Stealing the Show: KSHV Hijacks Host RNA Regulatory Pathways to Promote Infection


{kind=link}
{kind=link}
Abstract
1. Introduction
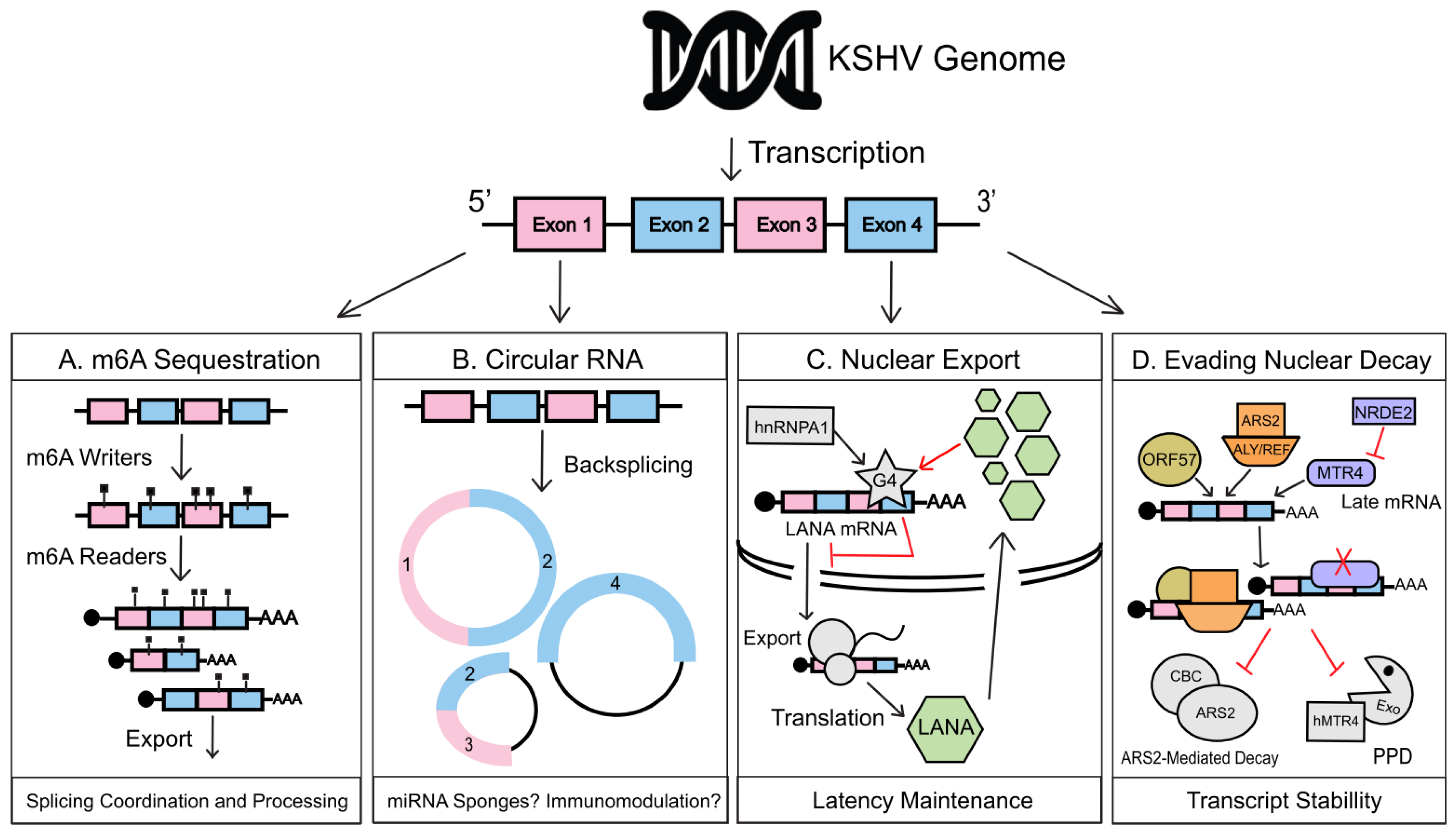
2. Nuclear Regulation of RNA during KSHV Infection
2.1. Co-Transcriptional Regulation
2.2. Nuclear Export
2.3. Nuclear Decay
3. Cytoplasmic Regulation of RNA during KSHV Infection
3.1. mRNA Translation
3.2. RNA Decay
4. Conclusions
Funding
Conflicts of Interest
References
- Chang, Y.; Cesarman, E.; Pessin, M.S.; Lee, F.; Culpepper, J.; Knowles, D.M.; Moore, P.S. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 1994, 266, 1865–1869. [Google Scholar] [CrossRef] [PubMed]
- Dupin, N.; Fisher, C.; Kellam, P.; Ariad, S.; Tulliez, M.; Franck, N.; van Marck, E.; Salmon, D.; Gorin, I.; Escande, J.P.; et al. Distribution of human herpesvirus-8 latently infected cells in Kaposi’s sarcoma, multicentric Castleman’s disease, and primary effusion lymphoma. Proc. Natl. Acad. Sci. USA 1999, 96, 4546–4551. [Google Scholar] [CrossRef] [PubMed]
- Soulier, J.; Grollet, L.; Oksenhendler, E.; Cacoub, P.; Cazals-Hatem, D.; Babinet, P.; d’Agay, M.F.; Clauvel, J.P.; Raphael, M.; Degos, L. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman’s disease. Blood 1995, 86, 1276–1280. [Google Scholar] [CrossRef] [PubMed]
- Giffin, L.; Damania, B. KSHV: Pathways to tumorigenesis and persistent infection. Adv. Virus Res. 2014, 88, 111–159. [Google Scholar]
- Arvin, A.; Campadelli-Fiume, G.; Mocarski, E.; Moore, P.S.; Roizman, B.; Whitley, R.; Yamanishi, K.; Lukac, D.M.; Yuan, Y. Reactivation and lytic replication of KSHV. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Russo, J.J.; Bohenzky, R.A.; Chien, M.C.; Chen, J.; Yan, M.; Maddalena, D.; Parry, J.P.; Peruzzi, D.; Edelman, I.S.; Chang, Y.; et al. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc. Natl. Acad. Sci. USA 1996, 93, 14862–14867. [Google Scholar] [CrossRef] [PubMed]
- Ganem, D. KSHV and Kaposi’s sarcoma: The end of the beginning? Cell 1997, 91, 157–160. [Google Scholar] [CrossRef]
- Arias, C.; Weisburd, B.; Stern-Ginossar, N.; Mercier, A.; Madrid, A.S.; Bellare, P.; Holdorf, M.; Weissman, J.S.; Ganem, D. KSHV 2.0: A comprehensive annotation of the Kaposi’s sarcoma-associated herpesvirus genome using next-generation sequencing reveals novel genomic and functional features. PLoS Pathog. 2014, 10, e1003847. [Google Scholar] [CrossRef]
- Dahan, N.; Choder, M. The eukaryotic transcriptional machinery regulates mRNA translation and decay in the cytoplasm. Biochim. Biophys. Acta 2013, 1829, 169–173. [Google Scholar] [CrossRef]
- Haimovich, G.; Choder, M.; Singer, R.H.; Trcek, T. The fate of the messenger is pre-determined: A new model for regulation of gene expression. Biochim. Biophys. Acta 2013, 1829, 643–653. [Google Scholar] [CrossRef]
- Liu, J.; Yue, Y.; Han, D.; Wang, X.; Fu, Y.; Zhang, L.; Jia, G.; Yu, M.; Lu, Z.; Deng, X.; et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 2014, 10, 93–95. [Google Scholar] [CrossRef]
- Warda, A.S.; Kretschmer, J.; Hackert, P.; Lenz, C.; Urlaub, H.; Höbartner, C.; Sloan, K.E.; Bohnsack, M.T. Human METTL16 is a N6-methyladenosine (m6A) methyltransferase that targets pre-mRNAs and various non-coding RNAs. EMBO Rep. 2017, 18, 2004–2014. [Google Scholar] [CrossRef] [PubMed]
