Upregulation of GLS1 Isoforms KGA and GAC Facilitates Mitochondrial Metabolism and Cell Proliferation in Epstein–Barr Virus Infected Cells

Abstract
1. Introduction
2. Materials and Methods
2.1. Cells
2.2. Virus
2.3. Antibodies and Reagents
2.4. Lentivirus Production
2.5. Glutamate Assay
2.6. Alpha-Ketoglutarate Assay
2.7. Quantitative Real-Time Reverse-Transcription PCR
2.8. Western Blot
2.9. Immunofluorescence Assay
2.10. BrdU Cell Proliferation Assay
2.11. MTT Assay
3. Results
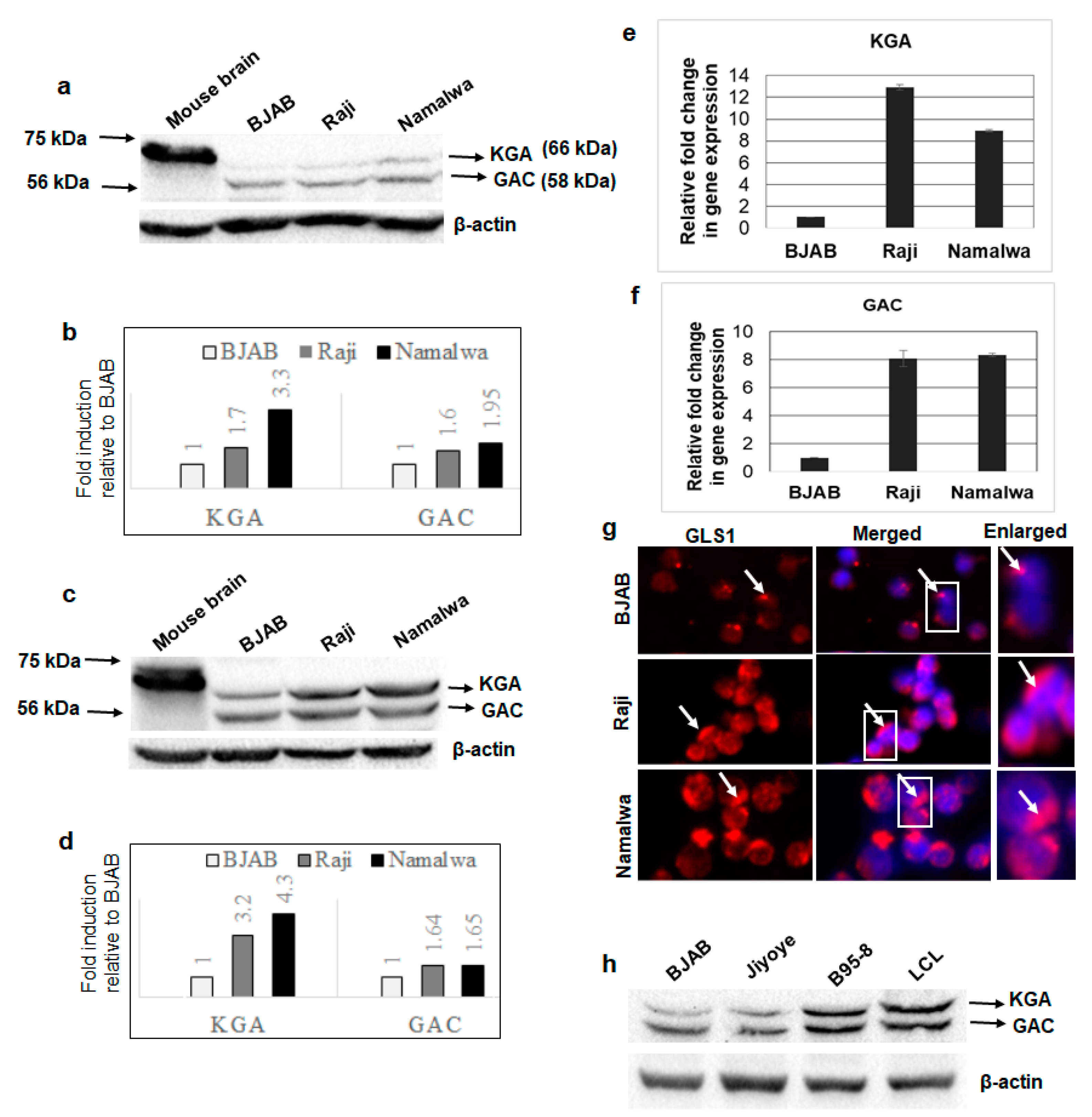
3.1. GLS1 Isoforms KGA and GAC Are Expressed at High Levels in Latently EBV-Infected Lymphoma Cells
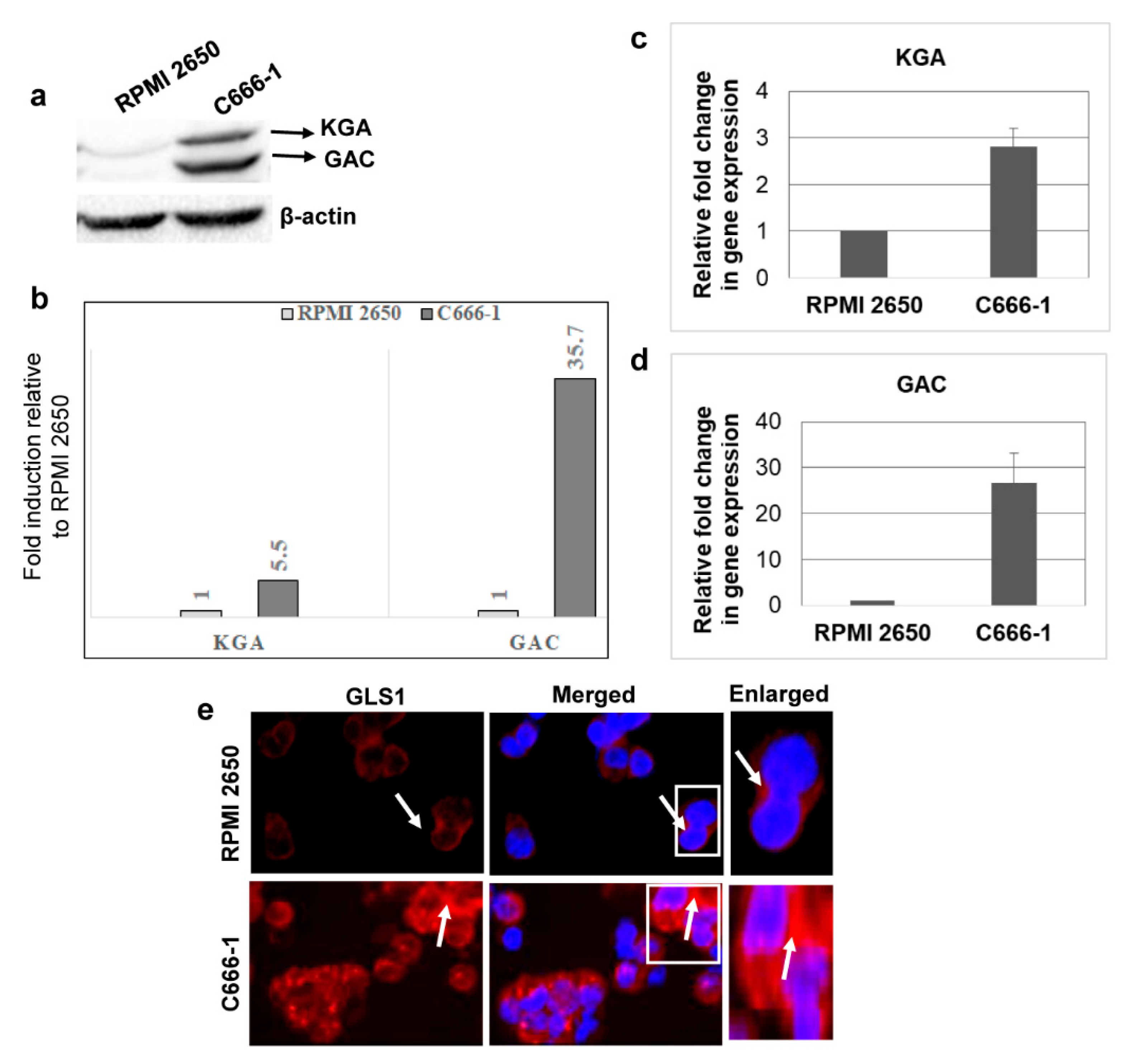
3.2. KGA and GAC Isoforms of GLS1 Are Upregulated in EBV-Infected Nasopharyngeal Carcinoma Cells
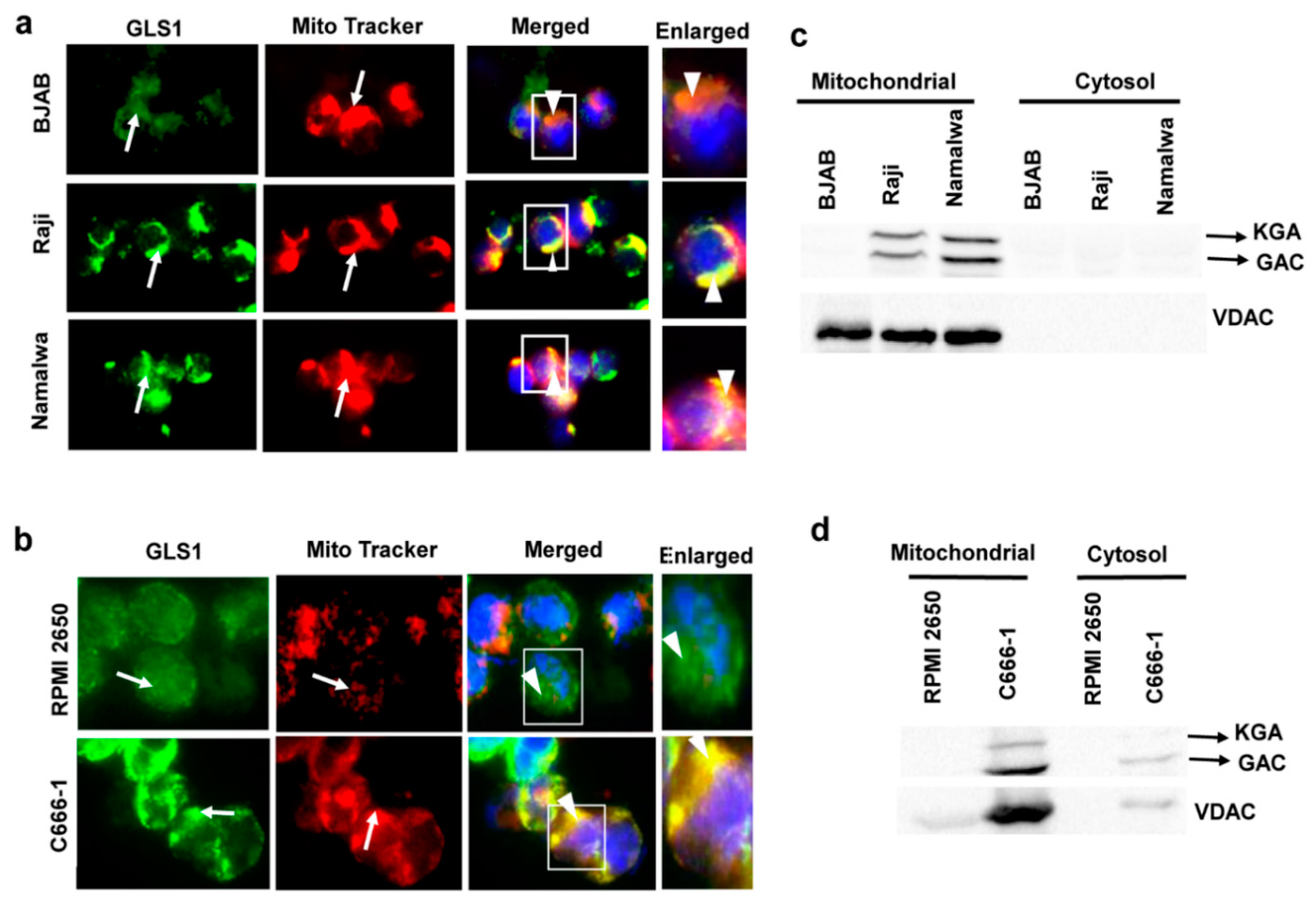
3.3. KGA and GAC Localized to the Mitochondria of EBV-Infected Cancer Cells
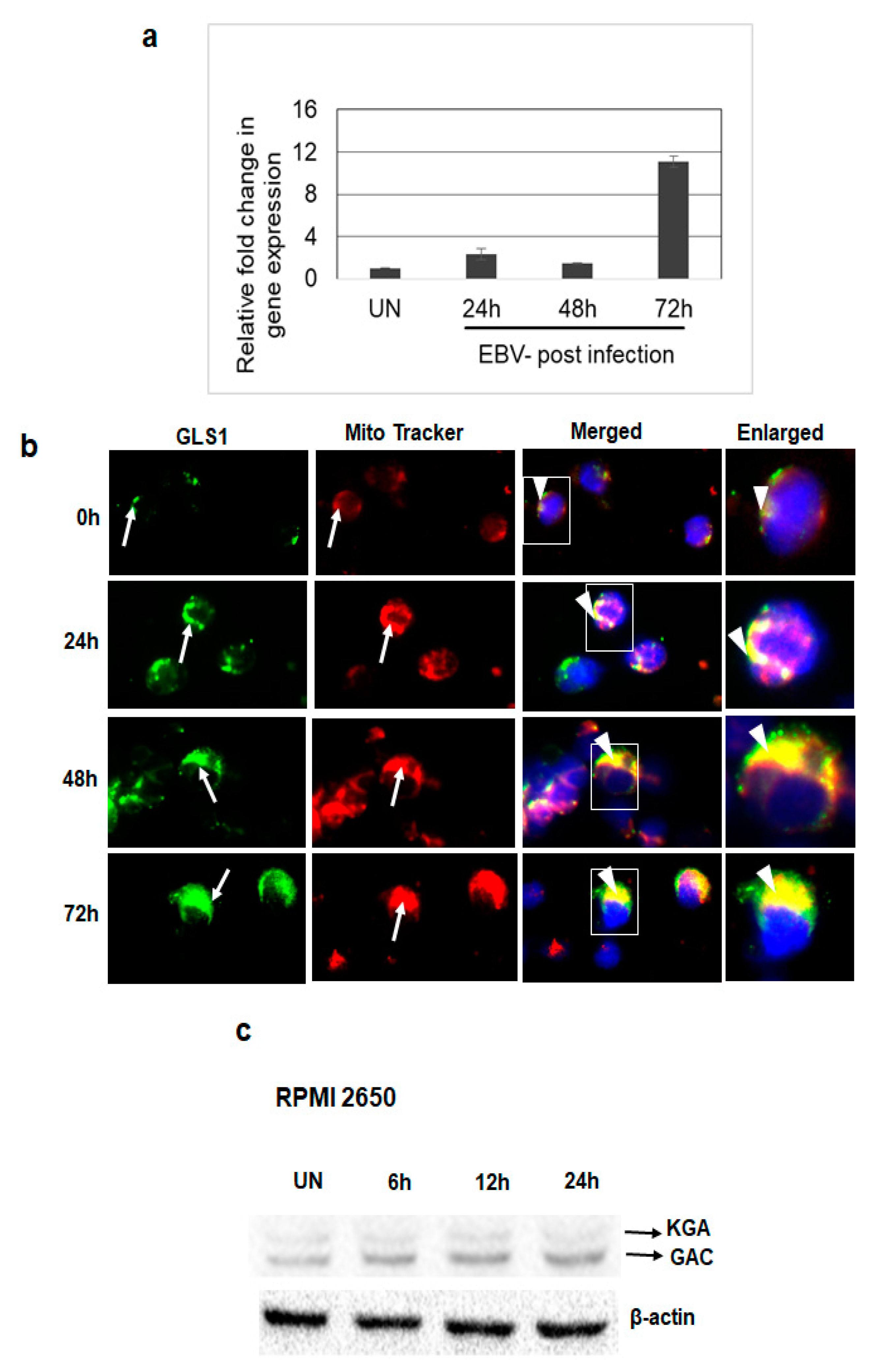
3.4. De Novo EBV Infection Induces GLS1 Expression and Mitochondrial Localization
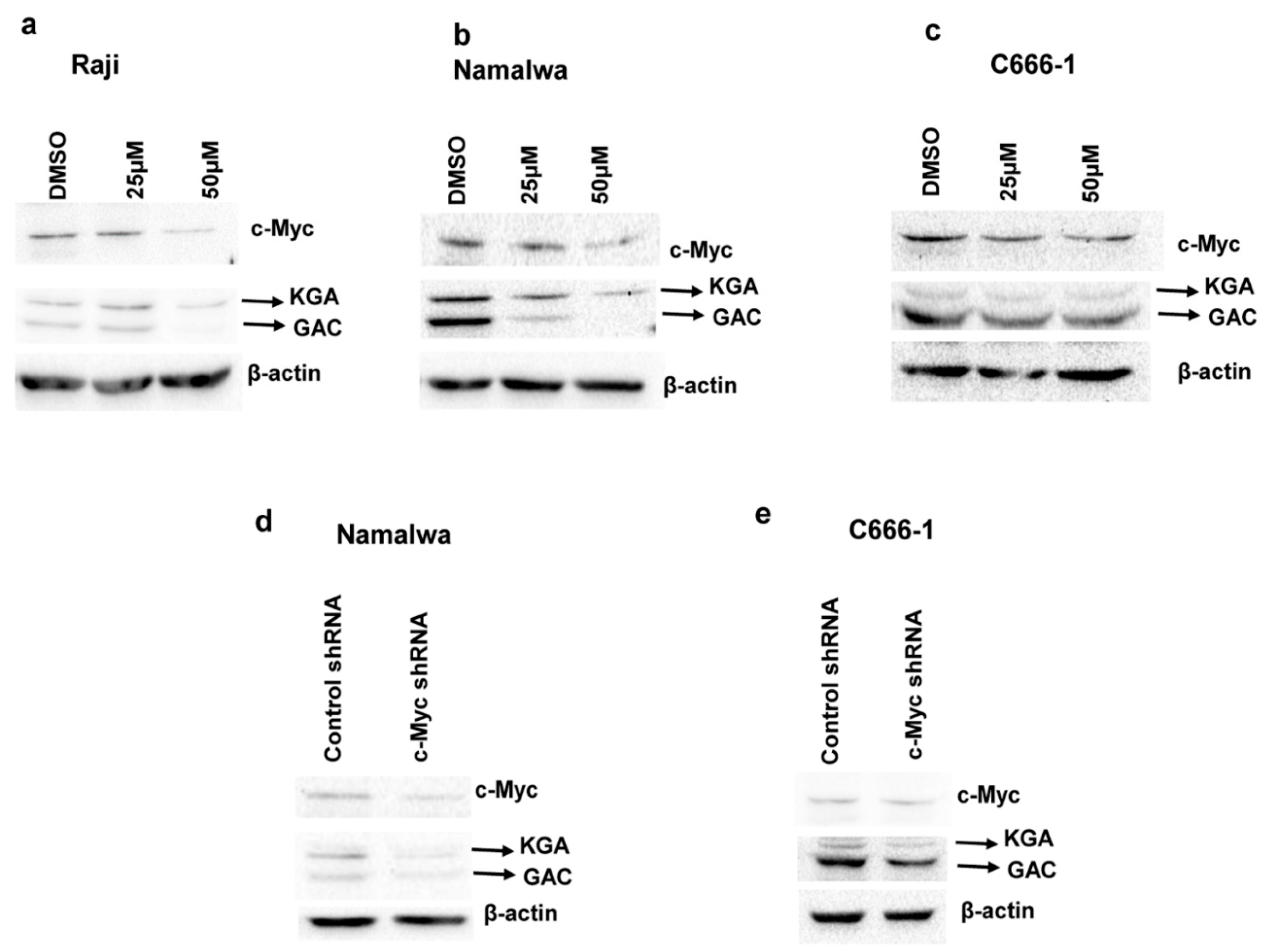
3.5. c-Myc Regulates the Expression of KGA and GAC in EBV-Infected Cells
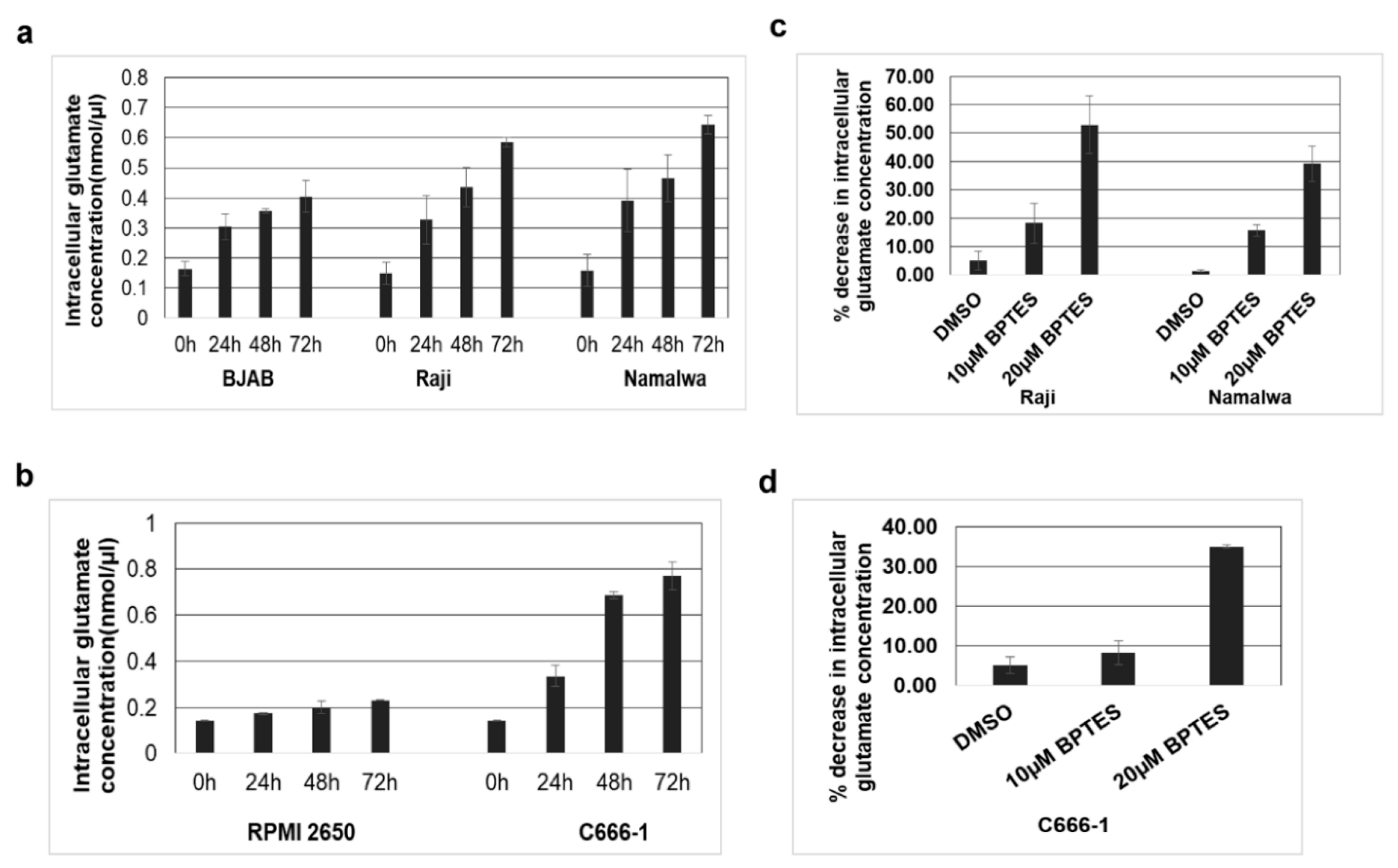
3.6. KGA and GAC Expression Increases Intracellular Glutamate Levels in EBV-Infected Cells
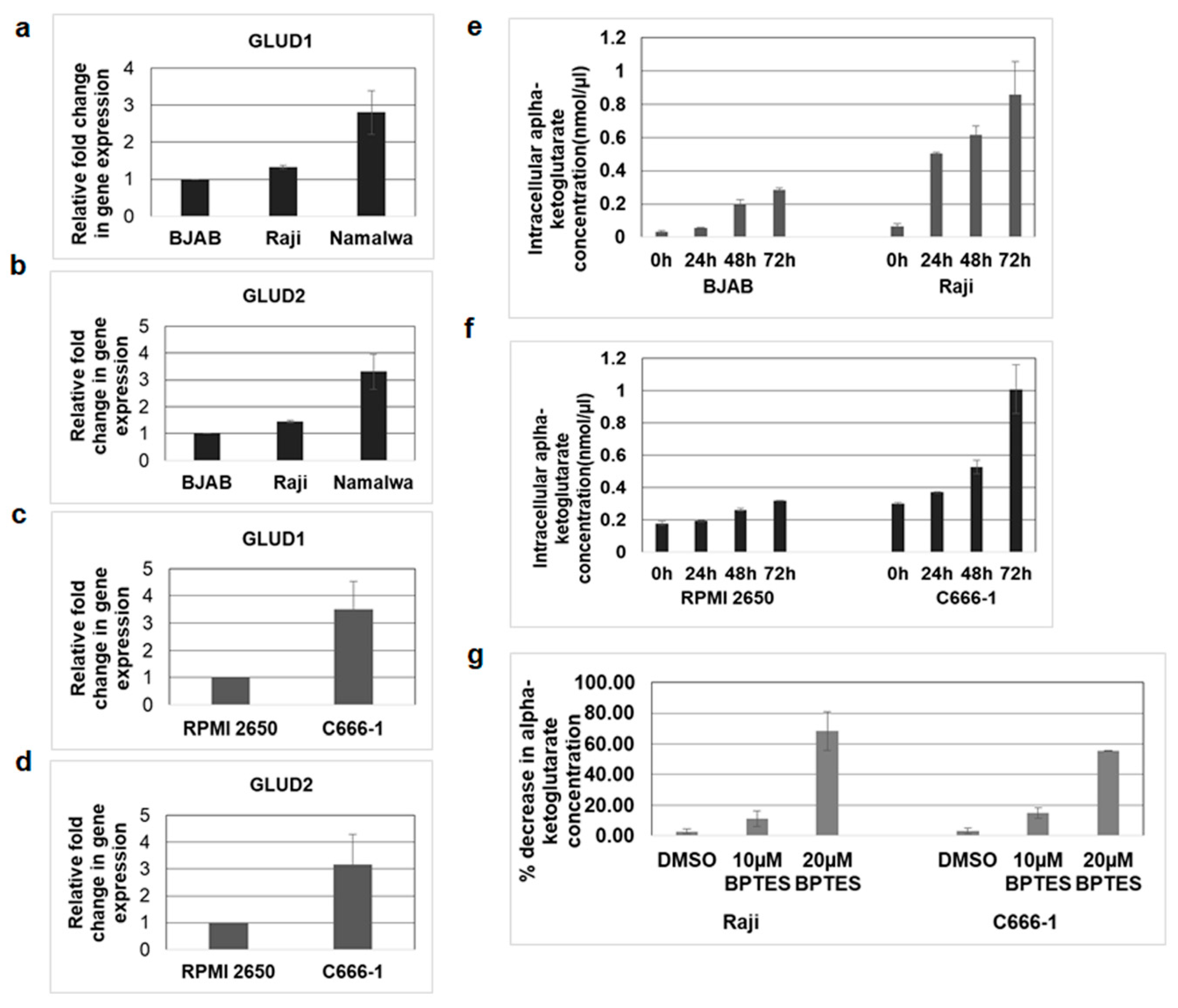
3.7. Mitochondrial GLUD1 and GLUD2 Were Upregulated and Alpha-Ketoglutarate Levels Increased in EBV-Infected Cells
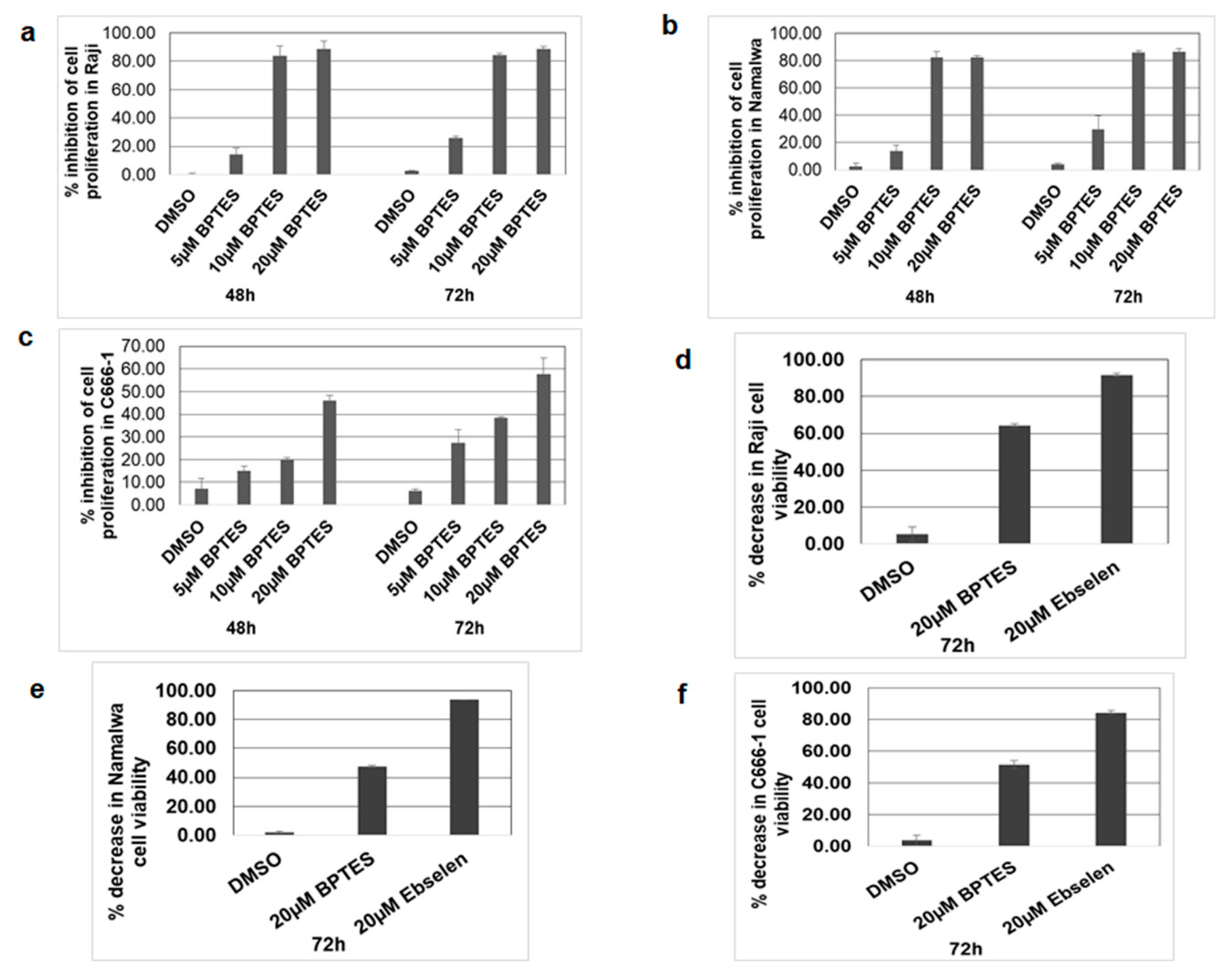
3.8. KGA, GAC Inhibitor, and GLUD1 Inhibitor Blocked Proliferation and Viability of EBV-Infected Cancer Cells
