Molecular Evolution and Structural Mapping of N-Terminal Domain in Spike Gene of Middle East Respiratory Syndrome Coronavirus (MERS-CoV)

 , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethical Considerations
2.2. Biosafety Considerations
2.3. Study Specimens
2.4. Laboratory Investigations
2.5. RNA Extraction
2.6. Real-Time PCR Assay—Upstream of E Gene (upE Assay)
2.7. Confirmatory Real-Time PCR Assay in ORF 1A (1A Assay)
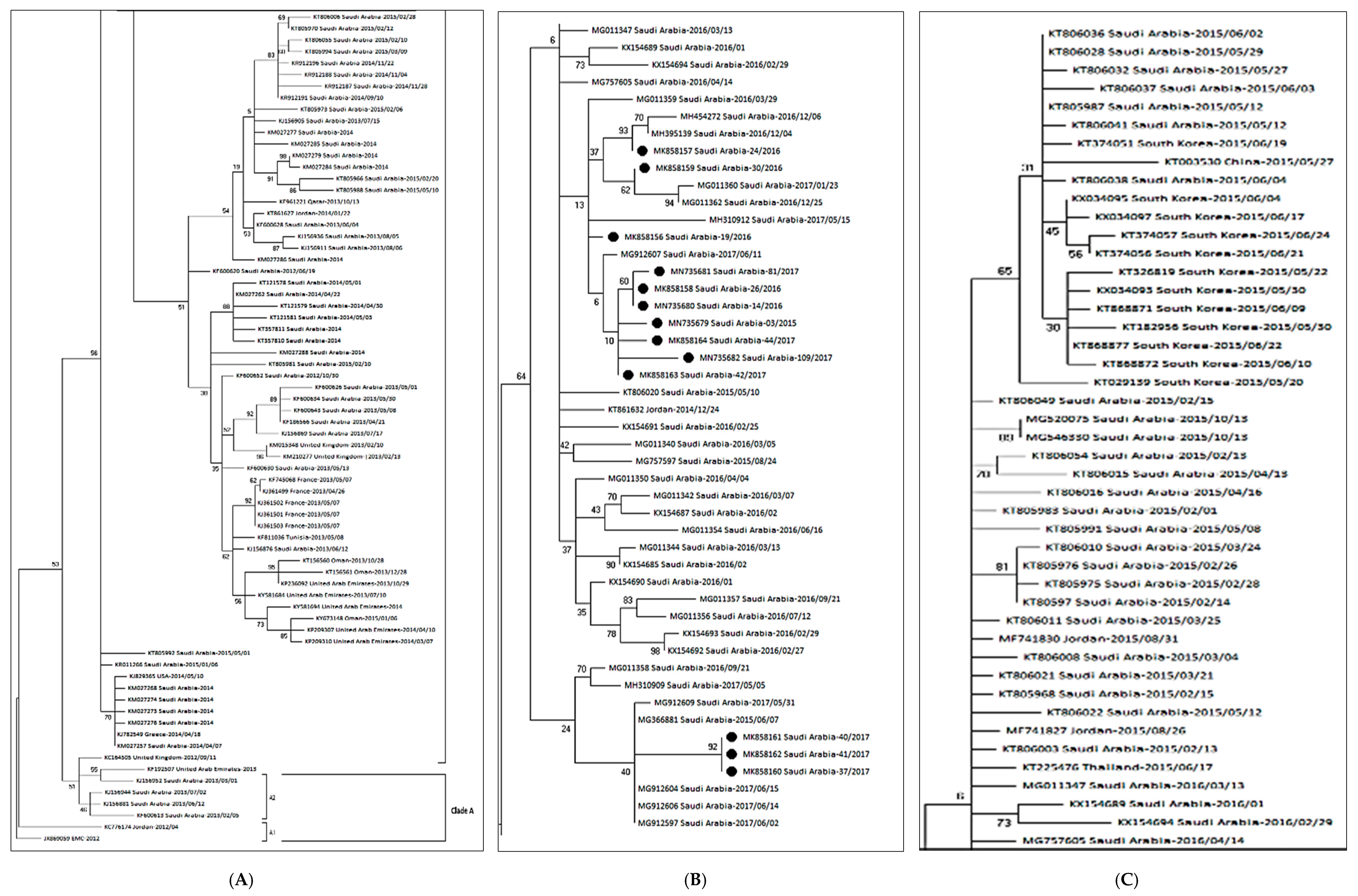
2.8. Genetic Sequencing & Phylogenetic Analyses
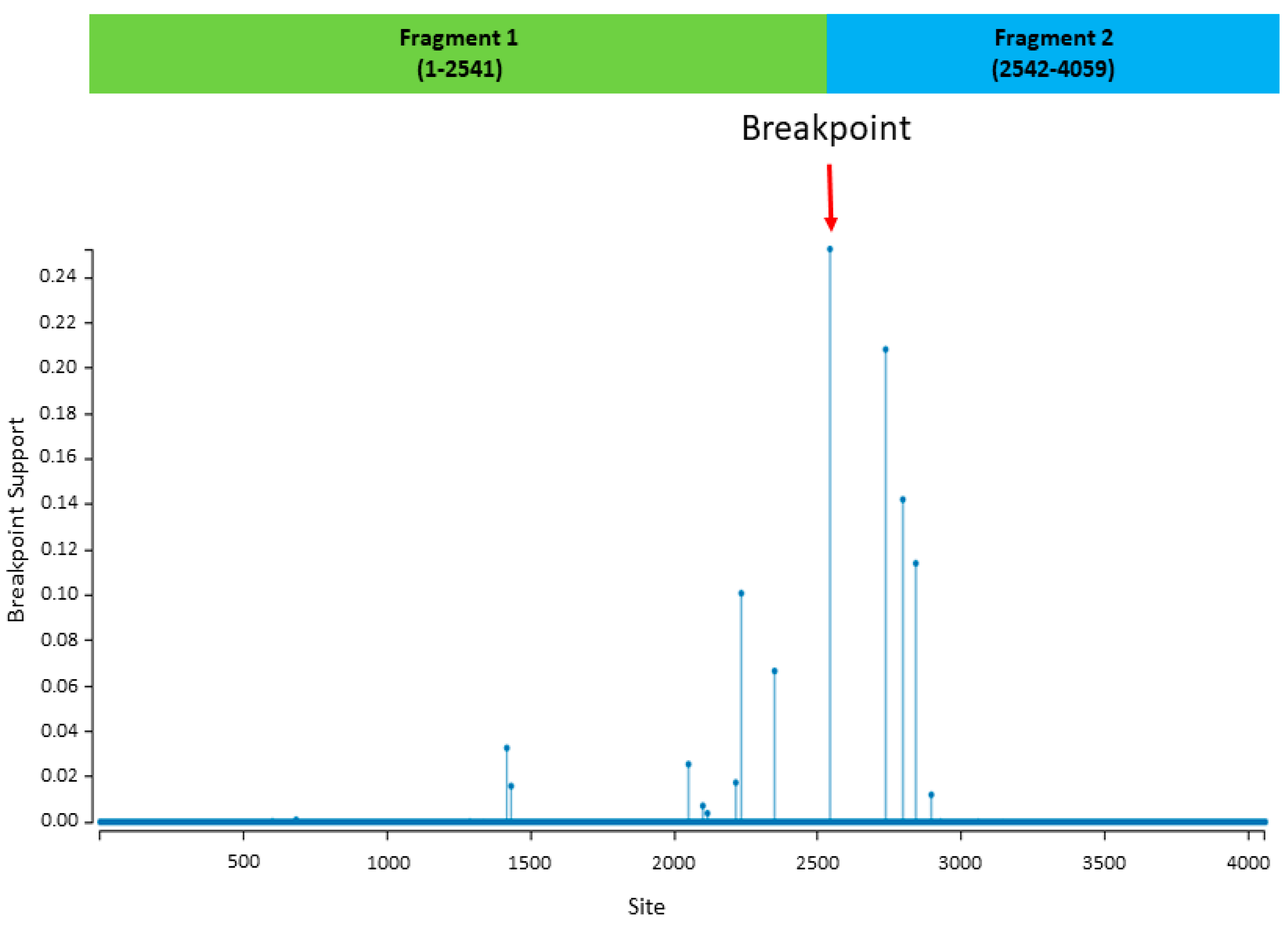
2.9. Recombination Analysis and Phylogenetic Reconstruction Based on Recombinant Fragments
2.10. Measurement of Selection Pressure
2.11. Prediction of Glycosylation Sites
2.12. MERS-CoV Viral Load Assay
2.13. Statistical Analysis
3. Results
3.1. Selective Pressure Analysis