- Zaccara, S.; Ries, R.J.; Jaffrey, S.R. Reading, writing and erasing mRNA methylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 608–624. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Hsu, P.J.; Chen, Y.-S.; Yang, Y.-G. Dynamic transcriptomic m6A decoration: Writers, erasers, readers and functions in RNA metabolism. Cell Res. 2018, 28, 616–624. [Google Scholar] [CrossRef] [PubMed]
- Ke, S.; Pandya-Jones, A.; Saito, Y.; Fak, J.J.; Vågbø, C.B.; Geula, S.; Hanna, J.H.; Black, D.L.; Darnell, J.E.; Darnell, R.B. m6A mRNA modifications are deposited in nascent pre-mRNA and are not required for splicing but do specify cytoplasmic turnover. Genes Dev. 2017, 31, 990–1006. [Google Scholar] [CrossRef]
- Slobodin, B.; Han, R.; Calderone, V.; Vrielink, J.A.F.O.; Loayza-Puch, F.; Elkon, R.; Agami, R. Transcription Impacts the Efficiency of mRNA Translation via Co-transcriptional N6-adenosine Methylation. Cell 2017, 169, 326–337. [Google Scholar] [CrossRef]
- Zheng, G.; Dahl, J.A.; Niu, Y.; Fedorcsak, P.; Huang, C.-M.; Li, C.J.; Vågbø, C.B.; Shi, Y.; Wang, W.-L.; Song, S.-H.; et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell 2013, 49, 18–29. [Google Scholar] [CrossRef]
- Wei, J.; Liu, F.; Lu, Z.; Fei, Q.; Ai, Y.; He, P.C.; Shi, H.; Cui, X.; Su, R.; Klungland, A.; et al. Differential m6A, m6Am, and m1A Demethylation Mediated by FTO in the Cell Nucleus and Cytoplasm. Mol. Cell 2018, 71, 973–985. [Google Scholar] [CrossRef]
- Tan, B.; Liu, H.; Zhang, S.; da Silva, S.R.; Zhang, L.; Meng, J.; Cui, X.; Yuan, H.; Sorel, O.; Zhang, S.-W.; et al. Viral and cellular N6-methyladenosine and N6,2’-O-dimethyladenosine epitranscriptomes in the KSHV life cycle. Nat. Microbiol. 2018, 3, 108–120. [Google Scholar] [CrossRef]
- Andreassi, C.; Riccio, A. To localize or not to localize: mRNA fate is in 3′UTR ends. Trends Cell Biol. 2009, 19, 465–474. [Google Scholar] [CrossRef]
- Moody, R.; Zhu, Y.; Huang, Y.; Cui, X.; Jones, T.; Bedolla, R.; Lei, X.; Bai, Z.; Gao, S.-J. KSHV microRNAs mediate cellular transformation and tumorigenesis by redundantly targeting cell growth and survival pathways. PLoS Pathog. 2013, 9, e1003857. [Google Scholar] [CrossRef]
- Ye, F.; Chen, E.R.; Nilsen, T.W. Kaposi’s Sarcoma-Associated Herpesvirus Utilizes and Manipulates RNA N6-Adenosine Methylation To Promote Lytic Replication. J. Virol. 2017, 91, 201. [Google Scholar] [CrossRef] [PubMed]
- Hesser, C.R.; Karijolich, J.; Dominissini, D.; He, C.; Glaunsinger, B.A. N6-methyladenosine modification and the YTHDF2 reader protein play cell type specific roles in lytic viral gene expression during Kaposi’s sarcoma-associated herpesvirus infection. PLoS Pathog. 2018, 14, e1006995. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Wang, X.; Liu, K.; Roundtree, I.A.; Tempel, W.; Li, Y.; Lu, Z.; He, C.; Min, J. Structural basis for selective binding of m6A RNA by the YTHDC1 YTH domain. Nat. Chem. Biol. 2014, 10, 927–929. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Roundtree, I.A.; Wang, P.; Wang, X.; Wang, L.; Sun, C.; Tian, Y.; Li, J.; He, C.; Xu, Y. Crystal structure of the YTH domain of YTHDF2 reveals mechanism for recognition of N6-methyladenosine. Cell Res. 2014, 24, 1493–1496. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.; Sun, H.; Xu, C. YTH Domain: A Family of N6-methyladenosine (m6A) Readers. Genom. Proteom. Bioinform. 2018, 16, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhao, D.; Wu, J.; Shi, Y. Structure of the YTH domain of human YTHDF2 in complex with an m(6)A mononucleotide reveals an aromatic cage for m(6)A recognition. Cell Res. 2014, 24, 1490–1492. [Google Scholar] [CrossRef]
