Abstract
Approximately 10 percent of the mouse genome consists of endogenous retroviruses (ERVs), relics of ancient retroviral infections that are classified based on their relatedness to exogenous retroviral genera. Because of the ability of ERVs to retrotranspose, as well as their cis-acting regulatory potential due to functional elements located within the elements, mammalian ERVs are generally subject to epigenetic silencing by DNA methylation and repressive histone modifications. The mobilisation and expansion of ERV elements is strain-specific, leading to ERVs being highly polymorphic between inbred mouse strains, hinting at the possibility of the strain-specific regulation of ERVs. In this review, we describe the existing evidence of mouse strain-specific epigenetic control of ERVs and discuss the implications of differential ERV regulation on epigenetic inheritance models. We consider Krüppel-associated box domain (KRAB) zinc finger proteins as likely candidates for strain-specific ERV modifiers, drawing on insights gained from the study of the strain-specific behaviour of transgenes. We conclude by considering the coevolution of KRAB zinc finger proteins and actively transposing ERV elements, and highlight the importance of cross-strain studies in elucidating the mechanisms and consequences of strain-specific ERV regulation.
1. Introduction
Endogenous retroviruses (ERVs), a subclass of transposable element (TE), constitute approximately 10 percent of the mouse genome and arise either as a result of the successful integration of an ancient exogenous retrovirus (XRV) into the germline of the host or, more commonly, due to the retrotransposition of a previously integrated proviral sequence [1,2]. ERVs are classified based on the sequence of their reverse transcriptase gene and their relatedness to the seven XRV genera—Gamma- and Epsilonretrovirus; Alpha-, Beta-, and Deltaretrovirus; and Spumaretrovirus—into class I, II, and III ERVs, respectively [3,4,5,6]. In the mouse, class I ERVs include murine leukaemia viruses (MLVs), class II ERVs include early transposon/Mus musculus type D retrovirus (ETn/MusD) and intracisternal A-type particle (IAP) elements, and class III ERVs include mouse endogenous retrovirus type L (MERV-L) elements [5].
Full-length ERVs consist of 5′ and 3′ long terminal repeats (LTRs) that flank internal viral genes (gag, pol, and, in some elements, env) which are both essential and necessary for autonomous retrotransposition [7,8]. Non-autonomous elements, such as ETn elements, lack the reverse transcription and integrase machinery required for transposition and thus, mobilisation of these elements relies on trans-acting transposases encoded by other TEs. In fact, after IAPs, non-autonomous ETn elements are responsible for the second highest number of murine germline mutations of any transposon type and mobilise using the machinery of autonomous MusD elements [9,10]. Similarly, the non-autonomous IΔ1-type IAP, which has a 1.9kb deletion in gag-pol, accounts for the majority of IAP insertional mutations [11]. Whilst the majority of ERVs exist as solo LTRs which arise through inter-LTR homologous recombination, these fragmented elements still pose a significant threat to genomic integrity. This is due to the functional regulatory sequences contained within LTRs, as well as their mobilisation potential through “hijacking” the machinery of transposition-competent full-length elements [12]. In total, ERVs mainly of class I and II types are responsible for up to 12% of germline mutations in mice [11]. Given the mutagenic potential of ERVs, both full-length elements and solo LTRs are targeted for silencing via epigenetic mechanisms, as outlined in further detail below.
In inbred laboratory strains of mice, there has been a historic, and ongoing, expansion of ERV families, most notably of IAP and ETn/MusD elements, which are highly polymorphic between strains [13,14,15,16]. The susceptibility of the mouse genome to IAP mobilisation is strain-specific, with 84% of germline IAP mutations (for which a strain of origin for the TE mutation could be determined) occurring in the C3H genetic background [9]. Indeed, strain-specific diverse regions (SSDRs), which show a higher diversity between strains than is normally seen between mouse and rat and account for between 0.5% and 2.8% of the mouse genome, are enriched for recent long interspersed nuclear element (LINE) and LTR insertions [17,18]. While there is little evidence so far that strain-specific TE variants act as causal effectors of strain-specific gene expression changes or quantitative trait loci (QTL), it is perhaps notable that intronic TE variants are more frequently associated with differentially expressed genes than would be expected by chance [15].
The term “modifier gene” defines genetic variants which alter the phenotypic outcome of an independent locus, but which have no phenotypic consequence of their own [19,20]. Strain-specific morphological, physiological, and behavioural differences are well recognised, but the mechanisms underlying inter-strain variation and the causative modifier loci remain largely uncharacterised due to technical and practical limitations.
In this review, we discuss the key players in the silencing of ERVs, the existing evidence for the mouse strain-specific epigenetic control of ERVs, and the implications of differential ERV regulation on epigenetic inheritance. We reflect on lessons learned from the strain-specific behaviour of transgenes and discuss the potential mechanisms by which the strain-specific epigenetic silencing of ERVs is likely to occur.
2. Epigenetic Regulation of ERVs
Specific ERV classes are silenced by distinct epigenetic mechanisms in the early embryo and embryonic stem cells (ESCs). In embryonic cells, class I and II ERVs are enriched for H3K9me3, a mark deposited by the histone methyltransferase SETDB1 [21,22]. These ERVs are targeted for silencing in a sequence-dependent manner by Krüppel-associated box domain zinc-finger proteins (KRAB-ZFPs), which make up a large family of DNA binding proteins whose sequence specificity is determined through their C-terminal zinc finger arrays [23,24]. Through their KRAB domain, KRAB-ZFPs recruit KRAB-associated protein 1 (KAP1), which acts as a scaffold for other components of the transcriptional silencing machinery, including SETDB1, HP1 (heterochromatin protein 1), NuRD/HDAC (nucleosome remodelling and deacetylase complex), and DNA methyltransferases [25,26,27]. For a more detailed description of KRAB-ZFP structure and function, we refer the reader to the following comprehensive reviews [27,28]. In mouse ESCs and primordial germ cells (PGCs), whilst DNA methylation is dispensable for ERV silencing as evident from bulk ERV type analysis, knocking out SETDB1 or KAP1 results in the upregulation of several class I and II ERVs [21,22,29,30]. In contrast, DNA methylation is essential for ERV silencing in differentiated cell types and later embryonic time points, though the extent of this is not known [31,32,33]. The silencing mechanisms for class III ERVs are less clear; in ESCs, class III ERVs are largely devoid of H3K9me3 except at MERV-Ls, whose silencing and H3K9me2/3 deposition is dependent on G9a/GLP activity [34,35]. A role for the lysine-specific histone demethylase LSD1/KDM1A in MERV-L silencing in early embryos and ESCs has been proposed, but is less well defined [36,37].
In the mouse, as in other mammals, there are tightly regulated periods of epigenetic reprogramming whereby epigenetic modifications are globally erased and developmental potency is re-established. This occurs immediately post fertilisation in the zygote and in PGCs and is generally recapitulated in ESCs in vitro [38]. During development, the genome-wide removal of DNA methylation results in the transient transcriptional activation of certain classes of ERVs, notably class III MERV-Ls, which play a role in zygotic genome activation (ZGA) at the 2-cell stage in mice [39,40,41]. Retrotransposition inhibition and the transcriptional control of other classes of ERVs is maintained during epigenetic reprogramming implicating additional silencing pathways [42,43,44,45,46]. Other epigenetic mechanisms associated with ERV silencing involve 3′ tRNA fragments [47], piRNA pathways [48], and histone variants [49,50,51,52].
3. Evidence of Strain-Specific ERV Control
The high prevalence of polymorphic ERVs raises many questions about the similarities and differences in transposon regulation between strains. The strain-specific expansion of ERVs in inbred mice hints at the possibility of strain-specific ERV silencing, or lack thereof, and subsequent mobilisation. Thus far, no unbiased or genome-wide screens to assess strain-specific ERV control have been carried out. The few documented instances of strain-specific modifiers were identifiable due to obvious and observable phenotypic differences. These are discussed in more detail below.
3.1. Dactylaplasia-Causing MusD Insertions at Fbxw4
Dactylaplasia is an inherited limb malformation which manifests as the absence of phalangeal bones in the middle digits of each foot in mice. The first identified mutation (Dac1j) arose in the SM7B/SC inbred strain and was found to be a homozygous lethal dominant allele [53]; a second dactylaplasia-causing mutation (Dac2j) was reported several years later on the MRL/MpJ genetic background [54]. Fine mapping and sequencing experiments established that both Dac1j and Dac2j are due to independent, full length, highly similar (99.6% identical), and polymorphic MusD element insertions, which lie either 10kb upstream (Dac1j) or within an intron (Dac2j) of the Fbxw4 gene locus, a member of the F-box/WD40 gene family involved in protein ubiquitination and degradation [54,55,56,57] (Figure 1A, top and middle panel). The mechanism by which these MusD insertions cause dactylaplasia remains unknown [56,57].
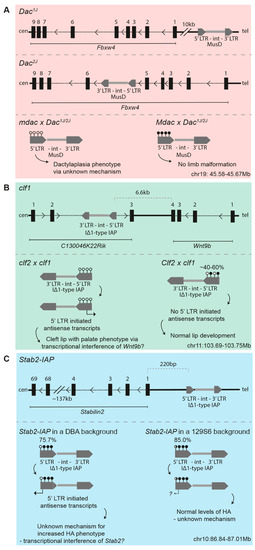
Figure 1.
Schematics for the insertion sites of endogenous retroviruses (ERVs) subject to strain-specific regulation (upper panel) and the effects of strain-specific modifier activity (lower panel) for (A) Dac1j and Dac2j, (B) clf1, and (C) Stab2-IAP. Sticks with closed circles represent methylated CpGs in the long terminal repeat (LTR) of the ERV; sticks with open circles represent unmethylated CpGs. Black dotted lines depict introns; thick black lines depict intergenic regions. The information is based on the latest patch release of the 2011 mouse assembly on the UCSC Genome Browser (GRCm38.p6); the coordinates given are mm10.