3.2. Prediction of Glycosylation Sites
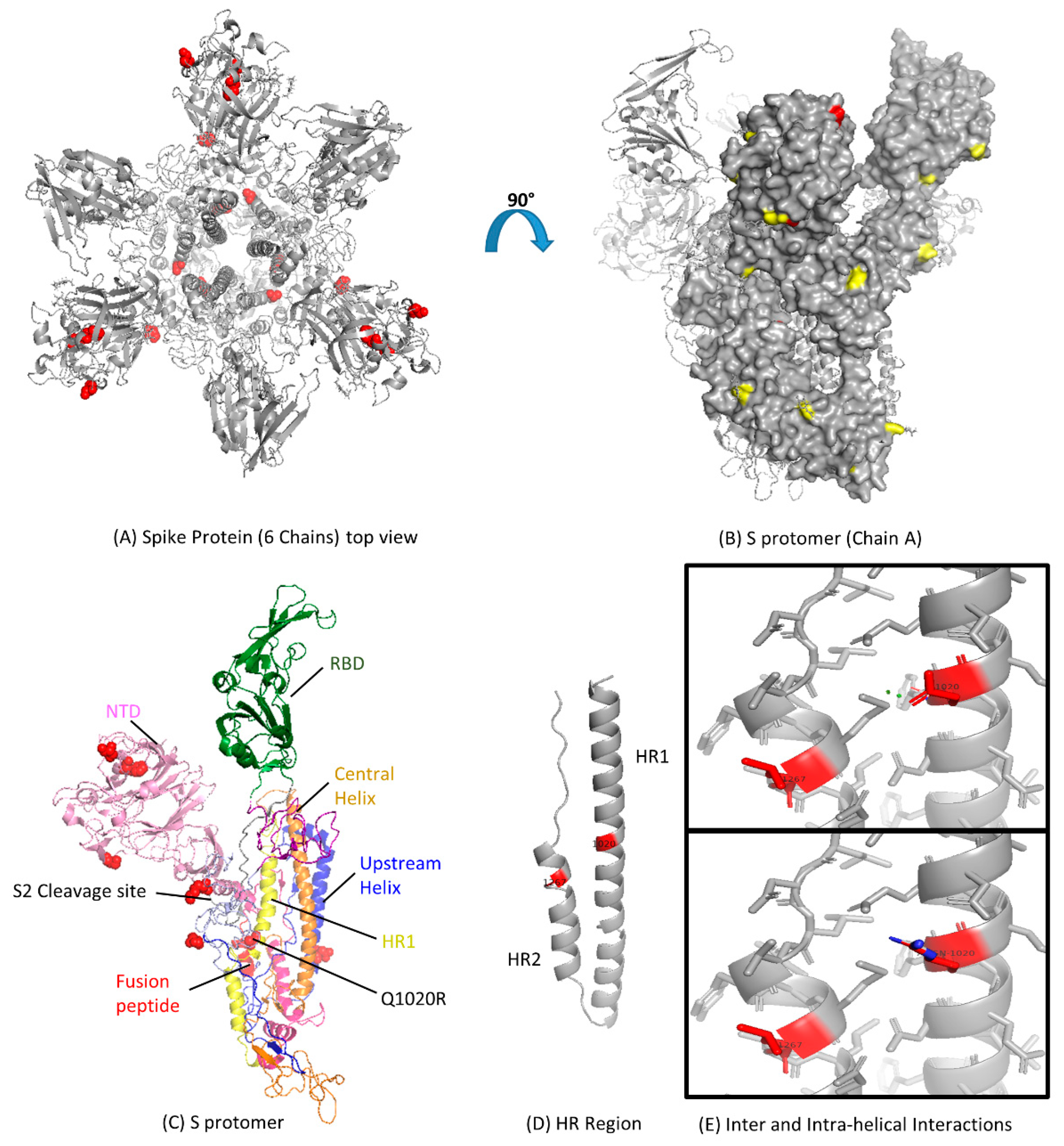
3.3. Modeling of Spike Protein
3.4. Analysis of MERS-CoV HR Variation
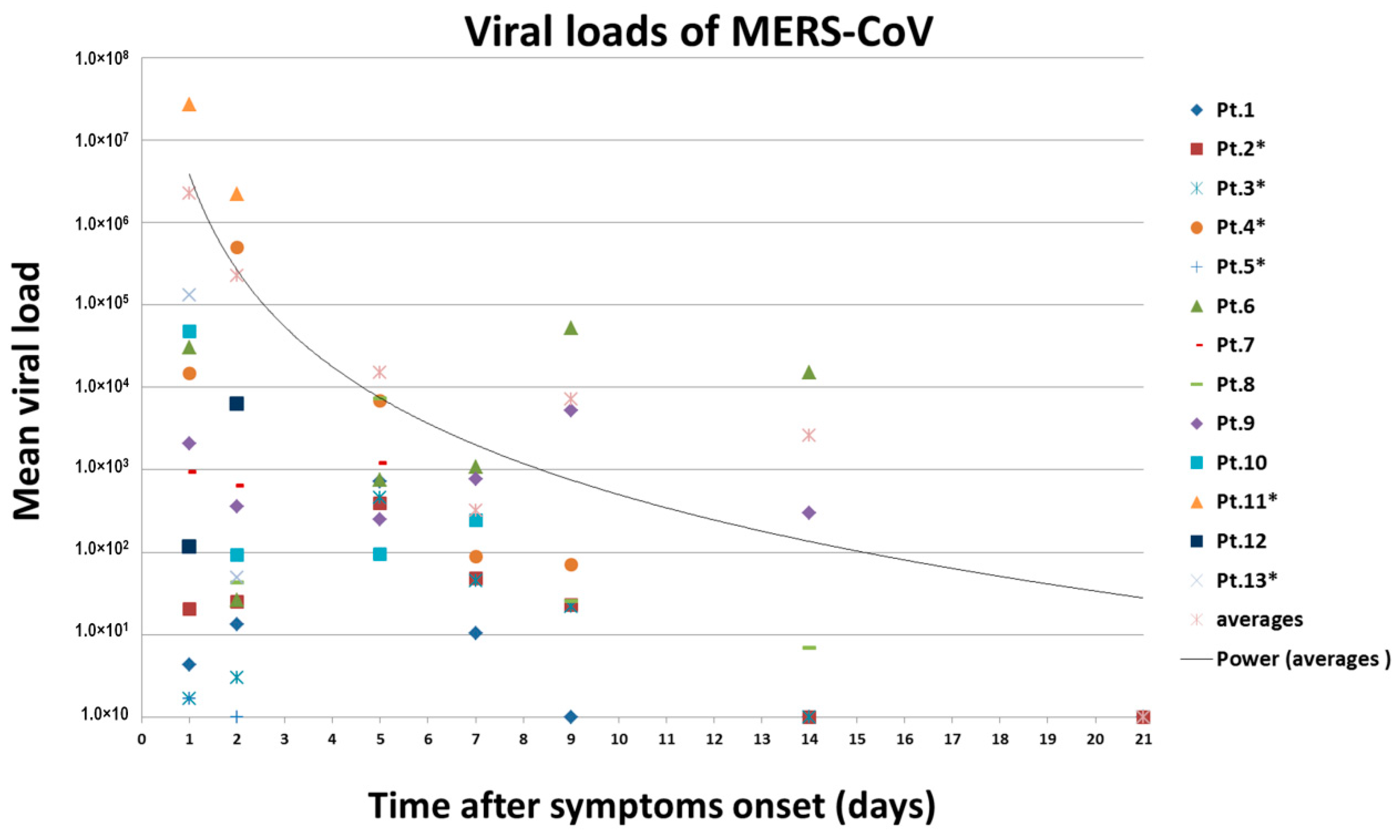
3.5. Viral Loads Assay
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MERS-CoV | Middle East Respiratory Syndrome Coronavirus |
| DPP4 | dipeptidyl peptidase 4 |
| RBD | Receptor binding domain |
| NTD | N terminal domain |
| HR1 | Heptad repeat 1 |
| S | Spike |
| Q | Glutamine |
| R | Arginine |
| RT-PCR | Reverse Transcription Polymerase Chain Reaction |
| KFMC | King Fahad Medical City |
| IRB | Institutional Review Board |
References
- Zaki, A.M.; van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.; Fouchier, R.A. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef] [PubMed]
- De Groot, R.J.; Baker, S.C.; Baric, R.S.; Brown, C.S.; Drosten, C.; Enjuanes, L.; Fouchier, R.A.M.; Galiano, M.; Gorbalenya, A.E.; Memish, Z.A.; et al. Middle East respiratory syndrome coronavirus (MERS-CoV): Announcement of the Coronavirus Study Group. J. Virol. 2013, 87, 7790–7792. [Google Scholar] [CrossRef] [PubMed]
- Al-Abdallat, M.M.; Payne, D.C.; Alqasrawi, S.; Rha, B.; Tohme, R.A.; Abedi, G.R.; Nsour, M.A.; Iblan, I.; Jarour, N.; Farag, N.H.; et al. Hospital-associated outbreak of middle East respiratory syndrome coronavirus: A serologic, epidemiologic, and clinical description. Clin. Infect. Dis. 2014, 59, 1225–1233. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Update and Clarification on Recent MERS Cases Reported by the Kingdom of Saudi Arabia. 2019. Available online: http://who.int/emergencies/mers-cov/saudi-arabia-update/en/ (accessed on 24 December 2019).