- Xiao, W.; Adhikari, S.; Dahal, U.; Chen, Y.-S.; Hao, Y.-J.; Sun, B.-F.; Sun, H.-Y.; Li, A.; Ping, X.-L.; Lai, W.-Y.; et al. Nuclear m(6)A Reader YTHDC1 Regulates mRNA Splicing. Mol. Cell 2016, 61, 507–519. [Google Scholar] [CrossRef]
- Roundtree, I.A.; Luo, G.-Z.; Zhang, Z.; Wang, X.; Zhou, T.; Cui, Y.; Sha, J.; Huang, X.; Guerrero, L.; Xie, P.; et al. YTHDC1 mediates nuclear export of N6-methyladenosine methylated mRNAs. eLife 2017, 6, e31311. [Google Scholar] [CrossRef]
- Patil, D.P.; Chen, C.-K.; Pickering, B.F.; Chow, A.; Jackson, C.; Guttman, M.; Jaffrey, S.R. m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature 2016, 537, 369–373. [Google Scholar] [CrossRef]
- Baquero-Perez, B.; Antanaviciute, A.; Yonchev, I.D.; Carr, I.M.; Wilson, S.A.; Whitehouse, A. The Tudor SND1 protein is an m6A RNA reader essential for replication of Kaposi’s sarcoma-associated herpesvirus. eLife 2019, 8, e47261. [Google Scholar] [CrossRef]
- Jeck, W.R.; Sorrentino, J.A.; Wang, K.; Slevin, M.K.; Burd, C.E.; Liu, J.; Marzluff, W.F.; Sharpless, N.E. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 2013, 19, 141–157. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Z. Efficient backsplicing produces translatable circular mRNAs. RNA 2015, 21, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Salzman, J.; Gawad, C.; Wang, P.L.; Lacayo, N.; Brown, P.O. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS ONE 2012, 7, e30733. [Google Scholar] [CrossRef]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef]
- Ashwal-Fluss, R.; Meyer, M.; Pamudurti, N.R.; Ivanov, A.; Bartok, O.; Hanan, M.; Evantal, N.; Memczak, S.; Rajewsky, N.; Kadener, S. circRNA biogenesis competes with pre-mRNA splicing. Mol. Cell 2014, 56, 55–66. [Google Scholar] [CrossRef]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Huang, C.; Bao, C.; Chen, L.; Lin, M.; Wang, X.; Zhong, G.; Yu, B.; Hu, W.; Dai, L.; et al. Exon-intron circular RNAs regulate transcription in the nucleus. Nat. Struct. Mol. Biol. 2015, 22, 256–264. [Google Scholar] [CrossRef]
- Liu, C.-X.; Li, X.; Nan, F.; Jiang, S.; Gao, X.; Guo, S.-K.; Xue, W.; Cui, Y.; Dong, K.; Ding, H.; et al. Structure and Degradation of Circular RNAs Regulate PKR Activation in Innate Immunity. Cell 2019, 177, 865–880. [Google Scholar] [CrossRef] [PubMed]
- Toptan, T.; Abere, B.; Nalesnik, M.A.; Swerdlow, S.H.; Ranganathan, S.; Lee, N.; Shair, K.H.; Moore, P.S.; Chang, Y. Circular DNA tumor viruses make circular RNAs. Proc. Natl. Acad. Sci. USA 2018, 115, E8737–E8745. [Google Scholar] [CrossRef] [PubMed]
- Tagawa, T.; Gao, S.; Koparde, V.N.; Gonzalez, M.; Spouge, J.L.; Serquiña, A.P.; Lurain, K.; Ramaswami, R.; Uldrick, T.S.; Yarchoan, R.; et al. Discovery of Kaposi’s sarcoma herpesvirus-encoded circular RNAs and a human antiviral circular RNA. Proc. Natl. Acad. Sci. USA 2018, 115, 12805–12810. [Google Scholar] [CrossRef]
- Liang, D.; Wilusz, J.E. Short intronic repeat sequences facilitate circular RNA production. Genes Dev. 2014, 28, 2233–2247. [Google Scholar] [CrossRef]
- Wang, J.; Wang, L. Deep learning of the back-splicing code for circular RNA formation. Bioinformatics 2019, 35, 5235–5242. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Fan, X.; Mao, M.; Song, X.; Wu, P.; Zhang, Y.; Jin, Y.; Yang, Y.; Chen, L.-L.; Wang, Y.; et al. Extensive translation of circular RNAs driven by N6-methyladenosine. Cell Res. 2017, 27, 626–641. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Lee, E.E.; Kim, J.; Yang, R.; Chamseddin, B.; Ni, C.; Gusho, E.; Xie, Y.; Chiang, C.-M.; Buszczak, M.; et al. Transforming activity of an oncoprotein-encoding circular RNA from human papillomavirus. Nat. Commun. 2019, 10, 2300–2312. [Google Scholar] [CrossRef]