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Young, L.S.; Dawson, C.W. Epstein-Barr virus and nasopharyngeal carcinoma. Chin. J. Cancer 2014, 33, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Shannon-Lowe, C.; Rickinson, A.B.; Bell, A.I. Epstein-Barr virus-associated lymphomas. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160271. [Google Scholar] [CrossRef] [PubMed]
- Rickinson, A.B. Co-infections, inflammation and oncogenesis: Future directions for EBV research. Semin. Cancer Biol. 2014, 26, 99–115. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.S.; Kieff, E. Epstein-Barr virus latent genes. Exp. Mol. Med. 2015, 47, e131. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.; Robertson, E.S. Epstein-Barr virus-associated B-cell lymphomas: Pathogenesis and clinical outcomes. Clin. Cancer Res. 2011, 17, 3056–3063. [Google Scholar] [CrossRef]
- Thorley-Lawson, D.A.; Hawkins, J.B.; Tracy, S.I.; Shapiro, M. The pathogenesis of Epstein-Barr virus persistent infection. Curr. Opin. Virol. 2013, 3, 227–232. [Google Scholar] [CrossRef]
- Piccaluga, P.P.; Weber, A.; Ambrosio, M.R.; Ahmed, Y.; Leoncini, L. Epstein-Barr Virus-Induced Metabolic Rearrangements in Human B-Cell Lymphomas. Front. Microbiol. 2018, 9, 1233. [Google Scholar] [CrossRef]
- Ruf, I.K.; Rhyne, P.W.; Yang, H.; Borza, C.M.; Hutt-Fletcher, L.M.; Cleveland, J.L.; Sample, J.T. Epstein-barr virus regulates c-MYC, apoptosis, and tumorigenicity in Burkitt lymphoma. Mol. Cell. Biol. 1999, 19, 1651–1660. [Google Scholar] [CrossRef]
- Polack, A.; Hortnagel, K.; Pajic, A.; Christoph, B.; Baier, B.; Falk, M.; Mautner, J.; Geltinger, C.; Bornkamm, G.W.; Kempkes, B. c-myc activation renders proliferation of Epstein-Barr virus (EBV)-transformed cells independent of EBV nuclear antigen 2 and latent membrane protein 1. Proc. Natl. Acad. Sci. USA 1996, 93, 10411–10416. [Google Scholar] [CrossRef]