The effects of both Dac1j and Dac2j were found to be modified by an unlinked allele Mdac (modifier of Dac), which resulted in highly polymorphic phenotypes between inbred mouse strains [53,54,55]. The strains carrying the Mdac allele are hypermethylated and enriched for H3K9me3 at the 5′ LTRs of Dac1j and Dac2j, resulting in the loss of aberrant MusD expression at the apical ectodermal ridge in the limb bud seen in mdac strains and enabling normal limb development to occur [56] (Figure 1A, bottom panel). The Mdac locus was first mapped to a 28Mb interval on Chromosome 13 and was later refined to a 9.4Mb region containing 125 genes, including many known to be important for limb development—e.g., Ror2, Msx2, Fgfr4, and Patched [55,56] (Figure 2). This 9.4Mb region contains a KRAB-ZFP cluster of six KRAB-ZFP genes which, given their known role in ERV epigenetic targeting, are possible Mdac candidates [58]. It is worth noting, however, that a recent study where this KRAB-ZFP cluster (Chr13.1-cl KO) was deleted did not lead to a global upregulation of MusD elements in ESCs [59]. This is in line with the finding that a control MusD element was not differentially regulated according to the mdac/Mdac genotype [56].

Figure 2.
The mapped intervals of four strain-specific ERV modifiers—Mdac, Clf2, Snerv1 and Snerv2 and Stab2-modifier—on Chromosome 13. The underlying genes are separated into Krüppel-associated box domain zinc-finger protein (KRAB-ZFP) genes and non-KRAB-ZFP genes; the KRAB-ZFP clusters are highlighted and named as in [59]. Multiple isoforms are not shown.
3.2. Cleft Lip Palate-Causing IAP Insertions at Wnt9b
Nonsyndromic cleft lip with palate (CL/P) defects arise when the medial and frontal nasal prominences fail to fuse; CL/P causes neonatal lethality in mice, as the pups are unable to suckle. CL/P spontaneously occurs with a 20–30% frequency in the inbred mouse strain A/WySn, making it a commonly used model to study clefting [60]. Similarly to the dactylaplasia phenotype, the CL/P phenotype in A/WySn mice involves two unlinked genes, a recessive mutation (clf1), and a second semi-dominant modifier locus (Clf2) [61]. The clf1 mutation was identified as an IAP element insertion into a ncRNA, C130046K22Rik, located 6.6kb downstream of Wnt9b [62,63] (Figure 1B, upper panel). The insertion is only present in “A” strains and is a IΔ1-type IAP, the same type responsible for the Agouti viable yellow (Avy) and Axin-fused (AxinFu) metastable epialleles (discussed below). Wnt9b has been implicated in clefting previously: Wnt9b null embryos have deficient growth of the facial prominences, resulting in CL/P, which possibly manifests via the downregulation of Fibroblast Growth Factor (FGF) signalling in these mutants [64].
In A/WySn embryos with CL/P, the 5′ LTR of the clf1 IAP element is unmethylated [63,65,66,67] (Figure 1B, lower panel). 5′ LTR initiated antisense IAP transcripts and reduced Wnt9b levels are detected in CL/P A/WySn embryos compared to phenotypically normal A/WySn embryos, but the mechanism by which this occurs is unknown [63,67]. On a C57BL/6J (B6J) genetic background with the Clf2 modifier, the clf1 IAP element is more highly methylated, no IAP transcripts are detectable, and Wnt9b expression is normal [66]. As assessed by Combined Bisulfite and Restriction Analysis (COBRA), phenotypically normal A/WySn embryos appear to exhibit variable DNA methylation at the 5′ LTR of the clf1 IAP element. This suggests this IAP is a metastable epiallele and it was thus redefined as Wnt9bIAP by Juriloff et al. [63].
Once again, the modifier responsible for the strain-specific methylation of the CL/P-inducing IAP, Clf2, has not yet been identified but has been mapped to a 3Mb region on Chromosome 13; this region contains 48 genes and includes a known KRAB-ZFP cluster of more than 30 KRAB-ZFPs [58,66] (Figure 2). Many of the KRAB-ZFP genes in this cluster contain divergent non-synonymous single nucleotide polymorphisms (SNPs) between the B6J and A/WySn strains [28,66]. Though both are on Chromosome 13, this KRAB-ZFP cluster is distinct from that identified in the mapping experiments of the Mdac candidate.
3.3. Non-Ecotropic ERV Activation Links to Lupus
The mouse strains commonly used as models for human systemic lupus erythematosus (SLE)—New Zealand Black (NZB), New Zealand White (NZW), and 129—have high gene expression and protein levels of non-ecotropic ERV (NEERV) envelope glycoprotein gp70, concomitant with nephritis. However, a causative link between NEERV dysregulation and lupus pathology has not been established, and the mechanism for NEERV dysregulation is unknown. Previously, independent QTL analyses in the NZB/NZW and 129 strains mapped the loci (Sgp3 in NZB and Gv-1 in 129) responsible for the gp70 autoantigen expression to large intervals on Chromosome 13 [68,69].
A recent comparative RNA-seq study between B6J and C57BL/6N (B6N) found that the majority of differentially expressed loci between these two sub-strains were NEERVs; the ERV envelope protein and NEERV gene expression were significantly higher in B6N compared to B6J [70]. F1 hybrid mice showed low NEERV gene expression and ERV envelope protein levels that were comparable to B6J mice, indicating the presence of a dominant NEERV repressor in the B6J sub-strain. A QTL analysis revealed a single QTL locus on Chromosome 13 responsible for NEERV dysregulation; an inter-strain comparison and copy number analysis revealed a 1Mb deletion specific to B6N in the mapped interval. This 1Mb region in B6J contains two genes, four non-coding RNAs, and four pseudogenes (Figure 2). The knockouts of the two genes in this interval on a B6J genetic background phenocopied the NEERV dysregulation seen in the B6N strain. The two genes are the KRAB-ZFP genes 2410141K09Rik and Gm10324, renamed suppressor of NEERV (Snerv) 1 and 2.
The previously mapped intervals for Sgp3 and Gv-1 in NZB and 129, respectively, include Snerv1 and Snerv2. Additionally, the 1Mb deletion in B6N also appears to be deleted in NZB and 129, indicating that the previously mapped Sgp3 and Gv-1 loci may be the same modifiers as Snerv1 and 2. Complementation experiments found that hybrid F1 mice (Snerv1/2-/- X NZB/129) were unable to rescue the NEERV repression phenotype, indicating that the strain-specific absence of these KRAB-ZFPs, previously identified as Sgp3 in NZB and Gv-1 in 129, may drive the NEERV dysregulation and lupus pathology in lupus-prone strains, NZB and 129.
3.4. IAP-Driven Stabilin2 Expression in DBA/2J Mice
Stabilin2 (Stab2) encodes a type I transmembrane receptor which functions via clathrin-mediated endocytosis as a scavenger receptor for hyaluronans (HA), heparin, and pro-collagen peptides, amongst other macromolecules [71,72]. The main phenotype of Stab2 null mice, which were generated on B6J and BALB/cJ genetic backgrounds, is a 10-fold higher plasma HA concentration compared to wild type [73,74]. Independently, one study reported that wild-type DBA/2J (DBA) mice have a more than 10-fold higher plasma HA concentration than 129S6 or B6J mice, a phenotype which was mapped to the Stab2 locus [75]. Recently, it was shown that a 5.5kb IΔ1-type IAP element inserted 220bp upstream of the canonical Stab2 transcription start site (TSS), providing an alternative TSS which drives the ectopic expression of Stab2 [76] (Figure 1c, upper panel). The IAP element (Stab2-IAP), alternative TSS, and ectopic Stab2 expression are unique to the DBA genetic background.
In B6J x DBA (BxD) F1 hybrid heart tissue, IAP-driven Stab2 expression is completely abrogated, indicative of a single dominant modifier in the B6J strain targeting the Stab2-IAP for silencing and preventing aberrant transcription. A congenic line homozygous for the DBA-specific Stab2-IAP in an otherwise 129S6/Sv (129S6) genetic background exhibited Stab2 expression levels that were significantly reduced compared to DBA but that were higher than a pure 129S6 genetic background. The lack of the complete transcriptional repression of the IAP-driven transcripts suggests that an additional locus or loci are responsible for targeting the DBA-specific IAP on the 129S6 genetic background. The 5′ LTR of the Stab2-IAP is highly methylated on the DBA and 129S6 genetic background, as assessed by clonal bisulfite sequencing (75.7% vs. 85.0%, respectively), indicating that a mechanism besides DNA methylation may also be involved in the strain-specific behaviour of this IAP element [76] (Figure 1C, lower panel). The methylation status of the Stab2-IAP was not assessed on a B6J background. The small increase in DNA methylation at the Stab2-IAP on a 129S6 genetic background may be a secondary consequence of other repressive epigenetic modifications, such as increased H3K9me3, which prevent ectopic transcription initiating from the LTR. Besides DNA methylation, additional epigenetic modifications at the Stab2-IAP were not assayed.
Utilising the BxD recombinant inbred lines and gene expression data from the Hybrid Diversity Panel, the most prominent trans expression QTL (eQTL) locus was mapped to Chromosome 13 and refined to 59.7–73Mb [76] (Figure 2). The region overlaps the modifiers (and KRAB-ZFP clusters) mapped for Mdac (56–65 Mb), Clf2 (64.95 -67.9 Mb) and Snerv1/2 (65.66–66.7 Mb). In the other three examples of strain-specific regulation discussed so far, the modifiers appear to be single dominant loci. This seems to hold true for the Stab2-IAP in a B6J F1 background. However, on a 129S6 F1 background, the Stab2-IAP is not fully repressed, as assessed by the elevated Stab2 expression and the methylation status of the 5′ LTR. In this regard, the strain-specific modifier acting on Stab2-IAP is particularly interesting, as it exhibits both strain-specific absence/presence polymorphism (B6J and 129S6 vs. DBA) and a strain-specific mode of action (B6J vs. 129S6). It is worth pointing out that when a single locus is mapped, there may be multiple modifiers that are always inherited together capable of recognising the target ERV, making it seem like the Mendelian segregation of a single gene. This is especially pertinent given that all of the mapped intervals contain KRAB-ZFP clusters, which are known to expand through segmental duplication, resulting in individual KRAB-ZFPs with highly redundant roles [59].
3.5. Epigenetic Inheritance of Metastable Epialleles, Avy and AxinFu
Metastable epialleles are regions of the genome which display variable epigenetic states between genetically identical individuals [77,78]. The most comprehensively studied metastable epialleles to date, Avy and AxinFu, result from the variable silencing of IAP element insertions upstream of, or within, endogenous gene loci, and were identified due to observable phenotypic differences between littermates [79,80,81,82,83]. Whilst no strain-specific modifiers have been identified that specifically act on the IAP elements responsible for the Avy or AxinFu alleles, it is clear that genetic background influences the heritability of these loci as well as the susceptibility of these alleles to environmental stimuli, two features for which these loci are particularly well known.