- Liu, Q.; Shuai, M.; Xia, Y. Knockdown of EBV-encoded circRNA circRPMS1 suppresses nasopharyngeal carcinoma cell proliferation and metastasis through sponging multiple miRNAs. Cancer Manag. Res. 2019, 11, 8023–8031. [Google Scholar] [CrossRef] [PubMed]
- Abere, B.; Li, J.; Zhou, H.; Toptan, T.; Moore, P.S.; Chang, Y. Kaposi’s Sarcoma-Associated Herpesvirus-Encoded circRNAs Are Expressed in Infected Tumor Tissues and Are Incorporated into Virions. MBio 2020, 11, 1865. [Google Scholar] [CrossRef]
- Zhang, H.; Ni, G.; Damania, B. ADAR1 Facilitates KSHV Lytic Reactivation by Modulating the RLR-Dependent Signaling Pathway. Cell Rep. 2020, 31, 107564. [Google Scholar] [CrossRef]
- Zhang, Y.; Liang, W.; Zhang, P.; Chen, J.; Qian, H.; Zhang, X.; Xu, W. Circular RNAs: Emerging cancer biomarkers and targets. J. Exp. Clin. Cancer Res. 2017, 36, 152. [Google Scholar] [CrossRef]
- Rhodes, D.; Lipps, H.J. G-quadruplexes and their regulatory roles in biology. Nucleic Acids Res. 2015, 43, 8627–8637. [Google Scholar] [CrossRef]
- Lavezzo, E.; Berselli, M.; Frasson, I.; Perrone, R.; Palù, G.; Brazzale, A.R.; Richter, S.N.; Toppo, S. G-quadruplex forming sequences in the genome of all known human viruses: A comprehensive guide. PLoS Comput Biol 2018, 14, e1006675. [Google Scholar] [CrossRef]
- Dabral, P.; Babu, J.; Zareie, A.; Verma, S.C. LANA and hnRNP A1 Regulate the Translation of LANA mRNA through G-Quadruplexes. J. Virol. 2020, 94, 1865. [Google Scholar] [CrossRef] [PubMed]
- Meola, N.; Domanski, M.; Karadoulama, E.; Chen, Y.; Gentil, C.; Pultz, D.; Vitting-Seerup, K.; Lykke-Andersen, S.; Andersen, J.S.; Sandelin, A.; et al. Identification of a Nuclear Exosome Decay Pathway for Processed Transcripts. Mol. Cell 2016, 64, 520–533. [Google Scholar] [CrossRef]
- Ruiz, J.C.; Hunter, O.V.; Conrad, N.K. Kaposi’s sarcoma-associated herpesvirus ORF57 protein protects viral transcripts from specific nuclear RNA decay pathways by preventing hMTR4 recruitment. PLoS Pathog. 2019, 15, e1007596. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, J.C.; Devlin, A.M.; Kim, J.; Conrad, N.K. Kaposi’s Sarcoma-Associated Herpesvirus Fine-Tunes the Temporal Expression of Late Genes by Manipulating a Host RNA Quality Control Pathway. J. Virol. 2020, 94, 3165. [Google Scholar] [CrossRef] [PubMed]
- Majerciak, V.; Yamanegi, K.; Nie, S.H.; Zheng, Z.-M. Structural and functional analyses of Kaposi sarcoma-associated herpesvirus ORF57 nuclear localization signals in living cells. J. Biol. Chem. 2006, 281, 28365–28378. [Google Scholar] [CrossRef]
- Stubbs, S.H.; Hunter, O.V.; Hoover, A.; Conrad, N.K. Viral factors reveal a role for REF/Aly in nuclear RNA stability. Mol. Cell. Biol. 2012, 32, 1260–1270. [Google Scholar] [CrossRef]
- Rossetto, C.C.; Pari, G.S. PAN’s Labyrinth: Molecular biology of Kaposi’s sarcoma-associated herpesvirus (KSHV) PAN RNA, a multifunctional long noncoding RNA. Viruses 2014, 6, 4212–4226. [Google Scholar] [CrossRef]
- Shi, M.; Zhang, H.; Wu, X.; He, Z.; Wang, L.; Yin, S.; Tian, B.; Li, G.; Cheng, H. ALYREF mainly binds to the 5′ and the 3′ regions of the mRNA in vivo. Nucleic Acids Res. 2017, 45, 9640–9653. [Google Scholar] [CrossRef]