- Fan, C.S.; Wong, N.; Leung, S.F.; To, K.F.; Lo, K.W.; Lee, S.W.; Mok, T.S.; Johnson, P.J.; Huang, D.P. Frequent c-myc and Int-2 overrepresentations in nasopharyngeal carcinoma. Hum. Pathol. 2000, 31, 169–178. [Google Scholar] [CrossRef]
- Hui, A.B.; Lo, K.W.; Teo, P.M.; To, K.F.; Huang, D.P. Genome wide detection of oncogene amplifications in nasopharyngeal carcinoma by array based comparative genomic hybridization. Int. J. Oncol. 2002, 20, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Xiao, J.; Tao, Z.; Li, X. Detection of c-myc gene expression in nasopharyngeal carcinoma by nonradioactive in situ hybridization and immunohistochemistry. Chin. Med. J. (Engl.) 1997, 110, 229–232. [Google Scholar] [PubMed]
- Zhao, B.; Zou, J.; Wang, H.; Johannsen, E.; Peng, C.W.; Quackenbush, J.; Mar, J.C.; Morton, C.C.; Freedman, M.L.; Blacklow, S.C.; et al. Epstein-Barr virus exploits intrinsic B-lymphocyte transcription programs to achieve immortal cell growth. Proc. Natl. Acad. Sci. USA 2011, 108, 14902–14907. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V.; Le, A.; Gao, P. MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin. Cancer Res. 2009, 15, 6479–6483. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef]
- Jin, L.; Alesi, G.N.; Kang, S. Glutaminolysis as a target for cancer therapy. Oncogene 2016, 35, 3619–3625. [Google Scholar] [CrossRef]
- Elgadi, K.M.; Meguid, R.A.; Qian, M.; Souba, W.W.; Abcouwer, S.F. Cloning and analysis of unique human glutaminase isoforms generated by tissue-specific alternative splicing. Physiol. Genomics 1999, 1, 51–62. [Google Scholar] [CrossRef]