- Alosaimi, B.; Hamed, M.; Naeem, A. Middle East Respiratory Syndrome Coronavirus (MERS-CoV): Transmission Dynamics and Prospects for Vaccine Development. EC Microbiol. J. 2019, 15, 109–119. [Google Scholar]
- Masters, P.S.; Perlman, S. Coronaviridae. In Fields Virology; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 825–858. [Google Scholar]
- van Boheemen, S.; de Graaf, M.; Lauber, C.; Bestebroer, T.M.; Raj, V.S.; Zaki, A.M.; Osterhaus, A.D.M.E.; Haagmans, B.L.; Gorbalenya, A.E.; Snijder, E.J.; et al. Genomic characterization of a newly discovered coronavirus associated with acute respiratory distress syndrome in humans. mBio 2012, 3, e00473-12. [Google Scholar] [CrossRef] [PubMed]
- Raj, V.S.; Mou, H.; Smits, S.L.; Dekkers, D.H.; Muller, M.A.; Dijkman, R.; Muth, D.; Demmers, J.A.A.; Zaki, A.; Fouchier, R.A.M.; et al. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature 2013, 495, 251–254. [Google Scholar] [CrossRef]
- Lu, G.; Hu, Y.; Wang, Q.; Qi, J.; Gao, F.; Li, Y.; Zhang, Y.; Zhang, W.; Yuan, Y.; Bao, J.; et al. Molecular basis of binding between novel human coronavirus MERS-CoV and its receptor CD26. Nature 2013, 500, 227. [Google Scholar] [CrossRef]
- Wang, N.; Shi, X.; Jiang, L.; Zhang, S.; Wang, D.; Tong p Guo, D.; Fu, L.; Cui, Y.; Liu, X.; Arledge, K.C.; et al. Structure of MERS-CoV spike receptor-binding domain complexed with human receptor DPP4. Cell Res. 2013, 23, 986–993. [Google Scholar] [CrossRef]
- Lu, L.; Liu, Q.; Zhu, Y.; Chan, K.H.; Qin, L.; Li, Y.; Wang, Q.; Chan, J.F.; Du, L.; Yu, F.; et al. Structure-based discovery of Middle East respiratory syndrome coronavirus fusion inhibitor. Nat. Commun. 2014, 5, 3067. [Google Scholar] [CrossRef]
- Drosten, C.; Muth, D.; Corman, V.M.; Hussain, R.; Al Masri, M.; HajOmar, W.; Landt, O.; Assiri, A.; Eckerle, I.; Al Shangiti, A.; et al. An observational, laboratory-based study of outbreaks of middle East respiratory syndrome coronavirus in Jeddah and Riyadh, Kingdom of Saudi Arabia, 2014. Clin. Infect. Dis. 2015, 60, 369. [Google Scholar] [CrossRef]
- Assiri, A.; McGeer, A.; Perl, T.M.; Price, C.S.; Al Rabeeah, A.A.; Cummings, D.A.; Alabdullatif, Z.N.; Assad, M.; Almulhim, A.; Makhdoom, H.; et al. Hospital outbreak of Middle East respiratory syndrome coronavirus. N. Engl. J. Med. 2013, 369, 407. [Google Scholar] [CrossRef] [PubMed]
- Memish, Z.A.; Zumla, A.I.; Al-Hakeem, R.F.; Al-Rabeeah, A.A.; Stephens, G.M. Family cluster of Middle East respiratory syndrome coronavirus infections. N. Engl. J. Med. 2013, 368, 2487–2494. [Google Scholar] [CrossRef] [PubMed]
- Omrani, A.S.; Matin, M.A.; Haddad, Q.; Al-Nakhli, D.; Memish, Z.A.; Albarrak, A.M. A family cluster of Middle East Respiratory Syndrome Coronavirus infections related to a likely unrecognized asymptomatic or mild case. Int. J. Infect. Dis. 2013, 17, e668–e672. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.K.; Lee, P.; Tsang, A.K.L.; Yip, C.C.Y.; Tse, H.; Lee, R.A.; So, L.-Y.; Lau, Y.-L.; Chan, K.-H.; Woo, P.C.Y.; et al. Molecular epidemiology of human coronavirus OC43 reveals evolution of different genotypes over time and recent emergence of a novel genotype due to natural recombination. J. Virol. 2011, 85, 11325. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.K.; Woo, P.C.Y.; Yip, C.C.Y.; Fan, R.Y.Y.; Huang, Y.; Wang, M.; Guo, R.; Lam, C.S.F.; Tsang, A.K.L.; Lai, K.K.Y.; et al. Isolation and characterization of a novel Betacoronavirus subgroup A coronavirus, rabbit coronavirus HKU14, from domestic rabbits. J. Virol. 2012, 86, 5481. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, D.; Shi, W.; Lu, R.; Wang, W.; Zhao, Y.; Deng, Y.; Zhou, W.; Ren, H.; Wu, J.; et al. Origin and Possible Genetic Recombination of the Middle East Respiratory Syndrome Coronavirus from the First Imported Case in China: Phylogenetics and Coalescence Analysis. MBio 2015, 6, e1215. [Google Scholar] [CrossRef]
- Cotton, M.; Watson, S.J.; Kellam p Al-Rabeeah, A.A.; Makhdoom, H.Q.; Assiri, A.; Al-Tawfiq, J.A.; Alhakeem, R.F.; Madani, H.; AlRabiah, F.A.; Al Hajjar, S.; et al. Transmission and evolution of the Middle East respiratory syndrome coronavirus in Saudi Arabia: A descriptive genomic study. Lancet 2013, 382, 1993. [Google Scholar] [CrossRef]
- Muth, D.; Corman, V.M.; Meyer, B.; Assiri, A.; Al-Masri, M.; Farah, M.; Steinhagen, K.; Lattwein, E.; Al-Tawfiq, J.A.; Albarrak, A.; et al. Infectious Middle East Respiratory Syndrome Coronavirus Excretion and Serotype Variability Based on Live Virus Isolates from Patients in Saudi Arabia. J. Clin. Microbiol. 2015, 53, 2951–2955. [Google Scholar] [CrossRef]