The Avy-causing IAP insertion first arose spontaneously on a C3H/HeJ (C3H) genetic background in pseudoexon 1A of the coat colour gene Agouti, 100kb upstream of the coding exons [79,84] (Figure 3A, upper panel). In wild-type mice, the Agouti expression is regulated by a hair cycle-specific promoter; transient expression at the beginning of each hair follicle cycle results in a sub-apical yellow band on an otherwise black hair follicle and the “agouti” coat pattern. In Avy mice, transcription originating from a cryptic promoter within the LTR of the IΔ1-type IAP element drives the ectopic expression of Agouti (Figure 3A, lower panel). The excess paracrine signalling molecule causes hair follicle melanocytes to constitutively synthesise yellow pigment (phaeomelanin), resulting in mice with yellow coats as well as adult-onset obesity, diabetes, and an increased susceptibility to tumours [85,86]. Isogenic individuals have variable DNA methylation at the IAP element, which inversely correlates with ectopic Agouti expression levels: highly-methylated individuals retain endogenous levels of expression and are indistinguishable from wild type (termed pseudoagouti); lowly-methylated individuals have high levels of ectopic expression and have a yellow coat, diabetes, and obesity; intermediately methylated individuals have an intermediate mottled coat-colour phenotype [80,84] (Figure 3A, lower panel).
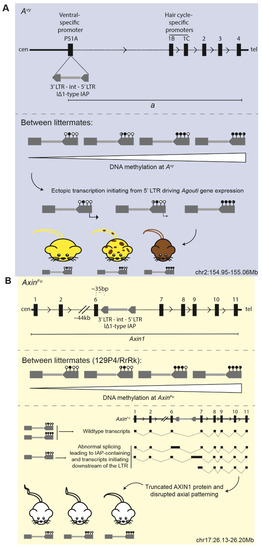
Figure 3.
Schematics depicting the insertion sites of the metastable epialleles (A) Avy and (B) AxinFu. Upper panel shows the intracisternal A-type particle (IAP) insertion relative to the affected gene; lower panel shows the functional consequence of the variably methylated IAP element. Coordinates are mm10. Sticks with closed circles represent methylated CpGs in the LTR of the ERV; sticks with open circles represent unmethylated CpGs. Black dotted lines depict introns; thick black lines depict intergenic regions.
Similarly, the AxinFu allele resulted from a spontaneous IΔ1-type IAP element insertion in the sixth intron of Axin1 in the Bussey Institution stock of mixed genetic backgrounds [81,82] (Figure 3B, upper panel). At a low rate, the intronic IAP causes aberrant splicing, which results in both wild-type and mutant transcripts which contain the IAP; the inclusion of the AxinFu IAP in the mRNA is predicted to generate a truncated AXIN1 protein [82] (Figure 3B, lower panel). Transcripts initiating within 100 bp downstream of the 3′ LTR of the AxinFu IAP have also been detected and may result in truncated peptides consisting of intron 6 and exons 7–10 [83]. The resultant kinked tail phenotype is attributed to the abnormal development of the posterior somites and axial duplications, which leads to vertebral fusions via atypical Wnt signalling [82,83]. Among isogenic individuals, tails range from kinked to completely normal, with the phenotypic severity inversely correlating with the DNA methylation status of the intronic AxinFu IAP element [83] (Figure 3B, middle and lower panels).
Hybrid experiments to assess whether the Avy mutation was pleiotropic provided the first evidence that the Avy IAP was sensitive to genetic background; 12% of the B6JxVY-Avy hybrids were pseudoagouti, compared to 58% of the AKR/LwNIcr (AKR)xVY-Avy hybrids [87] (Figure 4A, lower panel). This may be indicative of an AKR-specific modifier(s) that more robustly recognises the Avy IAP for silencing and “pushes” the offspring towards the pseudoagouti end of the phenotypic spectrum. Subsequent studies showed that the phenotypic distribution and physiological outcomes of Avy mice shifted dependent on genetic background [86,88,89,90,91]; similar findings have been reported for the AxinFu locus [92,93]. In addition, the phenotypic shifts in the offspring of Avy dams subjected to methyl-supplemented diets differ depending on the genetic background of the dam, likely reflecting differences in methyl metabolism between strains [90,91]. Figure 4A summarises the breeding experiments on Avy conducted by Wolff spanning almost 30 years.
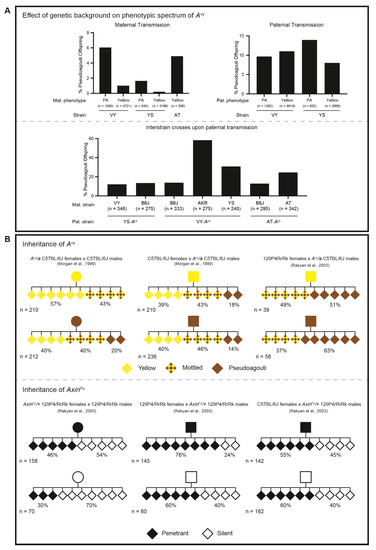
Figure 4.
The effect of genetic background on the phenotypic spectrum and inheritance of Avy upon maternal or paternal transmission. (A—upper) Maternal and paternal transmission of the Avy allele on VY/Wf (VY), YS/ChWf (YS), or AT/Wf (AT) genetic backgrounds. The percentage of pseudoagouti (PA) offspring depends upon the maternal coat colour phenotype, but not the paternal coat colour phenotype. Paternal transmission of Avy results in a higher percentage of PA offspring than the maternal transmission of Avy. Both maternally and paternally transmitted alleles are sensitive to genetic background effects, albeit in different ways. (A—lower) Paternal transmission of Avy is largely influenced by the genetic background of the dam. Paternal coat colour information is not included, as it is not available for all of the inter-strain crosses. B6J = C57BL/6J; AKR = AKR/LwNIcr. The data shown in the upper and lower panel are combined (based on the genotype and parent-of-origin) and adapted from [87,89,90]. (B—upper) On a C57BL/6J background, the maternal coat colour phenotype influences the coat colour of the Avy/a offspring, but the paternal coat colour phenotype has no effect on the phenotypic distribution of the offspring. When the Avy allele is paternally inherited through a 129P4/RrRk-fertilised oocyte, the paternal coat colour phenotype influences the coat colour of the Avy/a offspring; the percentage of PA and mottled is increased after passage through a 129P4/RrRk-fertilised oocyte compared to a C57BL/6J-fertilised oocyte. (B—lower) On a 129P4/RrRk background, the maternal and paternal tail kink phenotype influences the tail kink phenotype in the AxinFu/+ offspring. The phenotypic distributions are different upon the maternal versus paternal transmission of the allele. When the AxinFu allele is paternally inherited through a C57BL/6J-fertilised oocyte, the tail kink phenotype of the sire has no bearing on the phenotypic distribution in the AxinFu/+ offspring. The data shown in the upper and lower panel are adapted from [80,83]. Pedigrees: circle—female; square—male; diamond—unspecified.
Both the Avy and AxinFu alleles display strain-specific epigenetic inheritance across generations (Figure 4B). On a B6J genetic background, Avy displays partial inheritance when transmitted maternally, whilst the AxinFu allele on a 129P4/RfRk (129P4) genetic background is inherited upon both maternal and paternal transmission [80,83]. Interestingly, Avy also displays paternal inheritance when Avy/a B6J males are crossed with AxinFu/+ 129P4 females (Figure 4B, upper panel). Conversely, AxinFu is no longer paternally transmitted when AxinFu/+ 129P4 males are crossed with Avy/a B6J females, indicating that the inheritance of the AxinFu allele is strain-specific, rather than an intrinsic property of the locus [83] (Figure 4B, lower panel). These findings suggest that paternally inherited alleles are subject to the full erasure of DNA methylation during epigenetic reprogramming by B6J-fertilised oocytes, but not 129P4-fertilised oocytes. Indeed, immediately following fertilisation and in line with the rest of the genome, the paternal Avy allele undergoes rapid demethylation, whereas the maternal Avy allele does not in the B6J strain [94,95,96]. However, importantly, both the paternal and maternal alleles exhibit a comparable absence of DNA methylation at Avy at the blastocyst stage, which suggests that DNA methylation is not the inherited epigenetic mark [96].
Our group recently performed a genome-wide systematic screen of IAP elements in B6J to identify novel metastable epialleles, which we termed variably methylated IAP elements (VM-IAPs) [97]. Although VM-IAPs are not commonly associated with transcriptional changes, nor do they retain a memory of the parental methylation level in the offspring, we do find that VM-IAPs are sensitive to genetic background and parent-of-origin effects, in line with the Avy and AxinFu alleles [98]. Given that VM-IAPs are naturally occurring alleles that are polymorphic between strains, they represent an attractive model in which to study the modifiers and mechanisms involved in the strain-specific epigenetic regulation of IAP elements. Taken together, in depth cross-strain analyses on Avy, AxinFu, and VM-IAPs are likely to provide mechanistic insight into the establishment and maintenance of these unique loci.
4. Lessons from Transgenes
The similarities between metastable epialleles and the epigenetic targeting of transgenes have been highlighted previously [77,78,99,100,101,102]. Methylated transgenes and endogenous metastable epialleles are loci with varying degrees of DNA methylation that can exhibit parent-of-origin effects upon transmission. In addition, the genetic background in which transgenes and metastable epialleles are studied affects their methylation state and heritability, suggesting that strain-specific modifiers are acting on these loci.