- Cassago, A.; Ferreira, A.P.; Ferreira, I.M.; Fornezari, C.; Gomes, E.R.; Greene, K.S.; Pereira, H.M.; Garratt, R.C.; Dias, S.M.; Ambrosio, A.L. Mitochondrial localization and structure-based phosphate activation mechanism of Glutaminase C with implications for cancer metabolism. Proc. Natl. Acad. Sci. USA 2012, 109, 1092–1097. [Google Scholar] [CrossRef]
- Lan, K.; Verma, S.C.; Murakami, M.; Bajaj, B.; Robertson, E.S. Epstein-Barr Virus (EBV): Infection, propagation, quantitation, and storage. Curr. Protoc. Microbiol. 2007, 6, 14E.2.1–14E.2.21. [Google Scholar] [CrossRef]
- Tiscornia, G.; Singer, O.; Verma, I.M. Production and purification of lentiviral vectors. Nat. Protoc. 2006, 1, 241–245. [Google Scholar] [CrossRef]
- Li, B.; Cao, Y.; Meng, G.; Qian, L.; Xu, T.; Yan, C.; Luo, O.; Wang, S.; Wei, J.; Ding, Y.; et al. Targeting glutaminase 1 attenuates stemness properties in hepatocellular carcinoma by increasing reactive oxygen species and suppressing Wnt/beta-catenin pathway. EBioMedicine 2019, 39, 239–254. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef] [PubMed]
- Yap, J.L.; Wang, H.; Hu, A.; Chauhan, J.; Jung, K.Y.; Gharavi, R.B.; Prochownik, E.V.; Fletcher, S. Pharmacophore identification of c-Myc inhibitor 10074-G5. Bioorg. Med. Chem. Lett. 2013, 23, 370–374. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.M.; McBryant, S.J.; Tsukamoto, T.; Rojas, C.; Ferraris, D.V.; Hamilton, S.K.; Hansen, J.C.; Curthoys, N.P. Novel mechanism of inhibition of rat kidney-type glutaminase by bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES). Biochem. J. 2007, 406, 407–414. [Google Scholar] [CrossRef]
- Huang, Q.; Stalnecker, C.; Zhang, C.; McDermott, L.A.; Iyer, P.; O’Neill, J.; Reimer, S.; Cerione, R.A.; Katt, W.P. Characterization of the interactions of potent allosteric inhibitors with glutaminase C, a key enzyme in cancer cell glutamine metabolism. J. Biol. Chem. 2018, 293, 3535–3545. [Google Scholar] [CrossRef]