- Assiri, A.M.; Midgley, C.M.; Abedi, G.R.; Bin Saeed, A.; Almasri, M.M.; Lu, X.; Al-Abdely, H.M.; Abdalla, O.; Mohammed, M.; Algarni, H.S.; et al. Epidemiology of a Novel Recombinant Middle East Respiratory Syndrome Coronavirus in Humans in Saudi Arabia. J. Infect. Dis. 2016, 214, 712–721. [Google Scholar] [CrossRef]
- Park, Y.S.; Lee, C.; Kim, K.M.; Kim, S.W.; Lee, K.J.; Ahn, J.; Ki, M. The first case of the 2015 Korean Middle East Respiratory Syndrome outbreak. Epidemiol. Health 2015, 37, e2015049. [Google Scholar] [CrossRef]
- Guery, B.; Poissy, J.; el Mansouf, L.; Séjourné, C.; Ettahar, N.; Lemaire, X.; Vuotto, F.; Goffard, A.; Behillil, S.; Enouf, V.; et al. Clinical features and viral diagnosis of two cases of infection with Middle East Respiratory Syndrome coronavirus: A report of nosocomial transmission. Lancet 2013, 381, 2265. [Google Scholar] [CrossRef]
- Abroug, F.; Slim, A.; Ouanes-Besbes, L.; Hadj Kacem, M.A.; Dachraoui, F.; Ouanes, I.; Lu, X.; Tao, Y.; Paden, C.; Caidi, H.; et al. Family cluster of Middle East respiratory syndrome coronavirus infections, Tunisia, 2013. Emerg. Infect. Dis. 2014, 20, 1527. [Google Scholar] [CrossRef] [PubMed]
- Raj, V.S.; Farag, E.A.; Reusken, C.B.; Lamers, M.M.; Pas, S.D.; Voermans, J.; Smits, S.L.; Osterhaus, A.D.; Al-Mawlawi, N.; Al-Romaihi, H.E.; et al. Isolation of MERS coronavirus from a dromedary camel, Qatar, 2014. Emerg. Infect. Dis. 2014, 20, 1339. [Google Scholar] [CrossRef] [PubMed]
- Hui, D.S.; Perlman, S.; Zumla, A. Spread of MERS to South Korea and China. Lancet Respir. Med. 2015, 3, 509. [Google Scholar] [CrossRef]
- Park, H.Y.; Lee, E.J.; Ryu, Y.W.; Kim, Y.; Kim, H.; Lee, H.; Yi, S.J. Epidemiological investigation of MERS-CoV spread in a single hospital in South Korea, May to June 2015. Eurosurveillance 2015, 20, 1. [Google Scholar] [CrossRef] [PubMed]
- Hunter, J.C.; Nguyen, D.; Aden, B.; Al Bandar, Z.; Al Dhaheri, W.; Abu Elkheir, K.; Khudair, A.; Al Mulla, M.; El Saleh, F.; Imambaccus, H.; et al. Transmission of Middle East Respiratory Syndrome Coronavirus Infections in Healthcare Settings, Abu Dhabi. Emerg. Infect. Dis. 2016, 22, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Drosten, C.; Meyer, B.; Müller, M.A.; Corman, V.M.; Al-Masri, M.; Hossain, R.; Madani, H.; Sieberg, A.; Bosch, B.J.; Lattwein, E.; et al. Transmission of MERS-coronavirus in household contacts. N. Engl. J. Med. 2014, 371, 828–835. [Google Scholar] [CrossRef]
- Corman, V.M.; Eckerle, I.; Bleicker, T.; Zaki, A.; Landt, O.; Eschbach-Bludau, M.; van Boheemen, S.; Gopal, R.; Ballhause, M.; Bestebroer, T.M.; et al. Detection of a novel human coronavirus by real-time reverse-transcription polymerase chain reaction. Eurosurveillance 2012, 17, 20285. [Google Scholar] [CrossRef]
- Corman, V.M.; Müller, M.A.; Costabel, U.; Timm, J.; Binger, T.; Meyer, B.; Kreher p Lattwein, E.; Eschbach-Bludau, M.; Nitsche, A.; Bleicker, T.; et al. Assays for laboratory confirmation of novel human coronavirus (hCoV-EMC) infections. Eurosurveillance 2012, 17, 20334. [Google Scholar] [CrossRef]
- Drosten, C.; Günther, S.; Preiser, W.; van der Werf, S.; Brodt, H.R.; Becker, S.; Rabenau, H.; Panning, M.; Kolesnikova, L.; Fouchier, R.A.; et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1967–1976. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Dong, W.; Milewska, A.; Golda, A.; Qi, Y.; Zhu, Q.K.; Marasco, W.A.; Baric, R.S.; Sims, A.C.; Pyrc, K.; et al. Human Coronavirus HKU1 Spike Protein Uses O-Acetylated sialic acid as an attachment receptor determinant and employs hemagglutinin-esterase protein as a receptor-destroying enzyme. J. Virol. 2015, 89, 7202–7213. [Google Scholar] [CrossRef] [PubMed]
- Delport, W.; Poon, A.F.; Frost, S.D.; Pond, K.; Datamonkey, S.L. 2010: A suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 2010, 26, 2455–2457. [Google Scholar] [CrossRef] [PubMed]
- Arenas, M.; Posada, D. The effect of recombination on the reconstruction of ancestral sequences. Genetics 2010, 184, 1133–1139. [Google Scholar] [CrossRef]
- Naeem, A.; Hosomi, T.; Nishimura, Y.; Alam, M.M.; Oka, T.; Zaidi, S.S.Z.; Shimizu, H. Genetic diversity of circulating Saffold viruses in Pakistan and Afghanistan. J. Gen. Virol. 2014, 95, 1945–1957. [Google Scholar] [CrossRef]
- Gupta, R.; Jung, E.; Brunak, S. Prediction of N-glycosylation Sites in Human Proteins, 2004, Database: NetNGlyc 1.0. Available online: http://www.cbs.dtu.dk/services/NetNGlyc/ (accessed on 4 February 2020).