The strain-specific silencing of ERVs is particularly reminiscent of the decades-spanning work on a transgene designed to study the rearrangement of immunoglobulin genes in the Storb lab. The transgene was designed as a marker of V-J recombination: the construct contains a mouse metallothionein-1 promoter upstream of the bacterial xanthine/guanine phosphoribosyltransferase (gpt) gene, whose translation is dependent on V-J rearrangement to form an upstream in-frame initiation codon [103]. The transgene was named HRD (heavy chain enhancer, rearrangement by deletion), and was found to recombine 100% of the time in a transfected pre-B cell line [103,104]. When maintained on the DBA genetic background, the HRD transgene is unmethylated. Upon crossing the founder transgenic mice to B6J, the HRD transgene becomes highly methylated and no longer undergoes V-J recombination in the lymphoid organs of the offspring [105]. The further crossing of B6J mice with the HRD transgene to DBA or SJL/J (SJL) mice resulted in offspring with a methylated HRD transgene. Crossing these F1 hybrids (B6Jx DBA or SJL) back to DBA or SJL mice resulted in offspring (N1) with an unmethylated, partially methylated, or methylated HRD transgene, suggestive of a dominant B6J modifier that had been lost in some of the backcrossed N1 individuals. The single dominant B6J allele responsible for HRD transgene methylation, Strain-specific modifier 1 in C57BL/6J (Ssm1b), was found to be concordant with the B allele of Fv-1, a resistance allele to the Friend leukaemia virus, which mapped Ssm1b to Chromosome 4 [105]. Ssm1b was later fine-mapped to a 0.5Mb interval on Chromosome 4, a region containing ~12 genes, six of which are KRAB-ZFPs [106]. Several overlapping bacterial artificial chromosomes (BACs) covering the mapped region were injected into fertilised eggs carrying the HRD transgene on an unmethylated genetic background (C3H x DBA hybrids). Two of the BACs containing a single gene in common resulted in the methylation of the HRD transgene, enabling the identification of Ssm1b as Zfp979 (NCBI designation is 2610305D13Rik) [106].
Zfp979 has a KRAB-A box and three functional C2H2 zinc fingers interspersed with three non-functional zinc fingers. It resides in a cluster which contains 20 other KRAB-ZFPs on Chromosome 4. Zfp979 is widely expressed during embryonic development until embryonic day 8.5 (E8.5). Whilst ESCs on a DBA background have 0% DNA methylation at the HRD transgene, B6J ESCs have ~40% DNA methylation, indicating that the methylation of the HRD transgene likely occurs peri-implantation, coincidental with the rest of the genome [107]. In a B6J or hybrid BxD background, methylation at the HRD transgene increases from 40% to almost 100% between E8.5 and E9.5 and relies on the de novo methyltransferase DNMT3B [106]. The direct binding of ZFP979 to the HRD transgene and the existence of a ZFP979:KAP1 interaction have not yet been shown. A recent analysis of KRAB-ZFP clusters established that ZFP979/2610305D13Rik binds to IAPEY-int elements [59].
Ssm1b was originally identified due to the differential methylation of the HRD transgene in B6J (methylated) and DBA (unmethylated) genetic backgrounds. In total, when maintained on an F1 background with DBA or SJL, the HRD transgene is methylated in seven strains of mice (C57BL/6J, FVB/NJ, C57L/J, LP/J, 129/SvJ, BALB/cJ, and A/J) and unmethylated in six strains of mice (DBA/2J, C3H/HeJ, SJL/J, CBA/J, SM/J, and AKR/J) [108]. The reference genomes for 16 strains were recently generated but there remain significant gaps in this region, likely due to the repetitive nature of the KRAB-ZFPs within this cluster, which are predicted to have arisen through segmental gene duplications [17,59,109]. This makes inter-strain sequence alignments of Zfp979 currently impossible. The generation of new reference genomes using long-read sequencing technologies will alleviate these issues and enable inter-strain comparisons at KRAB-ZFP clusters and other repeat-dense regions in the future.
Indeed, the variable epigenetic state of both transgenes and metastable epialleles has been utilised to screen for modifiers involved in their epigenetic targeting: large-scale N-ethyl-N-nitrosourea (ENU) mutagenesis screens have identified dominant and recessive genes capable of modifying GFP transgene variability in mice [102,110,111,112,113,114,115,116]. The hits from these screens have been named Modifiers of Murine Metastable Epialleles (Mommes), and the effect of some of these mutants on the phenotypic spectrum of Avy has been assessed [111,112]. Many of the candidates from the screen are known components of the ERV epigenetic silencing pathway, but it is not yet known if, and to what extent, these modifiers act in a strain-specific manner. In this regard, a screen focussing on the strain-specific modifiers of ERV silencing would be timely.
5. KRAB-ZFPs as Effectors of Strain-Specific ERV Regulation
Thus far, the most compelling examples of strain-specific ERV regulation involve IΔ1-type IAP elements, with the exception of the dactylaplasia-causing MusD insertion at Fbxw4. This is perhaps unsurprising, given the strain-specific expansions of IAP and MusD/ETn elements [9,11,13,15]. Whilst the two IΔ1-type IAP elements whose modifiers map to overlapping regions, clf1 and Stab2-IAP, are permissive in different strains, they are both targeted for silencing on a B6J genetic background, raising the possibility that their respective modifiers, Clf2 and Stab2-modifier, may in fact be the same (Table 1). As these strain-specific modifiers have been discovered due to observable phenotypes rather than specially designed screens, only dominant-acting alleles have been detected so far. Aside from the NEERV effectors, 2410141K09Rik and Gm10324, specific modifiers for the other ERVs subject to strain-specific regulation are yet to be identified. It is worth noting that many of the modifiers linked to strain-specific ERV regulation reside on Chromosome 13 and overlap known KRAB-ZFP clusters, one of which includes KRAB-ZFPs Rsl1 and Rsl2 (Figure 2 and Table 1) [66,70,76,117]. Rsl1 and Rsl2 are strain-specific KRAB-ZFPs that regulate sexually dimorphic gene expression in the liver. One of the target genes of Rsl1, sex-limited protein (Slp), lies 2kb downstream of an ancient ERV [118,119]. In KAP1 knockout livers, there is an upregulation of Rsl1/Rsl2-target cytochrome P450 genes, implicating the KRAB-ZFP/KAP1 pathway, with its already established role in ERV silencing, in the functional mechanism of RSL1 and RSL2 [27,120]. The KRAB-ZFP cluster (Chr13.2-cl) containing Rsl1 and Rsl2 has previously been identified as being highly variable between mouse strains [121]. It is possible that certain clusters contain KRAB-ZFPs with particularly rapid evolution in response to the amplification of active ERV elements, making them more polymorphic across mouse strains than other clusters. This may explain the strain-specific modifiers mapping predominantly to Chr13.2-cl, as well as the Ssm1b cluster, Chr4-cl.

Table 1.
Overview of mapped strain-specific modifiers.
KRAB-ZFPs are attractive candidates for strain-specific modifiers of ERVs due to the sequence specificity endowed by their C-terminal ZFPs, which are under strong positive selection [123,124,125]. Indeed, it is worth noting that many of the ERVs subject to strain-specific regulation are the same type of TE (IΔ1-type IAP: clf1, Stab2-IAP, Avy, and AxinFu), providing support for the role of sequence in the strain-specific epigenetic targeting of these elements. Furthermore, the positive correlation that exists between the number of LTR retrotransposons and the number of zinc finger domains across mammalian species is indicative of concurrent waves of KRAB-ZFP and TE expansion [124,125]. The varying KRAB-ZFP gene content between strains, and even sub-strains of mice with less than 75 years divergence in the case of B6N and B6J, may underlie differing ERV activity and epigenetic regulation between strains of inbred laboratory mice [17,70,126,127]. In particular, the C3H strain is particularly susceptible to IAP mobilisation, and it is tempting to speculate that this may be explained by a KRAB-ZFP cluster or gene deletion, as has occurred in the B6N, 129, and NZB strains, causing NEERV dysregulation [9,70]. The coexistence of strain-specific ERVs and strain-specific KRAB-ZFPs provides intriguing complexity to the KRAB-ZFP/TE coevolution debate in light of the non-mutually exclusive “arms race” and “domestication” models [27,28,128]. Future attempts to functionally characterise strain-specific KRAB-ZFPs in detail may be difficult. In addition to the high prevalence of gaps currently in the reference genomes at KRAB-ZFP clusters and repetitive regions, the high level of redundancy in the KRAB-ZFP-targeting of ERVs will make the identification and validation of candidate modifiers challenging [23,125,129]. This high level of redundancy may explain why KRAB-ZFPs were not identified in the Mommes mutagenesis screen [102,116].
It is not yet clear whether modifier loci are required for the establishment or maintenance of strain-specific epigenetic states at ERVs and transgenes. The identification and emphasis of KRAB-ZFPs as strain-specific modifiers so far in this review suggests the focus may be on the strain-specific “establishment” of an epigenetic state, although some KRAB-ZFPs are known to play a protective role maintaining germline-derived DNA methylation marks during early embryonic development, notably ZFP57 and ZFP445 at imprinted loci [32,130,131,132]. Whilst 75% of ZFP57 binding sites are located in ERVs, the loss of this protein does not affect H3K9me3 deposition or DNA methylation at ERVs, nor does it lead to the loss of transcriptional repression of ERVs in ESCs [130,131,133]. This perhaps reflects the highly redundant nature of KRAB-ZFP-mediated ERV repression, or it may suggest that ZFP57 does not play a role at these TEs. Interestingly, instances of strain-specific ZFP57 binding have been reported previously, conferred by genetic variation either in the ZFP57 binding motif itself or in neighbouring regions between strains, causing strain-specific differential methylation and subsequent ZFP57 binding [131]. Recently, the strain-specific loss of imprints in ESCs (129 v B6J) was mapped by QTL analysis to an interval spanning 52Mb-67.7Mb on Chromosome 13, overlapping entirely or partially with the Mdac, Clf2, Snerv1/2, and Stab2-modifier loci [134]. Needless to say, the establishment of a strain-specific epigenetic state may occur via mechanisms outside of the KRAB-ZFP targeting pathway. Whilst our understanding of these processes is limited, it is possible that strain-specific epigenetic states could arise through the strain-specific protection (by KRAB-ZFPs or other proteins) or the strain-specific removal of epigenetic modifications during epigenetic reprogramming or at other time points in development.
6. Concluding Remarks
It is well established that the genetic background of mouse models can result in large phenotypic differences not attributable directly to the phenotype-associated genetic locus itself. Historically, these strain differences have been largely overlooked due to the technical challenges associated with identifying the underlying modifier genes. This has resulted in specific strains of mice being used in the study of certain traits, as is the case with the “A” strain mice and clefting. We note that while attention to genetic background as a variable across experiments is necessary to ensure experimental reproducibility as well as cross-strain, and potentially cross-species, generalisability, we hope that here we have emphasised the biological and mechanistic value of cross-strain experiments. Currently, the annotations of KRAB-ZFP clusters are poor even in the C57BL/6J reference genome. Advances in long-read sequencing technologies and the resultant high-quality mouse strain reference genomes with full coverage over KRAB-ZFP clusters and their target TEs will be required to enable large-scale inter-strain experiments and provide further mechanistic insight into the complex relationship between repetitive elements, the KRAB-ZFP machinery, and their coevolution.