- Melko, M.; Winczura, K.; Rouvière, J.O.; Oborská-Oplová, M.; Andersen, P.K.; Heick Jensen, T. Mapping domains of ARS2 critical for its RNA decay capacity. Nucleic Acids Res. 2020, 48, 6943–6953. [Google Scholar] [CrossRef]
- Fan, J.; Kuai, B.; Wu, G.; Wu, X.; Chi, B.; Wang, L.; Wang, K.; Shi, Z.; Zhang, H.; Chen, S.; et al. Exosome cofactor hMTR4 competes with export adaptor ALYREF to ensure balanced nuclear RNA pools for degradation and export. EMBO J. 2017, 36, 2870–2886. [Google Scholar] [CrossRef]
- Jackson, B.R.; Boyne, J.R.; Noerenberg, M.; Taylor, A.; Hautbergue, G.M.; Walsh, M.J.; Wheat, R.; Blackbourn, D.J.; Wilson, S.A.; Whitehouse, A. An interaction between KSHV ORF57 and UIF provides mRNA-adaptor redundancy in herpesvirus intronless mRNA export. PLoS Pathog. 2011, 7, e1002138. [Google Scholar] [CrossRef]
- Wang, J.; Chen, J.; Wu, G.; Zhang, H.; Du, X.; Chen, S.; Zhang, L.; Wang, K.; Fan, J.; Gao, S.; et al. NRDE2 negatively regulates exosome functions by inhibiting MTR4 recruitment and exosome interaction. Genes Dev. 2019, 33, 536–549. [Google Scholar] [CrossRef] [PubMed]
- Majerciak, V.; Yamanegi, K.; Allemand, E.; Kruhlak, M.; Krainer, A.R.; Zheng, Z.-M. Kaposi’s sarcoma-associated herpesvirus ORF57 functions as a viral splicing factor and promotes expression of intron-containing viral lytic genes in spliceosome-mediated RNA splicing. J. Virol. 2008, 82, 2792–2801. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.-H.B.; Li, L.; Silva, L.; Sandri-Goldin, R.M. ICP27 recruits Aly/REF but not TAP/NXF1 to herpes simplex virus type 1 transcription sites although TAP/NXF1 is required for ICP27 export. J. Virol. 2005, 79, 3949–3961. [Google Scholar] [CrossRef] [PubMed]
- Kwun, H.J.; da Silva, S.R.; Qin, H.; Ferris, R.L.; Tan, R.; Chang, Y.; Moore, P.S. The central repeat domain 1 of Kaposi’s sarcoma-associated herpesvirus (KSHV) latency associated-nuclear antigen 1 (LANA1) prevents cis MHC class I peptide presentation. Virology 2011, 412, 357–365. [Google Scholar] [CrossRef]
- Thakker, S.; Purushothaman, P.; Gupta, N.; Challa, S.; Cai, Q.; Verma, S.C. Kaposi’s Sarcoma-Associated Herpesvirus Latency-Associated Nuclear Antigen Inhibits Major Histocompatibility Complex Class II Expression by Disrupting Enhanceosome Assembly through Binding with the Regulatory Factor X Complex. J. Virol. 2015, 89, 5536–5556. [Google Scholar] [CrossRef]
- Park, O.H.; Ha, H.; Lee, Y.; Boo, S.H.; Kwon, D.H.; Song, H.K.; Kim, Y.K. Endoribonucleolytic Cleavage of m6A-Containing RNAs by RNase P/MRP Complex. Mol. Cell 2019, 74, 494–507. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, B.S.; Roundtree, I.A.; Lu, Z.; Han, D.; Ma, H.; Weng, X.; Chen, K.; Shi, H.; He, C. N(6)-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell 2015, 161, 1388–1399. [Google Scholar] [CrossRef]
- Grosswendt, S.; Filipchyk, A.; Manzano, M.; Klironomos, F.; Schilling, M.; Herzog, M.; Gottwein, E.; Rajewsky, N. Unambiguous identification of miRNA:target site interactions by different types of ligation reactions. Mol. Cell 2014, 54, 1042–1054. [Google Scholar] [CrossRef]
- Qin, J.; Li, W.; Gao, S.-J.; Lu, C. KSHV microRNAs: Tricks of the Devil. Trends Microbiol. 2017, 25, 648–661. [Google Scholar] [CrossRef]
- Tagawa, T.; Serquiña, A.; Kook, I.; Ziegelbauer, J. Viral non-coding RNAs: Stealth strategies in the tug-of-war between humans and herpesviruses. Semin. Cell Dev. Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Stedman, W.; Yousef, M.; Renne, R.; Lieberman, P.M. Epigenetic regulation of Kaposi’s sarcoma-associated herpesvirus latency by virus-encoded microRNAs that target Rta and the cellular Rbl2-DNMT pathway. J. Virol. 2010, 84, 2697–2706. [Google Scholar] [CrossRef] [PubMed]