- Lau, S.K.P.; Wong, A.C.P.; Lau, T.C.K.; Woo, P.C.Y. Molecular Evolution of MERS Coronavirus: Dromedaries as a Recent Intermediate Host or Long-Time Animal Reservoir? Int. J. Mol. Sci. 2017, 18, 2138. [Google Scholar] [CrossRef]
- Corman, V.M.; Ithete, N.L.; Richards, L.R.; Schoeman, M.C.; Preiser, W.; Drosten, C.; Drexler, J.F. Rooting the phylogenetic tree of Middle East respiratory syndrome coronavirus by characterization of a conspecific virus from an African bat. J. Virol. 2014, 88, 11297–11303. [Google Scholar] [CrossRef]
- Kossyvakis, A.; Tao, Y.; Lu, X.; Pogka, V.; Tsiodras, S.; Emmanouil, M.; Mentis, A.F.; Tong, S.; Erdman, D.D.; Antoniadis, A. Laboratory investigation and phylogenetic analysis of an imported Middle East respiratory syndrome coronavirus case in Greece. PLoS ONE 2015, 10, e0125809. [Google Scholar] [CrossRef]
- Sabir, J.S.; Lam, T.T.; Ahmed, M.M.; Li, L.; Shen, Y.; Abo-Aba, S.E.; Qureshi, M.I.; Abu-Zeid, M.; Zhang, Y.; Khiyami, M.A.; et al. Co-circulation of three camel coronavirus species and recombination of MERS-CoVs in Saudi Arabia. Science 2016, 351, 81–84. [Google Scholar] [CrossRef]
- Kryazhimskiy, S.; Plotkin, J.B. The population genetics of dN/dS. PLoS Genet. 2008, 4, e1000304. [Google Scholar] [CrossRef]
- De Groot, R.J.; Luytjes, W.; Horzinek, M.C.; van der Zeijst, B.A.; Spaan, W.J.; Lenstra, J.A. Evidence for a coiled-coil structure in the spike proteins of coronaviruses. J. Mol. Biol. 1987, 196, 963–966. [Google Scholar] [CrossRef]
- Zhang, Z.; Shen, L.; Gu, X. Evolutionary dynamics of MERS-CoV: Potential recombination, positive selection and transmission. Sci. Rep. 2016, 6, 25049. [Google Scholar] [CrossRef] [PubMed]
- Naeem, A.; Elbakkouri, K.; Alfaiz, A.; Hamed, M.; Alsaran, H.; AlOtaiby, S.; Enani, M.; Alosaimi, B. Antigenic Drift of Hemagglutinin and Neuraminidase in seasonal H1N1 influenza Viruses from Saudi Arabia in 2014–2015. J. Med. Virol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.W.; Kim, Y.J.; Park, S.H.; Yun, M.R.; Yang, J.S.; Kang, H.J.; Han, Y.W.; Lee, H.S.; Kim, H.M.; Kim, H.; et al. Variations in spike glycoprotein gene of MERS-CoV, South Korea, 2015. Emerg. Infect. Dis. 2016, 22, 100–104. [Google Scholar] [CrossRef]
- Plipat, T.; Buathong, R.; Wacharapluesadee, S.; Siriarayapon, P.; Pittayawonganon, C.; Sangsajja, C.; Kaewpom, T.; Petcharat, S.; Ponpinit, T.; Jumpasri, J.; et al. Imported case of Middle East respiratory syndrome coronavirus (MERS-CoV) infection from Oman to Thailand, June 2015. Eurosurveillance 2017, 22, 30598. [Google Scholar] [CrossRef]
- Graham, R.L.; Baric, R.S. Recombination, reservoirs, and the modular spike: Mechanisms of coronavirus cross-species transmission. J. Virol. 2010, 84, 3134–3146. [Google Scholar] [CrossRef]
- Zhao, K.; Ye, C.; Chang, X.-B.; Jiang, C.-G.; Wang, S.-J.; Cai, X.-H.; Tong, G.; Tian, Z.; Shi, M.; An, T. Importation and recombination are responsible for the latest emergence of highly pathogenic PRRSV in China. J. Virol. 2015, 89, 10712–10716. [Google Scholar] [CrossRef]
- Assiri, A.M.; Biggs, H.M.; Abedi, G.R.; Lu, X.; Bin Saeed, A.; Abdalla, O.; Mohammed, M.; Al-Abdely, H.M.; Algarni, H.S.; Alhakeem, R.F.; et al. Increase in Middle East respiratory syndrome-coronavirus cases in Saudi Arabia linked to hospital outbreak with continued circulation of recombinant virus, July 1–August 31, 2015. Open Forum Infect. Dis. 2016, 3, ofw165. [Google Scholar] [CrossRef]
- Lamers, M.M.; Raj, V.S.; Shafei, M.; Ali, S.S.; Abdallh, S.M.; Gazo, M.; Nofal, S.; Lu, X.; Erdman, D.D.; Koopmans, M.P.; et al. Deletion variants of Middle East respiratory syndrome coronavirus from humans, Jordan, 2015. Emerg. Infect. Dis. 2016, 22, 716–719. [Google Scholar] [CrossRef]