The strain-specific modifiers outlined in this review were identified due to observable phenotypic differences between permissive and repressive strains. The extent to which polymorphic ERVs, and indeed polymorphic ERV regulation, act as drivers of phenotypic variation between inbred mouse strains remains unclear. However, it is important to note that inbred laboratory mouse strains suffer from severe inbreeding depression, which may put an unusual strain on the host defence mechanisms against TE mobilisation. Experiments using wild-derived mice alongside laboratory strains would help elucidate whether these effects are reflective of host-TE dynamics in natural populations. Additionally, the studies discussed in this review serve as an important reminder that seemingly complicated epigenetic phenomena are sometimes explained by underlying genetic differences, highlighting the mutual dependence and interrelatedness of genetic and epigenetic pathways.
Funding
This work was supported by grants from the MRC (MR/R009791/1), BBSRC (BB/R009996/1), and a Wellcome Trust Investigator Award (210757/Z/18/Z) to A.C.F.-S. and by a PhD award from the BBSRC (1795629) to J.L.E.
Acknowledgments
We are grateful to Tessa Bertozzi and Poppy Gould for valuable input and feedback during the preparation of the manuscript and to other members of the Ferguson-Smith lab for useful discussions.
Conflicts of Interest
The authors declare no conflicts of interest associated with this manuscript.
References
- Boeke, J.; Stoye, J. Retrotransposons, Endogenous Retroviruses, and the Evolution of Retroelements. In Retroviruses; Coffin, J.M., Hughes, S.H., Varmus, H.E., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1997; Volume 1, pp. 123–456. ISBN 0879695714. [Google Scholar]
- Waterston, R.H.; Lindblad-Toh, K.; Birney, E.; Rogers, J.; Abril, J.F.; Agarwal, P.; Agarwala, R.; Ainscough, R.; Alexandersson, M.; An, P.; et al. Initial sequencing and comparative analysis of the mouse genome. Nature 2002, 420, 520–562. [Google Scholar] [CrossRef] [PubMed]
- Jern, P.; Sperber, G.O.; Blomberg, J. Use of Endogenous Retroviral Sequences (ERVs) and structural markers for retroviral phylogenetic inference and taxonomy. Retrovirology 2005, 2, 50. [Google Scholar] [CrossRef] [PubMed]
- Gifford, R.; Kabat, P.; Martin, J.; Lynch, C.; Tristem, M. Evolution and Distribution of Class II-Related Endogenous Retroviruses. J. Virol. 2005, 79, 6478–6486. [Google Scholar] [CrossRef]
- Stocking, C.; Kozak, C.A. Endogenous retroviruses: Murine endogenous retroviruses. Cell. Mol. Life Sci. 2008, 65, 3383–3398. [Google Scholar] [CrossRef] [PubMed]
- Gifford, R.J.; Blomberg, J.; Coffin, J.M.; Fan, H.; Heidmann, T.; Mayer, J.; Stoye, J.; Tristem, M.; Johnson, W.E. Nomenclature for endogenous retrovirus (ERV) loci. Retrovirology 2018, 15, 59. [Google Scholar] [CrossRef]
- Mietz, J.A.; Grossman, Z.; Lueders, K.K.; Kuff, E.L. Nucleotide sequence of a complete mouse intracisternal A-particle genome: Relationship to known aspects of particle assembly and function. J. Virol. 1987, 61, 3020–3029. [Google Scholar] [CrossRef] [PubMed]
- Thompson, P.J.; Macfarlan, T.S.; Lorincz, M.C. Long Terminal Repeats: From Parasitic Elements to Building Blocks of the Transcriptional Regulatory Repertoire. Mol. Cell 2016, 62, 766–776. [Google Scholar] [CrossRef]
- Gagnier, L.; Belancio, V.P.; Mager, D.L. Mouse germ line mutations due to retrotransposon insertions. Mob. DNA 2019, 10, 1–22. [Google Scholar] [CrossRef]
- Ribet, D.; Dewannieux, M.; Heidmann, T. An active murine transposon family pair: Retrotransposition of “master” MusD copies and ETn trans-mobilization. Genome Res. 2004, 14, 2261–2267. [Google Scholar] [CrossRef]
- Maksakova, I.A.; Romanish, M.T.; Gagnier, L.; Dunn, C.A.; van de Lagemaat, L.N.; Mager, D.L. Retroviral Elements and Their Hosts: Insertional Mutagenesis in the Mouse Germ Line. PLoS Genet. 2006, 2, e2. [Google Scholar] [CrossRef]
- Bourque, G.; Burns, K.H.; Gehring, M.; Gorbunova, V.; Seluanov, A.; Hammell, M.; Imbeault, M.; Izsvák, Z.; Levin, H.L.; Macfarlan, T.S.; et al. Ten things you should know about transposable elements 06 Biological Sciences 0604 Genetics. Genome Biol. 2018, 19, 1–12. [Google Scholar] [CrossRef]
- Zhang, Y.; Maksakova, I.A.; Gagnier, L.; van de Lagemaat, L.N.; Mager, D.L. Genome-Wide Assessments Reveal Extremely High Levels of Polymorphism of Two Active Families of Mouse Endogenous Retroviral Elements. PLoS Genet. 2008, 4, e1000007. [Google Scholar] [CrossRef]
- Li, J.; Akagi, K.; Hu, Y.; Trivett, A.L.; Hlynialuk, C.J.W.; Swing, D.A.; Volfovsky, N.; Morgan, T.C.; Golubeva, Y.; Stephens, R.M.; et al. Mouse endogenous retroviruses can trigger premature transcriptional termination at a distance. Genome Res. 2012, 22, 870–884. [Google Scholar] [CrossRef] [PubMed]
- Nellåker, C.; Keane, T.M.; Yalcin, B.; Wong, K.; Agam, A.; Belgard, T.G.; Flint, J.; Adams, D.J.; Frankel, W.N.; Ponting, C.P. The genomic landscape shaped by selection on transposable elements across 18 mouse strains. Genome Biol. 2012, 13, R45. [Google Scholar] [CrossRef] [PubMed]
- Thybert, D.; Roller, M.; Navarro, F.C.P.; Fiddes, I.; Streeter, I.; Feig, C.; Martin-Galvez, D.; Kolmogorov, M.; Janoušek, V.; Akanni, W.; et al. Repeat associated mechanisms of genome evolution and function revealed by the Mus caroli and Mus pahari genomes. Genome Res. 2018, 28, 448–459. [Google Scholar] [CrossRef] [PubMed]
- Lilue, J.; Doran, A.G.; Fiddes, I.T.; Abrudan, M.; Armstrong, J.; Bennett, R.; Chow, W.; Collins, J.; Collins, S.; Czechanski, A.; et al. Sixteen diverse laboratory mouse reference genomes define strain-specific haplotypes and novel functional loci. Nat. Genet. 2018, 50, 1574–1583. [Google Scholar] [CrossRef]
- Lilue, J.; Shivalikanjli, A.; Adams, D.J.; Keane, T.M. Mouse protein coding diversity: What’s left to discover? PLoS Genet. 2019, 15, e1008446. [Google Scholar] [CrossRef]
- Hamilton, B.A.; Yu, B.D. Modifier Genes and the Plasticity of Genetic Networks in Mice. PLoS Genet. 2012, 8, e1002644. [Google Scholar] [CrossRef]
- Perry, M.N.; Bello, S.M.; Smith, C.L. Know Your Model: Why mouse inbred strain contribution matters. Lab Anim. 2020, 49, 133–134. [Google Scholar] [CrossRef]
- Matsui, T.; Leung, D.; Miyashita, H.; Maksakova, I.A.; Miyachi, H.; Kimura, H.; Tachibana, M.; Lorincz, M.C.; Shinkai, Y. Proviral silencing in embryonic stem cells requires the histone methyltransferase ESET. Nature 2010, 464, 927–931. [Google Scholar] [CrossRef]
- Karimi, M.M.; Goyal, P.; Maksakova, I.A.; Bilenky, M.; Leung, D.; Tang, J.X.; Shinkai, Y.; Mager, D.L.; Jones, S.; Hirst, M.; et al. DNA methylation and SETDB1/H3K9me3 regulate predominantly distinct sets of genes, retroelements, and chimeric transcripts in mescs. Cell Stem Cell 2011, 8, 676–687. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.; Goff, S.P. Embryonic stem cells use ZFP809 to silence retroviral DNAs. Nature 2009, 458, 1201–1204. [Google Scholar] [CrossRef] [PubMed]
- Wolf, G.; Yang, P.; Füchtbauer, A.C.; Füchtbauer, E.M.; Silva, A.M.; Park, C.; Wu, W.; Nielsen, A.L.; Pedersen, F.S.; Macfarlan, T.S. The KRAB zinc finger protein ZFP809 is required to initiate epigenetic silencing of endogenous retroviruses. Genes Dev. 2015, 29, 538–554. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.-T. KAPtain in charge of multiple missions: Emerging roles of KAP1. World J. Biol. Chem. 2014, 5, 308. [Google Scholar] [CrossRef] [PubMed]
- Goodier, J.L. Restricting retrotransposons: A review. Mob. DNA 2016, 7, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Ecco, G.; Imbeault, M.; Trono, D. KRAB zinc finger proteins. Development 2017, 144, 2719–2729. [Google Scholar] [CrossRef]
- Bruno, M.; Mahgoub, M.; Macfarlan, T.S. The Arms Race Between KRAB–Zinc Finger Proteins and Endogenous Retroelements and Its Impact on Mammals. Annu. Rev. Genet. 2019, 53, 393–416. [Google Scholar] [CrossRef]
- Rowe, H.M.; Jakobsson, J.; Mesnard, D.; Rougemont, J.; Reynard, S.; Aktas, T.; Maillard, P.V.; Layard-Liesching, H.; Verp, S.; Marquis, J.; et al. KAP1 controls endogenous retroviruses in embryonic stem cells. Nature 2010, 463, 237–240. [Google Scholar] [CrossRef]