- Dölken, L.; Malterer, G.; Erhard, F.; Kothe, S.; Friedel, C.C.; Suffert, G.; Marcinowski, L.; Motsch, N.; Barth, S.; Beitzinger, M.; et al. Systematic analysis of viral and cellular microRNA targets in cells latently infected with human gamma-herpesviruses by RISC immunoprecipitation assay. Cell Host Microbe 2010, 7, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Hussein, H.A.M.; Alfhili, M.A.; Pakala, P.; Simon, S.; Hussain, J.; McCubrey, J.A.; Akula, S.M. miRNAs and their roles in KSHV pathogenesis. Virus Res. 2019, 266, 15–24. [Google Scholar] [CrossRef]
- Morrison, K.; Manzano, M.; Chung, K.; Schipma, M.J.; Bartom, E.T.; Gottwein, E. The Oncogenic Kaposi’s Sarcoma-Associated Herpesvirus Encodes a Mimic of the Tumor-Suppressive miR-15/16 miRNA Family. Cell Rep. 2019, 29, 2961–2969. [Google Scholar] [CrossRef]
- Zhang, J.; Pu, X.-M.; Xiong, Y. kshv-mir-k12-1-5p promotes cell growth and metastasis by targeting SOCS6 in Kaposi’s sarcoma cells. Cancer Manag. Res. 2019, 11, 4985–4995. [Google Scholar] [CrossRef]
- Hussein, H.A.M.; Akula, S.M. Profiling of cellular microRNA responses during the early stages of KSHV infection. Arch. Virol. 2017, 162, 3293–3303. [Google Scholar] [CrossRef]
- Lin, X.; Liang, D.; He, Z.; Deng, Q.; Robertson, E.S.; Lan, K. miR-K12-7-5p encoded by Kaposi’s sarcoma-associated herpesvirus stabilizes the latent state by targeting viral ORF50/RTA. PLoS ONE 2011, 6, e16224. [Google Scholar] [CrossRef]
- Bellare, P.; Ganem, D. Regulation of KSHV lytic switch protein expression by a virus-encoded microRNA: An evolutionary adaptation that fine-tunes lytic reactivation. Cell Host Microbe 2009, 6, 570–575. [Google Scholar] [CrossRef]
- Li, W.; Jia, X.; Shen, C.; Zhang, M.; Xu, J.; Shang, Y.; Zhu, K.; Hu, M.; Yan, Q.; Qin, D.; et al. A KSHV microRNA enhances viral latency and induces angiogenesis by targeting GRK2 to activate the CXCR2/AKT pathway. Oncotarget 2016, 7, 32286–32305. [Google Scholar] [CrossRef]
- Ziegelbauer, J.M.; Sullivan, C.S.; Ganem, D. Tandem array-based expression screens identify host mRNA targets of virus-encoded microRNAs. Nat. Genet. 2009, 41, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.-R.; Ganem, D. Viral microRNA target allows insight into the role of translation in governing microRNA target accessibility. Proc. Natl. Acad. Sci. USA 2011, 108, 5148–5153. [Google Scholar] [CrossRef] [PubMed]
- Majerciak, V.; Ni, T.; Yang, W.; Meng, B.; Zhu, J.; Zheng, Z.-M. A viral genome landscape of RNA polyadenylation from KSHV latent to lytic infection. PLoS Pathog. 2013, 9, e1003749. [Google Scholar] [CrossRef] [PubMed]
- McClure, L.V.; Kincaid, R.P.; Burke, J.M.; Grundhoff, A.; Sullivan, C.S. Comprehensive mapping and analysis of Kaposi’s sarcoma-associated herpesvirus 3′ UTRs identify differential posttranscriptional control of gene expression in lytic versus latent infection. J. Virol. 2013, 87, 12838–12849. [Google Scholar] [CrossRef]
- Bai, Z.; Huang, Y.; Li, W.; Zhu, Y.; Jung, J.U.; Lu, C.; Gao, S.-J. Genomewide mapping and screening of Kaposi’s sarcoma-associated herpesvirus (KSHV) 3′ untranslated regions identify bicistronic and polycistronic viral transcripts as frequent targets of KSHV microRNAs. J. Virol. 2014, 88, 377–392. [Google Scholar] [CrossRef]