- Payne, D.C.; Biggs, H.M.; Al-Abdallat, M.M.; Alqasrawi, S.; Lu, X.; Abedi, G.R.; Haddadin, A.; Iblan, I.; Alsanouri, T.; Nsour, M.A.; et al. Multihospital outbreak of a Middle East respiratory syndrome coronavirus deletion variant, Jordan: A molecular, serologic, and epidemiologic investigation. Open Forum Infect Dis. 2018, 5, ofy095. [Google Scholar] [CrossRef]
- Schultze, B.; Gross, H.J.; Brossmer, R.; Herrler, G. The S protein of bovine coronavirus is a hemagglutinin recognizing 9-O-acetylated sialic acid as a receptor determinant. J. Virol. 1991, 65, 6232–6237. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Park, S.J.; Cho, S.Y.; Cha, R.H.; Jee, H.G.; Kim, G.; Shin, H.S.; Kim, Y.; Jung, Y.M.; Yang, J.S.; et al. Viral RNA in Blood as Indicator of Severe Outcome in Middle East Respiratory Syndrome Coronavirus Infection. Emerg. Infect. Dis. 2016, 22, 1813–1816. [Google Scholar] [CrossRef] [PubMed]
- Corman, V.M.; Albarrak, A.M.; Omrani, A.S.; Albarrak, M.M.; Farah, M.E.; Almasri, M.; Muth, D.; Sieberg, A.; Meyer, B.; Assiri, A.M.; et al. Viral Shedding and Antibody Response in 37 Patients With Middle East Respiratory Syndrome Coronavirus Infection. Clin. Infect. Dis. 2016, 62, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.C. Viral membrane fusion. Virology 2015, 479–480, 498–507. [Google Scholar] [CrossRef]
- Darren, P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar]
- Alosaimi, B.; Hamed, M.E.; Naeem, A.; Alsharef, A.A.; AlQahtani, S.Y.; AlDosari, K.M.; Alamri, A.A.; Al-Eisa, K.; Khojah, T.; Assiri, A.M.; et al. MERS-CoV infection is associated with downregulation of genes encoding Th1 and Th2 cytokines/chemokines and elevated inflammatory innate immune response in the lower respiratory tract. Cytokine 2019, 126, 154895. [Google Scholar] [CrossRef]
- Oh, M.D.; Park, W.B.; Choe, P.G.; Choi, S.J.; Kim, J.I.; Chae, J.; Park, S.S.; Kim, E.C.; Oh, H.S.; Kim, E.J.; et al. Viral Load Kinetics of MERS Coronavirus Infection. N. Engl. J. Med. 2016, 375, 1303–1305. [Google Scholar] [CrossRef]
- Peiris, J.S.; Poon, L.L. Detection of SARS coronavirus. Methods Mol. Biol. 2011, 665, 369–382. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient No. | Sample No. | Gender | Age | Specimen Type * | Onset Date | Date of Sample Collection | E-Gene Ct | Orf1a Ct | Health-Care Worker | Outcome |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1 | Female | 57 | NPA | 20-Sep-15 | 23-Sep-15 | 37 | 35 | NO | Recovered |
| 2 | Female | 57 | NPA | 20-Sep-15 | 24-Sep-15 | 34 | 35 | |||
| 3 ** | Female | 57 | NPA | 20-Sep-15 | 28-Sep-15 | 30 | 30 | |||
| 4 | Female | 57 | NPA | 20-Sep-15 | 29-Sep-15 | 35 | 36 | |||
| 2 | 1 | Female | 59 | NPA | 30-Aug-15 | 23-Sep-15 | 35 | 36 | NO | Died |
| 2 ** | Female | 59 | NPA | 30-Aug-15 | 28-Sep-15 | 32 | 32 | |||
| 3 | Female | 59 | NPA | 30-Aug-15 | 29-Sep-15 | 34 | 33 | |||
| 4 | Female | 59 | NPA | 30-Aug-15 | 01-Oct-15 | 35 | 36 | |||
| 3 | 1 ** | Male | 57 | NPA | 10-Sep-15 | 29-Sep-15 | 33 | 33 | NO | Died |
| 2 | Male | 57 | NPA | 10-Sep-15 | 01-Oct-15 | 37 | 37 | |||
| 4 | 1 | Female | 75 | NPA | 12-Sep-15 | 30-Sep-15 | 30 | 30 | NO | Died |
| 2 ** | Female | 75 | NPA | 12-Sep-15 | 02-Oct-15 | 28 | 29 | |||
| 5 | 1 | Male | 26 | NPA | 30-Jun-16 | 02-Jul-16 | 25 | 26 | NO | Died |
| 2 ** | Male | 26 | NPA | 30-Jun-16 | 04-Jul-16 | 22 | 22 | |||
| 6 | 1 ** | Male | 26 | NPA | 04-Jun-17 | 07-Jun-17 | 26 | 26 | YES | Recovered |
| 2 | Male | 26 | NPA | 04-Jun-17 | 16-Jun-17 | 34 | 36 | |||
| 3 | Male | 26 | NPA | 04-Jun-17 | 19-Jun-17 | 30 | 30 | |||
| 4 | Male | 26 | NPA | 04-Jun-17 | 23-Jun-17 | 27 | 27 | |||
| 5 | Male | 26 | NPA | 04-Jun-17 | 25-Jun-17 | 26 | 27 | |||
| 7 | 1 ** | Female | 27 | NPA | 08-Jun-17 | 08-Jun-17 | 27 | 27 | YES | Recovered |
| 2 | Female | 27 | NPA | 08-Jun-17 | 13-Jun-17 | 30 | 30 | |||
| 3 | Female | 27 | NPA | 08-Jun-17 | 16-Jun-17 | 31 | 31 | |||