- Liu, S.; Brind’Amour, J.; Karimi, M.M.; Shirane, K.; Bogutz, A.; Lefebvre, L.; Sasaki, H.; Shinkai, Y.; Lorincz, M.C. Setdb1 is required for germline development and silencing of H3K9me3-marked endogenous retroviruses in primordial germ cells. Genes Dev. 2014, 28, 2041–2055. [Google Scholar] [CrossRef]
- Walsh, C.P.; Chaillet, J.R.; Bestor, T.H. Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nat. Genet. 1998, 20, 116. [Google Scholar] [CrossRef]
- Quenneville, S.; Turelli, P.; Bojkowska, K.; Raclot, C.; Offner, S.; Kapopoulou, A.; Trono, D. The KRAB-ZFP/KAP1 System Contributes to the Early Embryonic Establishment of Site-Specific DNA Methylation Patterns Maintained during Development. Cell Rep. 2012, 2, 766–773. [Google Scholar] [CrossRef] [PubMed]
- Rowe, H.M.; Friedli, M.; Offner, S.; Verp, S.; Mesnard, D.; Marquis, J.; Aktas, T.; Trono, D. De novo DNA methylation of endogenous retroviruses is shaped by KRAB-ZFPs/KAP1 and ESET. Development 2013, 140, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Macfarlan, T.S.; Gifford, W.D.; Driscoll, S.; Lettieri, K.; Rowe, H.M.; Bonanomi, D.; Firth, A.; Singer, O.; Trono, D.; Pfaff, S.L. Embryonic stem cell potency fluctuates with endogenous retrovirus activity. Nature 2012, 487, 57–63. [Google Scholar] [CrossRef]
- Maksakova, I.A.; Thompson, P.J.; Goyal, P.; Jones, S.J.M.; Singh, P.B.; Karimi, M.M.; Lorincz, M.C. Distinct roles of KAP1, HP1 and G9a/GLP in silencing of the two-cell-specific retrotransposon MERVL in mouse ES cells. Epigenetics Chromatin 2013, 6, 15. [Google Scholar] [CrossRef] [PubMed]
- Macfarlan, T.S.; Gifford, W.D.; Agarwal, S.; Driscoll, S.; Lettieri, K.; Wang, J.; Andrews, S.E.; Franco, L.; Rosenfeld, M.G.; Ren, B. Endogenous retroviruses and neighboring genes are coordinately repressed by LSD1/KDM1A. Genes Dev. 2011, 25, 594–607. [Google Scholar] [CrossRef]
- Ancelin, K.; Syx, L.; Borensztein, M.; Ranisavljevic, N.; Vassilev, I.; Briseño-Roa, L.; Liu, T.; Metzger, E.; Servant, N.; Barillot, E.; et al. Maternal LSD1/KDM1A is an essential regulator of chromatin and transcription landscapes during zygotic genome activation. Elife 2016, 5. [Google Scholar] [CrossRef]
- Morgan, H.D.; Santos, F.; Green, K.; Dean, W.; Reik, W. Epigenetic reprogramming in mammals. Hum. Mol. Genet. 2005, 14, R47–R58. [Google Scholar] [CrossRef] [PubMed]
- Kigami, D.; Minami, N.; Takayama, H.; Imai, H. MuERV-L Is One of the Earliest Transcribed Genes in Mouse One-Cell Embryos1. Biol. Reprod. 2003, 68, 651–654. [Google Scholar] [CrossRef]
- Svoboda, P.; Stein, P.; Anger, M.; Bernstein, E.; Hannon, G.J.; Schultz, R.M. RNAi and expression of retrotransposons MuERV-L and IAP in preimplantation mouse embryos. Dev. Biol. 2004, 269, 276–285. [Google Scholar] [CrossRef]
- Fu, B.; Ma, H.; Liu, D. Endogenous retroviruses function as gene expression regulatory elements during mammalian pre-implantation embryo development. Int. J. Mol. Sci. 2019, 20, 790. [Google Scholar] [CrossRef]
- Rowe, H.M.; Trono, D. Dynamic control of endogenous retroviruses during development. Virology 2011, 411, 273–287. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.C.; Lorincz, M.C. Silencing of endogenous retroviruses: When and why do histone marks predominate? Trends Biochem. Sci. 2012, 37, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.; Teissandier, A.; Pérez-Palacios, R.; Bourc’his, D. An epigenetic switch ensures transposon repression upon dynamic loss of DNA methylation in embryonic stem cells. Elife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Sharif, J.; Endo, T.A.; Nakayama, M.; Xie, H.; Lorincz, M.C.; Correspondence, H.K. Activation of Endogenous Retroviruses in Dnmt1-/-ESCs Involves Disruption of SETDB1-Mediated Repression by NP95 Binding to Hemimethylated DNA Accession Numbers GSE77781 Sharif et al. Stem Cell 2016, 19, 81–94. [Google Scholar] [CrossRef]
- Berrens, R.V.; Andrews, S.; Spensberger, D.; Santos, F.; Dean, W.; Gould, P.; Sharif, J.; Olova, N.; Chandra, T.; Koseki, H. An endosiRNA-based repression mechanism counteracts transposon activation during global DNA demethylation in embryonic stem cells. Cell Stem Cell 2017, 21, 694–703. [Google Scholar] [CrossRef]
- Schorn, A.J.; Martienssen, R. Tie-Break: Host and Retrotransposons Play tRNA. Trends Cell Biol. 2018, 28, 793–806. [Google Scholar] [CrossRef]
- Ozata, D.M.; Gainetdinov, I.; Zoch, A.; O’Carroll, D.; Zamore, P.D. PIWI-interacting RNAs: Small RNAs with big functions. Nat. Rev. Genet. 2019, 20, 89–108. [Google Scholar] [CrossRef]
- Elsässer, S.J.; Noh, K.M.; Diaz, N.; Allis, C.D.; Banaszynski, L.A. Histone H3.3 is required for endogenous retroviral element silencing in embryonic stem cells. Nature 2015, 522, 240–244. [Google Scholar] [CrossRef]
- Voon, H.P.J.; Wong, L.H. New players in heterochromatin silencing: Histone variant H3. 3 and the ATRX/DAXX chaperone. Nucleic Acids Res. 2016, 44, 1496–1501. [Google Scholar] [CrossRef]
- Wolf, G.; Rebollo, R.; Karimi, M.M.; Ewing, A.D.; Kamada, R.; Wu, W.; Wu, B.; Bachu, M.; Ozato, K.; Faulkner, G.J.; et al. On the role of H3.3 in retroviral silencing. Nature 2017, 548, E1–E3. [Google Scholar] [CrossRef]
- Hansen, J.C. Silencing the genome with linker histones. Proc. Natl. Acad. Sci. USA 2020, 202009513. [Google Scholar] [CrossRef] [PubMed]
- Chai, C.K. Dactylaplasia in mice: A two-locus model for developmental anomalies. J. Hered. 1981, 72, 234–237. [Google Scholar] [CrossRef] [PubMed]
- Sidow, A.; Bulotsky, M.S.; Kerrebrock, A.W.; Birren, B.W.; Altshuler, D.; Jaenisch, R.; Johnson, K.R.; Lander, E.S. A novel member of the F-box/WD40 gene family, encoding dactylin, is disrupted in the mouse dactylaplasia mutant. Nat. Genet. 1999, 23, 104–107. [Google Scholar] [CrossRef]
- Johnson, K.R.; Lane, P.W.; Ward-Bailey, P.; Davisson, M.T. Mapping the Mouse Dactylaplasia Mutation, Dac, and a Gene That Controls Its Expression, mdac. Genomics 1995, 29, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Kano, H.; Kurahashi, H.; Toda, T. Genetically regulated epigenetic transcriptional activation of retrotransposon insertion confers mouse dactylaplasia phenotype. Proc. Natl. Acad. Sci. USA 2007, 104, 19034–19039. [Google Scholar] [CrossRef]
- Friedli, M.; Nikolaev, S.; Lyle, R.; Arcangeli, M.; Duboule, D.; Spitz, F.; Antonarakis, S.E. Characterization of mouse Dactylaplasia mutations: A model for human ectrodactyly SHFM3. Mamm. Genome 2008, 19, 272–278. [Google Scholar] [CrossRef]
- Corsinotti, A.; Kapopoulou, A.; Gubelmann, C.; Imbeault, M.; Santoni de Sio, F.R.; Rowe, H.M.; Mouscaz, Y.; Deplancke, B.; Trono, D. Global and Stage Specific Patterns of Krüppel-Associated-Box Zinc Finger Protein Gene Expression in Murine Early Embryonic Cells. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Wolf, G.; de Iaco, A.; Sun, M.A.; Bruno, M.; Tinkham, M.; Hoang, D.; Mitra, A.; Ralls, S.; Trono, D.; Macfarlan, T.S. KRAB-zinc finger protein gene expansion in response to active retrotransposons in the murine lineage. Elife 2020, 9. [Google Scholar] [CrossRef]
- Juriloff, D.M. Differences in frequency of cleft lip among the a strains of mice. Teratology 1982, 25, 361–368. [Google Scholar] [CrossRef]
- Juriloff, D.M.; Harris, M.J.; Brown, C.J. Unravelling the complex genetics of cleft lip in the mouse model. Mamm. Genome 2001, 12, 426–435. [Google Scholar] [CrossRef]
- Juriloff, D.M.; Harris, M.J.; Dewell, S.L.; Brown, C.J.; Mager, D.L.; Gagnier, L.; Mah, D.G. Investigations of the genomic region that contains the clf1 mutation, a causal gene in multifactorial cleft lip and palate in mice. Birth Defects Res. Part A Clin. Mol. Teratol. 2005, 73, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Juriloff, D.M.; Harris, M.J.; Mager, D.L.; Gagnier, L. Epigenetic Mechanism Causes Wnt9B Deficiency and Nonsyndromic Cleft Lip and Palate in the A/WySn Mouse Strain. Birth Defects Res. Part A Clin. Mol. Teratol. 2014, 100, 772–788. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.R.; Han, X.H.; Taketo, M.M.; Yoon, J.K. Wnt9b-dependent FGF signaling is crucial for outgrowth of the nasal and maxillary processes during upper jaw and lip development. Development 2012, 139, 1821–1830. [Google Scholar] [CrossRef] [PubMed]