- McCormick, C.; Ganem, D. The kaposin B protein of KSHV activates the p38/MK2 pathway and stabilizes cytokine mRNAs. Science 2005, 307, 739–741. [Google Scholar] [CrossRef]
- McCormick, C.; Ganem, D. Phosphorylation and function of the kaposin B direct repeats of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2006, 80, 6165–6170. [Google Scholar] [CrossRef]
- Chang, H.-C.; Hsieh, T.-H.; Lee, Y.-W.; Tsai, C.-F.; Tsai, Y.-N.; Cheng, C.-C.; Wang, H.-W. c-Myc and viral cofactor Kaposin B co-operate to elicit angiogenesis through modulating miRNome traits of endothelial cells. BMC Syst. Biol. 2016, 10 (Suppl. S1), S1. [Google Scholar] [CrossRef]
- Shenoy, A.; Blelloch, R. Genomic analysis suggests that mRNA destabilization by the microprocessor is specialized for the auto-regulation of Dgcr. PLoS ONE 2009, 4, e6971. [Google Scholar] [CrossRef]
- Xing, L.; Kieff, E. cis-Acting effects on RNA processing and Drosha cleavage prevent Epstein-Barr virus latency III BHRF1 expression. J. Virol. 2011, 85, 8929–8939. [Google Scholar] [CrossRef]
- Karginov, F.V.; Cheloufi, S.; Chong, M.M.W.; Stark, A.; Smith, A.D.; Hannon, G.J. Diverse endonucleolytic cleavage sites in the mammalian transcriptome depend upon microRNAs, Drosha, and additional nucleases. Mol. Cell 2010, 38, 781–788. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-T.; Sullivan, C.S. Expanding the role of Drosha to the regulation of viral gene expression. Proc. Natl. Acad. Sci. USA 2011, 108, 11229–11234. [Google Scholar] [CrossRef] [PubMed]
- McNamara, R.P.; Chugh, P.E.; Bailey, A.; Costantini, L.M.; Ma, Z.; Bigi, R.; Cheves, A.; Eason, A.B.; Landis, J.T.; Host, K.M.; et al. Extracellular vesicles from Kaposi Sarcoma-associated herpesvirus lymphoma induce long-term endothelial cell reprogramming. PLoS Pathog. 2019, 15, e1007536. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Li, X.; Liang, D.; Lan, K. MicroRNAs and unusual small RNAs discovered in Kaposi’s sarcoma-associated herpesvirus virions. J. Virol. 2012, 86, 12717–12730. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.R.; Majerciak, V.; Kruhlak, M.J.; Yu, L.; Kang, J.G.; Yang, A.; Gu, S.; Fritzler, M.J.; Zheng, Z.-M. KSHV RNA-binding protein ORF57 inhibits P-body formation to promote viral multiplication by interaction with Ago2 and GW. Nucleic Acids Res. 2019, 47, 9368–9385. [Google Scholar] [CrossRef]
- Castle, E.L. Viral manipulation of a novel mechanoresponsive signaling axis disassembles processing bodies. bioRxiv 2020, 48. [Google Scholar] [CrossRef]
- Pitchiaya, S.; Mourao, M.D.A.; Jalihal, A.P.; Xiao, L.; Jiang, X.; Chinnaiyan, A.M.; Schnell, S.; Walter, N.G. Dynamic Recruitment of Single RNAs to Processing Bodies Depends on RNA Functionality. Mol. Cell 2019, 74, 521–533. [Google Scholar] [CrossRef]
- Covarrubias, S.; Richner, J.M.; Clyde, K.; Lee, Y.J.; Glaunsinger, B.A. Host shutoff is a conserved phenotype of gammaherpesvirus infection and is orchestrated exclusively from the cytoplasm. J. Virol. 2009, 83, 9554–9566. [Google Scholar] [CrossRef]
- Glaunsinger, B.A.; Ganem, D.E. Messenger RNA turnover and its regulation in herpesviral infection. Adv. Virus Res. 2006, 66, 337–394. [Google Scholar]
- Gaglia, M.M.; Rycroft, C.H.; Glaunsinger, B.A. Transcriptome-Wide Cleavage Site Mapping on Cellular mRNAs Reveals Features Underlying Sequence-Specific Cleavage by the Viral Ribonuclease SOX. PLoS Pathog. 2015, 11, e1005305. [Google Scholar] [CrossRef]