| 8 | 1 | Male | 39 | NPA | 11-Jun-17 | 11-Jun-17 | 30 | 31 | YES | Recovered |
| 2 | Male | 39 | NPA | 11-Jun-17 | 16-Jun-17 | 34 | 34 | |||
| 3 ** | Male | 39 | NPA | 11-Jun-17 | 19-Jun-17 | 27 | 27 | |||
| 4 | Male | 39 | NPA | 11-Jun-17 | 25-Jun-17 | 30 | 32 | |||
| 5 | Male | 39 | NPA | 11-Jun-17 | 29-Jun-17 | 32 | 32 | |||
| 6 | Male | 39 | NPA | 11-Jun-17 | 03-Jul-17 | 34 | 35 | |||
| 9 | 1 ** | Female | 35 | NPA | 31-May-17 | 13-Jun-17 | 26 | 26 | YES | Recovered |
| 2 | Female | 35 | NPA | 31-May-17 | 16-Jun-17 | 32 | 32 | |||
| 3 | Female | 35 | NPA | 31-May-17 | 19-Jun-17 | 29 | 30 | |||
| 4 | Female | 35 | NPA | 31-May-17 | 23-Jun-17 | 33 | 33 | |||
| 10 | 1 ** | Male | 39 | NPA | 16-Jun-17 | 19-Jun-17 | 25 | 25 | NO | Recovered |
| 2 | Male | 39 | NPA | 16-Jun-17 | 27-Jun-17 | 33 | 34 | |||
| 11 | 1 ** | Male | 75 | TA | 22-Jun-17 | 23-Jun-17 | 15 | 15 | NO | Died |
| 2 | Male | 75 | TA | 22-Jun-17 | 24-Jun-17 | 15 | 15 | |||
| 12 | 1 | Male | 77 | NPA | 05-Oct-17 | 08-Oct-17 | 34 | 35 | NO | Recovered |
| 2 ** | Male | 77 | NPA | 05-Oct-17 | 11-Oct-17 | 26 | 26 | |||
| 13 | 1 ** | Female | 75 | NPA | 07-Nov-17 | 12-Nov-17 | 25 | 25 | NO | Died |
| 2 | Female | 75 | NPA | 07-Nov-17 | 16-Nov-17 | 34 | 35 |
| S1 Subunit | S2 Subunit | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NTD | RBD | HR | ||||||||||||||
| MERS-CoV Strains | 32 | 95 | 126 | 200 | 222 | 228 | 473 | 556 | 588 | 700 | 710 | 745 | 848 | 1020 | 1224 | 1267 |
| JX869059-England-2012/06/13 * | E | T | S | S | N | F | F | A | L | R | P | L | N | Q | G | L |
| MN735680-Saudi Arabia-14-2016 | R | |||||||||||||||
| MN735679-Saudi Arabia-03-2015 | A | R | ||||||||||||||
| MN735681-Saudi Arabia-81-2017 | R | |||||||||||||||
| MN735682-Saudi Arabia-109-2017 | D | R | ||||||||||||||
| MK858156-Saudi Arabia-19-2016 | R | |||||||||||||||
| MK858157-Saudi Arabia-24-2016 | L | R | ||||||||||||||
| MK858158-Saudi Arabia-26-2016 | R | |||||||||||||||
| MK858159-Saudi Arabia-30-2016 | Y | R | ||||||||||||||
| MK858160-Saudi Arabia-37-2017 | A | L | F | R | ||||||||||||
| MK858161-Saudi Arabia-40-2017 | A | L | F | R | ||||||||||||
| MK858162-Saudi Arabia-41-2017 | A | L | F | R | ||||||||||||
| MK858163-Saudi Arabia-42-2017 | R | |||||||||||||||
| MK858164-Saudi Arabia-44-2017 | R | S | ||||||||||||||
| KT806006-Saudi Arabia-2015 * | Y | V | R | S | ||||||||||||
| KT805971-Saudi Arabia-2015 * | A | R | ||||||||||||||
| MG011342-Saudi Arabia-2016 * | I | I | H | R | ||||||||||||
| MG912606-Saudi Arabia-2017 * | A | L | S | F | R | |||||||||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naeem, A.; Hamed, M.E.; Alghoribi, M.F.; Aljabr, W.; Alsaran, H.; Enani, M.A.; Alosaimi, B. Molecular Evolution and Structural Mapping of N-Terminal Domain in Spike Gene of Middle East Respiratory Syndrome Coronavirus (MERS-CoV). Viruses 2020, 12, 502. https://doi.org/10.3390/v12050502
Naeem A, Hamed ME, Alghoribi MF, Aljabr W, Alsaran H, Enani MA, Alosaimi B. Molecular Evolution and Structural Mapping of N-Terminal Domain in Spike Gene of Middle East Respiratory Syndrome Coronavirus (MERS-CoV). Viruses. 2020; 12(5):502. https://doi.org/10.3390/v12050502
Chicago/Turabian StyleNaeem, Asif, Maaweya E. Hamed, Majed F. Alghoribi, Waleed Aljabr, Hadel Alsaran, Mushira A. Enani, and Bandar Alosaimi. 2020. "Molecular Evolution and Structural Mapping of N-Terminal Domain in Spike Gene of Middle East Respiratory Syndrome Coronavirus (MERS-CoV)" Viruses 12, no. 5: 502. https://doi.org/10.3390/v12050502
APA StyleNaeem, A., Hamed, M. E., Alghoribi, M. F., Aljabr, W., Alsaran, H., Enani, M. A., & Alosaimi, B. (2020). Molecular Evolution and Structural Mapping of N-Terminal Domain in Spike Gene of Middle East Respiratory Syndrome Coronavirus (MERS-CoV). Viruses, 12(5), 502. https://doi.org/10.3390/v12050502