- Juriloff, D.M.; Harris, M.J. Mouse genetic models of cleft lip with or without cleft palate. Birth Defects Res. Part A Clin. Mol. Teratol. 2008, 82, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Plamondon, J.A.; Harris, M.J.; Mager, D.L.; Gagnier, L.; Juriloff, D.M. The clf2 gene has an epigenetic role in the multifactorial etiology of cleft lip and palate in the A/WySn mouse strain. Birth Defects Res. Part A Clin. Mol. Teratol. 2011, 91, 716–727. [Google Scholar] [CrossRef] [PubMed]
- Green, R.M.; Leach, C.L.; Diewert, V.M.; Aponte, J.D.; Schmidt, E.J.; Cheverud, J.M.; Roseman, C.C.; Young, N.M.; Marcucio, R.S.; Hallgrimsson, B. Nonlinear gene expression-phenotype relationships contribute to variation and clefting in the A/WySn mouse. Dev. Dyn. 2019, 248, 1232–1242. [Google Scholar] [CrossRef]
- Oliver, P.L.; Stoye, J.P. Genetic Analysis of Gv1, a Gene Controlling Transcription of Endogenous Murine Polytropic Proviruses. J. Virol. 1999, 73, 8227–8234. [Google Scholar] [CrossRef]
- Laporte, C.; Ballester, B.; Mary, C.; Izui, S.; Reininger, L. The Sgp3 Locus on Mouse Chromosome 13 Regulates Nephritogenic gp70 Autoantigen Expression and Predisposes to Autoimmunity. J. Immunol. 2003, 171, 3872–3877. [Google Scholar] [CrossRef]
- Treger, R.S.; Pope, S.D.; Kong, Y.; Tokuyama, M.; Taura, M.; Iwasaki, A. The Lupus Susceptibility Locus Sgp3 Encodes the Suppressor of Endogenous Retrovirus Expression SNERV. Immunity 2019, 50, 334–347.e9. [Google Scholar] [CrossRef]
- Politz, O.; Gratchev, A.; McCourt, P.A.G.; Schledzewski, K.; Guillot, P.; Johansson, S.; Svineng, G.; Franke, P.; Kannicht, C.; Kzhyshkowska, J.; et al. Stabilin-1 and -2 constitute a novel family of fasciclin-like hyaluronan receptor homologues. Biochem. J. 2002, 362, 155–164. [Google Scholar] [CrossRef]
- Harris, E.N.; Weigel, J.A.; Weigel, P.H. The human hyaluronan receptor for endocytosis (HARE/stabilin-2) is a systemic clearance receptor for heparin. J. Biol. Chem. 2008, 283, 17341–17350. [Google Scholar] [CrossRef] [PubMed]
- Schledzewski, K.; Géraud, C.; Arnold, B.; Wang, S.; Gröne, H.J.; Kempf, T.; Wollert, K.C.; Straub, B.K.; Schirmacher, P.; Demory, A.; et al. Deficiency of liver sinusoidal scavenger receptors stabilin-1 and -2 in mice causes glomerulofibrotic nephropathy via impaired hepatic clearance of noxious blood factors. J. Clin. Investig. 2011, 121, 703–714. [Google Scholar] [CrossRef]
- Hirose, Y.; Saijou, E.; Sugano, Y.; Takeshita, F.; Nishimura, S.; Nonaka, H.; Chen, Y.R.; Sekine, K.; Kido, T.; Nakamura, T.; et al. Inhibition of Stabilin-2 elevates circulating hyaluronic acid levels and prevents tumor metastasis. Proc. Natl. Acad. Sci. USA 2012, 109, 4263–4268. [Google Scholar] [CrossRef] [PubMed]
- Kayashima, Y.; Makhanova, N.A.; Matsuki, K.; Tomita, H.; Bennett, B.J.; Maeda, N. Identification of Aortic Arch-Specific Quantitative Trait Loci for Atherosclerosis by an Intercross of DBA/2J and 129S6 Apolipoprotein E-Deficient Mice. PLoS ONE 2015, 10, e0117478. [Google Scholar] [CrossRef] [PubMed]
- Maeda-Smithies, N.; Hiller, S.; Dong, S.; Kim, H.S.; Bennett, B.J.; Kayashima, Y. Ectopic expression of the Stabilin2 gene triggered by an intracisternal A particle (IAP) element in DBA/2J strain of mice. Mamm. Genome 2020, 31, 2–16. [Google Scholar] [CrossRef]
- Rakyan, V.K.; Blewitt, M.E.; Druker, R.; Preis, J.I.; Whitelaw, E. Metastable epialleles in mammals. Trends Genet. 2002, 18, 348–351. [Google Scholar] [CrossRef]
- Bertozzi, T.M.; Ferguson-Smith, A.C. Metastable epialleles and their contribution to epigenetic inheritance in mammals. Semin. Cell Dev. Biol. 2020, 97, 93–105. [Google Scholar] [CrossRef]
- Dickies, M.M. A New Viable Yellow Mutation in the House Mouse. J. Hered. 1962, 53, 84–86. [Google Scholar] [CrossRef]
- Morgan, H.D.; Sutherland, H.G.E.; Martin, D.I.K.; Whitelaw, E. Epigenetic inheritance at the agouti locus in the mouse. Nat. Genet. 1999, 23, 314–318. [Google Scholar] [CrossRef]
- Reed, S.C. The inheritance and expression of Fused, a new mutation in the house mouse. Genetics 1937, 22, 1. [Google Scholar]
- Vasicek, T.J.; Zeng, L.; Guan, X.J.; Zhang, T.; Costantini, F.; Tilghman, S.M. Two dominant mutations in the mouse fused gene are the result of transposon insertions. Genetics 1997, 147, 777–786. [Google Scholar]
- Rakyan, V.K.; Chong, S.; Champ, M.E.; Cuthbert, P.C.; Morgan, H.D.; Luu, K.V.K.; Whitelaw, E. Transgenerational inheritance of epigenetic states at the murine AxinFu allele occurs after maternal and paternal transmission. Proc. Natl. Acad. Sci. USA 2003, 100, 2538–2543. [Google Scholar] [CrossRef] [PubMed]
- Duhl, D.M.J.; Vrieling, H.; Miller, K.A.; Wolff, G.L.; Barsh, G.S. Neomorphic agouti mutations in obese yellow mice. Nat. Genet. 1994, 8, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Wolff, G.L. Body composition and coat color correlation in different phenotypes of “viable yellow” mice. Science 1965, 147, 1145–1147. [Google Scholar] [CrossRef] [PubMed]
- Wolff, G.L.; Roberts, D.W.; Mountjoy, K.G. Physiological consequences of ectopic agouti gene expression: The yellow obese mouse syndrome. Physiol. Genomics 1999, 1, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Wolff, G.L. Genetic Modification of Homeostatic Regulation in the Mouse. Am. Nat. 1971, 105, 241–252. [Google Scholar] [CrossRef]
- Wolff, G.L.; Pitot, H.C. Variation of Hepatic Malic Enzyme Capacity with Hepatoma Susceptibility in Mice of Different Genotypes. Cancer Res. 1972, 32, 1861–1863. [Google Scholar]
- Wolff, G.L. Influence of maternal phenotype on metabolic differentiation of Agouti locus mutants in the mouse. Genetics 1978, 88, 529–539. [Google Scholar]
- Wolff, G.L.; Kodell, R.L.; Moore, S.R.; Cooney, C.A. Maternal epigenetics and methyl supplements affect agouti gene expression in Avy/a mice. FASEB J. 1998, 12, 949–957. [Google Scholar] [CrossRef]
- Cooney, C.A.; Dave, A.A.; Wolff, G.L. Maternal methyl supplements in mice affect epigenetic variation and DNA methylation of offspring. J. Nutr. 2002, 132, 2393S–2400S. [Google Scholar] [CrossRef]
- Belyaev, D.K.; Ruvinsky, A.O.; Borodin, P.M. Inheritance of alternative states of the fused gene in mice. J. Hered. 1981, 72, 107–112. [Google Scholar] [CrossRef]
- Ruvinsky, A.O.; Agulnik, A.I. Gametic imprinting and the manifestation of the fused gene in the house mouse. Dev. Genet. 1990, 11, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Oswald, J.; Engemann, S.; Lane, N.; Mayer, W.; Olek, A.; Fundele, R.; Dean, W.; Reik, W.; Walter, J. Active demethylation of the paternal genome in the mouse zygote. Curr. Biol. 2000, 10, 475–478. [Google Scholar] [CrossRef]
- Mayer, W.; Niveleau, A.; Walter, J.; Fundele, R.; Haaf, T. Embryogenesis: Demethylation of the zygotic paternal genome. Nature 2000, 403, 501–502. [Google Scholar] [CrossRef]
- Blewitt, M.E.; Vickaryous, N.K.; Paldi, A.; Koseki, H.; Whitelaw, E. Dynamic Reprogramming of DNA Methylation at an Epigenetically Sensitive Allele in Mice. PLoS Genet. 2006, 2, e49. [Google Scholar] [CrossRef] [PubMed]
- Kazachenka, A.; Bertozzi, T.M.; Sjoberg-Herrera, M.K.; Adams, S.; Adams, D.; Ferguson-Smith, A.C. Identification, Characterization, and Heritability of Murine Metastable Epialleles: Implications for Non-genetic Inheritance In Brief. Cell 2018, 175. [Google Scholar] [CrossRef]
- Bertozzi, T. Variable Methylation of Endogenous Retroviruses: Epigenetic Inheritance, Environmental Modulation, and Genetic Modifiers. Ph.D. Thesis, University of Cambridge, Cambridge, UK, 2020. [Google Scholar] [CrossRef]
- Allen, N.D.; Norris, M.L.; Surani, M.A. Epigenetic control of transgene expression and imprinting by genotype-specific modifiers. Cell 1990, 61, 853–861. [Google Scholar] [CrossRef]
- Chaillet, J.R. Genomic imprinting: Lessons from mouse transgenes. Mutat. Res. 1994, 307, 441–449. [Google Scholar] [CrossRef]
- Daxinger, L.; Whitelaw, E. Understanding transgenerational epigenetic inheritance via the gametes in mammals. Nat. Rev. Genet. 2012, 13, 153–162. [Google Scholar] [CrossRef]
- Blewitt, M.; Whitelaw, E. The use of mouse models to study epigenetics. Cold Spring Harb. Perspect. Biol. 2013, 5, a017939. [Google Scholar] [CrossRef]