- Duran, R.V.; Oppliger, W.; Robitaille, A.M.; Heiserich, L.; Skendaj, R.; Gottlieb, E.; Hall, M.N. Glutaminolysis activates Rag-mTORC1 signaling. Mol. Cell 2012, 47, 349–358. [Google Scholar] [CrossRef]
- Yu, Y.; Jin, Y.; Zhou, J.; Ruan, H.; Zhao, H.; Lu, S.; Zhang, Y.; Li, D.; Ji, X.; Ruan, B.H. Ebselen: Mechanisms of glutamate dehydrogenase and glutaminase enzyme inhibition. ACS Chem. Biol. 2017, 12, 3003–3011. [Google Scholar] [CrossRef]
- Young, L.S.; Murray, P.G. Epstein-Barr virus and oncogenesis: From latent genes to tumours. Oncogene 2003, 22, 5108–5121. [Google Scholar] [CrossRef]
- Saha, A.; Jha, H.C.; Upadhyay, S.K.; Robertson, E.S. Epigenetic silencing of tumor suppressor genes during in vitro Epstein-Barr virus infection. Proc. Natl. Acad. Sci. USA 2015, 112, E5199–E5207. [Google Scholar] [CrossRef]
- Mrozek-Gorska, P.; Buschle, A.; Pich, D.; Schwarzmayr, T.; Fechtner, R.; Scialdone, A.; Hammerschmidt, W. Epstein-Barr virus reprograms human B lymphocytes immediately in the prelatent phase of infection. Proc. Natl. Acad. Sci. USA 2019, 116, 16046–16055. [Google Scholar] [CrossRef] [PubMed]
- Sung, W.W.; Chu, Y.C.; Chen, P.R.; Liao, M.H.; Lee, J.W. Positive regulation of HIF-1A expression by EBV oncoprotein LMP1 in nasopharyngeal carcinoma cells. Cancer Lett. 2016, 382, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Daker, M.; Bhuvanendran, S.; Ahmad, M.; Takada, K.; Khoo, A.S. Deregulation of lipid metabolism pathway genes in nasopharyngeal carcinoma cells. Mol. Med. Rep. 2013, 7, 731–741. [Google Scholar] [CrossRef] [PubMed]