- Mendez, A.S.; Vogt, C.; Bohne, J.; Glaunsinger, B.A. Site specific target binding controls RNA cleavage efficiency by the Kaposi’s sarcoma-associated herpesvirus endonuclease SOX. Nucleic Acids Res. 2018, 46, 11968–11979. [Google Scholar] [CrossRef]
- Covarrubias, S.; Gaglia, M.M.; Kumar, G.R.; Wong, W.; Jackson, A.O.; Glaunsinger, B.A. Coordinated destruction of cellular messages in translation complexes by the gammaherpesvirus host shutoff factor and the mammalian exonuclease Xrn. PLoS Pathog. 2011, 7, e1002339. [Google Scholar] [CrossRef] [PubMed]
- Gilbertson, S.; Federspiel, J.D.; Hartenian, E.; Cristea, I.M.; Glaunsinger, B. Changes in mRNA abundance drive shuttling of RNA binding proteins, linking cytoplasmic RNA degradation to transcription. eLife 2018, 7, e37663. [Google Scholar] [CrossRef]
- Abernathy, E.; Gilbertson, S.; Alla, R.; Glaunsinger, B. Viral Nucleases Induce an mRNA Degradation-Transcription Feedback Loop in Mammalian Cells. Cell Host Microbe 2015, 18, 243–253. [Google Scholar] [CrossRef]
- Hartenian, E.; Gilbertson, S.; Federspiel, J.D.; Cristea, I.M.; Glaunsinger, B.A. RNA decay during gammaherpesvirus infection reduces RNA polymerase II occupancy of host promoters but spares viral promoters. PLoS Pathog. 2020, 16, e1008269. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, W.; Srivastav, K.; Muller, M. C19ORF66 Broadly Escapes Virus-Induced Endonuclease Cleavage and Restricts Kaposi’s Sarcoma-Associated Herpesvirus. J. Virol. 2019, 93, 9527. [Google Scholar] [CrossRef]
- Clyde, K.; Glaunsinger, B.A. Deep sequencing reveals direct targets of gammaherpesvirus-induced mRNA decay and suggests that multiple mechanisms govern cellular transcript escape. PLoS ONE 2011, 6, e19655. [Google Scholar] [CrossRef]
- Chandriani, S.; Ganem, D. Host transcript accumulation during lytic KSHV infection reveals several classes of host responses. PLoS ONE 2007, 2, e811. [Google Scholar] [CrossRef]
- Glaunsinger, B.; Ganem, D. Highly selective escape from KSHV-mediated host mRNA shutoff and its implications for viral pathogenesis. J. Exp. Med. 2004, 200, 391–398. [Google Scholar] [CrossRef]
- Muller, M.; Glaunsinger, B.A. Nuclease escape elements protect messenger RNA against cleavage by multiple viral endonucleases. PLoS Pathog. 2017, 13, e1006593. [Google Scholar] [CrossRef]
- Muller, M.; Hutin, S.; Marigold, O.; Li, K.H.; Burlingame, A.; Glaunsinger, B.A. A ribonucleoprotein complex protects the interleukin-6 mRNA from degradation by distinct herpesviral endonucleases. PLoS Pathog. 2015, 11, e1004899. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Macveigh-Fierro, D.; Rodriguez, W.; Miles, J.; Muller, M. Stealing the Show: KSHV Hijacks Host RNA Regulatory Pathways to Promote Infection. Viruses 2020, 12, 1024. https://doi.org/10.3390/v12091024
Macveigh-Fierro D, Rodriguez W, Miles J, Muller M. Stealing the Show: KSHV Hijacks Host RNA Regulatory Pathways to Promote Infection. Viruses. 2020; 12(9):1024. https://doi.org/10.3390/v12091024
Chicago/Turabian StyleMacveigh-Fierro, Daniel, William Rodriguez, Jacob Miles, and Mandy Muller. 2020. "Stealing the Show: KSHV Hijacks Host RNA Regulatory Pathways to Promote Infection" Viruses 12, no. 9: 1024. https://doi.org/10.3390/v12091024
APA StyleMacveigh-Fierro, D., Rodriguez, W., Miles, J., & Muller, M. (2020). Stealing the Show: KSHV Hijacks Host RNA Regulatory Pathways to Promote Infection. Viruses, 12(9), 1024. https://doi.org/10.3390/v12091024