three-Dimensional Organotypic Raft Equivalents of Human Skin" Viruses 13, no. 1: 138. https://doi.org/10.3390/v13010138
APA StyleLoke, A. S. W., Longley, B. J., Lambert, P. F., & Spurgeon, M. E. (2021). A Novel In Vitro Culture Model System to Study Merkel Cell Polyomavirus–Associated MCC Using Three-Dimensional Organotypic Raft Equivalents of Human Skin. Viruses, 13(1), 138. https://doi.org/10.3390/v13010138