- Lo, A.K.; Lo, K.W.; Ko, C.W.; Young, L.S.; Dawson, C.W. Inhibition of the LKB1-AMPK pathway by the Epstein-Barr virus-encoded LMP1 promotes proliferation and transformation of human nasopharyngeal epithelial cells. J. Pathol. 2013, 230, 336–346. [Google Scholar] [CrossRef]
- Li, Y.; Peer, J.; Zhao, R.; Xu, Y.; Wu, B.; Wang, Y.; Tian, C.; Huang, Y.; Zheng, J. Serial deletion reveals structural basis and stability for the core enzyme activity of human glutaminase 1 isoforms: Relevance to excitotoxic neurodegeneration. Transl. Neurodegener. 2017, 6, 10. [Google Scholar] [CrossRef]
- Levy, P.L.; Duponchel, S.; Eischeid, H.; Molle, J.; Michelet, M.; Diserens, G.; Vermathen, M.; Vermathen, P.; Dufour, J.F.; Dienes, H.P.; et al. Hepatitis C virus infection triggers a tumor-like glutamine metabolism. Hepatology 2017, 65, 789–803. [Google Scholar] [CrossRef]
- Valiya Veettil, M.; Dutta, D.; Bottero, V.; Bandyopadhyay, C.; Gjyshi, O.; Sharma-Walia, N.; Dutta, S.; Chandran, B. Glutamate secretion and metabotropic glutamate receptor 1 expression during Kaposi’s sarcoma-associated herpesvirus infection promotes cell proliferation. PLoS Pathog. 2014, 10, e1004389. [Google Scholar] [CrossRef]
- Thai, M.; Thaker, S.K.; Feng, J.; Du, Y.; Hu, H.; Ting Wu, T.; Graeber, T.G.; Braas, D.; Christofk, H.R. MYC-induced reprogramming of glutamine catabolism supports optimal virus replication. Nat. Commun. 2015, 6, 8873. [Google Scholar] [CrossRef]
- Xiang, Y.; Stine, Z.E.; Xia, J.; Lu, Y.; O’Connor, R.S.; Altman, B.J.; Hsieh, A.L.; Gouw, A.M.; Thomas, A.G.; Gao, P.; et al. Targeted inhibition of tumor-specific glutaminase diminishes cell-autonomous tumorigenesis. J. Clin. Investig. 2015, 125, 2293–2306. [Google Scholar] [CrossRef]
- Yang, J.; Guo, Y.; Seo, W.; Zhang, R.; Lu, C.; Wang, Y.; Luo, L.; Paul, B.; Yan, W.; Saxena, D.; et al. Targeting cellular metabolism to reduce head and neck cancer growth. Sci. Rep. 2019, 9, 4995. [Google Scholar] [CrossRef]