- Engler, P.; Storb, U. High-frequency deletional rearrangement of immunoglobulin kappa gene segments introduced into a pre-B-cell line. Proc. Natl. Acad. Sci. USA 1987, 84, 4949–4953. [Google Scholar] [CrossRef] [PubMed]
- Engler, P.; Roth, P.; Kim, J.Y.; Storb, U. Factors affecting the rearrangement efficiency of an Ig test gene. J. Immunol. 1991, 146, 2826–2835. [Google Scholar] [PubMed]
- Engler, P.; Haasch, D.; Pinkert, C.A.; Doglio, L.; Glymour, M.; Brinster, R.; Storb, U. A Strain-Specific Modifier on Mouse Chromosome 4 Controls the Methylation of Independent Transgene Loci. Cell 1991, 65, 939–947. [Google Scholar] [CrossRef]
- Ratnam, S.; Engler, P.; Bozek, G.; Mao, L.; Podlutsky, A.; Austad, S.; Martin, T.; Storb, U. Identification of Ssm1b, a novel modifier of DNA methylation, and its expression during mouse embryogenesis. Development 2014, 141, 2024–2034. [Google Scholar] [CrossRef][Green Version]
- Weng, A.; Magnuson, T.; Storb, U. Strain-specific transgene methylation occurs early in mouse development and can be recapitulated in embryonic stem cells. Development 1995, 121, 2853–2859. [Google Scholar]
- Ratnam, S.; Bozek, G.; Martin, T.; Gallagher, S.J.; Payne, C.J.; Storb, U. Ssm1b expression and function in germ cells of adult mice and in early embryos. Mol. Reprod. Dev. 2017, 84, 596–613. [Google Scholar] [CrossRef]
- Kauzlaric, A.; Ecco, G.; Cassano, M.; Duc, J.; Imbeault, M.; Trono, D. The mouse genome displays highly dynamic populations of KRAB-zinc finger protein genes and related genetic units. PLoS ONE 2017, 12, e0173746. [Google Scholar] [CrossRef]
- Preis, J.I.; Downes, M.; Oates, N.A.; Rasko, J.E.J.; Whitelaw, E. Sensitive flow cytometric analysis reveals a novel type of parent-of-origin effect in the mouse genome. Curr. Biol. 2003, 13, 955–959. [Google Scholar] [CrossRef]
- Blewitt, M.E.; Vickaryous, N.K.; Hemley, S.J.; Ashe, A.; Bruxner, T.J.; Preis, J.I.; Arkell, R.; Whitelaw, E. An N-ethyl-N-nitrosourea screen for genes involved in variegation in the mouse. Proc. Natl. Acad. Sci. USA 2005, 102, 7629–7634. [Google Scholar] [CrossRef]
- Chong, S.; Vickaryous, N.; Ashe, A.; Zamudio, N.; Youngson, N.; Hemley, S.; Stopka, T.; Skoultchi, A.; Matthews, J.; Scott, H.S.; et al. Modifiers of epigenetic reprogramming show paternal effects in the mouse. Nat. Genet. 2007, 39, 614–622. [Google Scholar] [CrossRef][Green Version]
- Ashe, A.; Morgan, D.K.; Whitelaw, N.C.; Bruxner, T.J.; Vickaryous, N.K.; Cox, L.L.; Butterfield, N.C.; Wicking, C.; Blewitt, M.E.; Wilkins, S.J.; et al. A genome-wide screen for modifiers of transgene variegation identifies genes with critical roles in development. Genome Biol. 2008, 9, R182. [Google Scholar] [CrossRef] [PubMed]
- Blewitt, M.E.; Gendrel, A.V.; Pang, Z.; Sparrow, D.B.; Whitelaw, N.; Craig, J.M.; Apedaile, A.; Hilton, D.J.; Dunwoodie, S.L.; Brockdorff, N.; et al. SmcHD1, containing a structural-maintenance-of-chromosomes hinge domain, has a critical role in X inactivation. Nat. Genet. 2008, 40, 663–669. [Google Scholar] [CrossRef]
- Daxinger, L.; Oey, H.; Apedaile, A.; Sutton, J.; Ashe, A.; Whitelaw, E. A forward genetic screen identifies eukaryotic translation initiation factor 3, subunit h (eiF3h), as an enhancer of variegation in the mouse. G3 Genes Genomes Genet. 2012, 2, 1393–1396. [Google Scholar] [CrossRef]
- Daxinger, L.; Harten, S.K.; Oey, H.; Epp, T.; Isbel, L.; Huang, E.; Whitelaw, N.; Apedaile, A.; Sorolla, A.; Yong, J.; et al. An ENU mutagenesis screen identifies novel and known genes involved in epigenetic processes in the mouse. Genome Biol. 2013, 14, R96. [Google Scholar] [CrossRef] [PubMed]
- Krebs, C.J.; Larkins, L.K.; Price, R.; Tullis, K.M.; Miller, R.D.; Robins, D.M. Regulator of sex-limitation (Rs1) encodes a pair of KRAB zinc-finger genes that control sexually dimorphic liver gene expression. Genes Dev. 2003, 17, 2664–2674. [Google Scholar] [CrossRef] [PubMed]
- Stavenhagen, J.B.; Robins, D.M. An ancient provirus has imposed androgen regulation on the adjacent mouse sex-limited protein gene. Cell 1988, 55, 247–254. [Google Scholar] [CrossRef]
- Krebs, C.J.; Schultz, D.C.; Robins, D.M. The KRAB Zinc Finger Protein RSL1 Regulates Sex- and Tissue-Specific Promoter Methylation and Dynamic Hormone-Responsive Chromatin Configuration. Mol. Cell. Biol. 2012, 32, 3732–3742. [Google Scholar] [CrossRef]
- Bojkowska, K.; Aloisio, F.; Cassano, M.; Kapopoulou, A.; Santoni de Sio, F.; Zangger, N.; Offner, S.; Cartoni, C.; Thomas, C.; Quenneville, S.; et al. Liver-specific ablation of Krüppel-associated box-associated protein 1 in mice leads to male-predominant hepatosteatosis and development of liver adenoma. Hepatology 2012, 56, 1279–1290. [Google Scholar] [CrossRef]
- Krebs, C.J.; Larkins, L.K.; Khan, S.M.; Robins, D.M. Expansion and diversification of KRAB zinc-finger genes within a cluster including Regulator of sex-limitation 1 and 2. Genomics 2005, 85, 752–761. [Google Scholar] [CrossRef]
- Juriloff, D.M.; Harris, M.J.; Dewell, S.L. A digenic cause of cleft lip in A-strain mice and definition of candidate genes for the two loci. Birth Defects Res. Part A Clin. Mol. Teratol. 2004, 70, 509–518. [Google Scholar] [CrossRef]
- Emerson, R.O.; Thomas, J.H. Adaptive evolution in zinc finger transcription factors. PLoS Genet. 2009, 5. [Google Scholar] [CrossRef]
- Thomas, J.H.; Schneider, S. Coevolution of retroelements and tandem zinc finger genes. Genome Res. 2011, 21, 1800–1812. [Google Scholar] [CrossRef] [PubMed]
- Imbeault, M.; Helleboid, P.Y.; Trono, D. KRAB zinc-finger proteins contribute to the evolution of gene regulatory networks. Nature 2017, 543, 550–554. [Google Scholar] [CrossRef] [PubMed]
- Mekada, K.; Abe, K.; Murakami, A.; Nakamura, S.; Nakata, H.; Moriwaki, K.; Obata, Y.; Yoshiki, A. Genetic differences among C57BL/6 substrains. Exp. Anim. 2009, 58, 141–149. [Google Scholar] [CrossRef]
- Simon, M.M.; Greenaway, S.; White, J.K.; Fuchs, H.; Gailus-Durner, V.; Wells, S.; Sorg, T.; Wong, K.; Bedu, E.; Cartwright, E.J.; et al. A comparative phenotypic and genomic analysis of C57BL/6J and C57BL/6N mouse strains. Genome Biol. 2013, 14, R82. [Google Scholar] [CrossRef]
- Helleboid, P.; Heusel, M.; Duc, J.; Piot, C.; Thorball, C.W.; Coluccio, A.; Pontis, J.; Imbeault, M.; Turelli, P.; Aebersold, R.; et al. The interactome of KRAB zinc finger proteins reveals the evolutionary history of their functional diversification. EMBO J. 2019, 38. [Google Scholar] [CrossRef]
- Ecco, G.; Cassano, M.; Kauzlaric, A.; Duc, J.; Coluccio, A.; Offner, S.; Imbeault, M.; Rowe, H.M.; Turelli, P.; Trono, D. Transposable Elements and Their KRAB-ZFP Controllers Regulate Gene Expression in Adult Tissues. Dev. Cell 2016, 36, 611–623. [Google Scholar] [CrossRef]
- Li, X.; Ito, M.; Zhou, F.; Youngson, N.; Zuo, X.; Leder, P.; Ferguson-Smith, A.C. A Maternal-Zygotic Effect Gene, Zfp57, Maintains Both Maternal and Paternal Imprints. Dev. Cell 2008, 15, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Strogantsev, R.; Krueger, F.; Yamazawa, K.; Shi, H.; Gould, P.; Goldman-Roberts, M.; McEwen, K.; Sun, B.; Pedersen, R.; Ferguson-Smith, A.C. Allele-specific binding of ZFP57 in the epigenetic regulation of imprinted and non-imprinted monoallelic expression. Genome Biol. 2015, 16, 112. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Coluccio, A.; Thorball, C.W.; Planet, E.; Shi, H.; Offner, S.; Turelli, P.; Imbeault, M.; Ferguson-Smith, A.C.; Trono, D. ZNF445 is a primary regulator of genomic imprinting. Genes Dev. 2019. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Strogantsev, R.; Takahashi, N.; Kazachenka, A.; Lorincz, M.C.; Hemberger, M.; Ferguson-Smith, A.C. ZFP57 regulation of transposable elements and gene expression within and beyond imprinted domains. Epigenet. Chromatin 2019. [Google Scholar] [CrossRef] [PubMed]
- Swanzey, E.; Mcnamara, T.F.; Apostolou, E.; Tahiliani, M.; Correspondence, M.S. A Susceptibility Locus on Chromosome 13 Profoundly Impacts the Stability of Genomic Imprinting in Mouse Pluripotent Stem Cells. Cell Rep. 2020, 30, 3597–3604.e3. [Google Scholar] [CrossRef] [PubMed]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).