- Thomas, A.G.; Rojas, C.; Tanega, C.; Shen, M.; Simeonov, A.; Boxer, M.B.; Auld, D.S.; Ferraris, D.V.; Tsukamoto, T.; Slusher, B.S. Kinetic characterization of ebselen, chelerythrine and apomorphine as glutaminase inhibitors. Biochem. Biophys. Res. Commun. 2013, 438, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Meng, G.; Zheng, M.; Zhang, Y.; Chen, A.; Wu, J.; Wei, J. The glutaminase-1 inhibitor 968 enhances dihydroartemisinin-mediated antitumor efficacy in hepatocellular carcinoma cells. PLoS ONE 2016, 11, e0166423. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Ruiz-Rodado, V.; Dowdy, T.; Huang, S.; Issaq, S.H.; Beck, J.; Wang, H.; Tran Hoang, C.; Lita, A.; Larion, M.; et al. Glutaminase-1 (GLS1) inhibition limits metastatic progression in osteosarcoma. Cancer Metab. 2020, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Gross, M.I.; Demo, S.D.; Dennison, J.B.; Chen, L.; Chernov-Rogan, T.; Goyal, B.; Janes, J.R.; Laidig, G.J.; Lewis, E.R.; Li, J.; et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol. Cancer Ther. 2014, 13, 890–901. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sl No. | Antibody | Make | Catalog No. |
|---|---|---|---|
| 1 | Rabbit anti-glutaminase antibody | Thermo Fisher Scientific | 710997 |
| 2 | Rabbit anti- KGA/GAC antibody | Proteintech | 12855-1-AP |
| 3 | Rabbit anti-β-actin antibody | Abcam | ab8227 |
| 4 | Mouse anti-c-Myc (9E10) antibody | Santa Cruz | sc-40 |
| 6 | Rabbit anti-VDAC antibody | Millipore | AB10527 |
| 7 | Anti-mouse secondary antibody linked to HRP | Thermo Fisher Scientific | 31430 |
| 8 | Anti-rabbit secondary antibody linked to HRP | Thermo Fisher Scientific | 31460 |
| 9 | Anti-rabbit secondary antibody conjugated to Alexa Flour 594 | Thermo Fisher Scientific | A11012 |
| 10 | Anti-rabbit secondary antibody conjugated to Alexa Flour 488 | Thermo Fisher Scientific | A11034 |
| Gene | Forward primer 5′-3′ | Reverse primer 5′-3′ |
|---|---|---|
| KGA | CTGGAAGCCTGCAAAGTAAAC | TGAGGTGTGTACTGGACTTGG |
| GAC | CCTCGAAGAGAAGGTGGTGATC | TGTCCTCATTTGACTCAGGTGAC |
| GLS1 | GTCACGATCTTGTTTCTCTGTG | GTCCAAAGAGCAGTGCTTCATCCATG |
| GLUD1 | GACACCAGGGTTTGGAGATAAA | TCAGACTCACCAACAGCAATAC |
| GLUD2 | ATCGGGTGCATCTGAGAAAG | CAGGTCCAATCCCAGGTTATAC |
| GAPDH | AGGGCTGCTTTTAACTCTGGT | CCCCACTTGATTTTGGAGGGA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krishna, G.; Soman Pillai, V.; Valiya Veettil, M. Upregulation of GLS1 Isoforms KGA and GAC Facilitates Mitochondrial Metabolism and Cell Proliferation in Epstein–Barr Virus Infected Cells. Viruses 2020, 12, 811. https://doi.org/10.3390/v12080811
Krishna G, Soman Pillai V, Valiya Veettil M. Upregulation of GLS1 Isoforms KGA and GAC Facilitates Mitochondrial Metabolism and Cell Proliferation in Epstein–Barr Virus Infected Cells. Viruses. 2020; 12(8):811. https://doi.org/10.3390/v12080811
Chicago/Turabian StyleKrishna, Gayathri, Vinod Soman Pillai, and Mohanan Valiya Veettil. 2020. "Upregulation of GLS1 Isoforms KGA and GAC Facilitates Mitochondrial Metabolism and Cell Proliferation in Epstein–Barr Virus Infected Cells" Viruses 12, no. 8: 811. https://doi.org/10.3390/v12080811
APA StyleKrishna, G., Soman Pillai, V., & Valiya Veettil, M. (2020). Upregulation of GLS1 Isoforms KGA and GAC Facilitates Mitochondrial Metabolism and Cell Proliferation in Epstein–Barr Virus Infected Cells. Viruses, 12(8), 811. https://doi.org/10.3390/v12080811