Human Endogenous Retrovirus K Rec Forms a Regulatory Loop with MITF that Opposes the Progression of Melanoma to an Invasive Stage

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Generating the Knockdown (KD)-Rec Cell Lines
2.2. Cell Culture
2.3. Transfection of Melanoma Cells and Cell Sorting
2.4. Quantitative Real-Time PCR and Semi-Quantitative PCR
2.5. RNA Sequencing and Data Analysis
2.6. Single-Cell RNA-Seq Data Processing
2.7. ATAC-Seq and ChIP-Seq Data Analyses
2.8. Cell Invasion Assay
2.9. The Accession of Datasets Used in This Study
- GSE60663: H3K27Ac Primary Melanoma
- GSE60663: H3K4Me3 Primary Melanoma
- GSE60663: MITF ChIP-seq Primary Melanoma
- GSE60664: RNA-seq Primary Melanoma
- GSE46817: RNA-seq of 7 distinct melanoma lines
- GSE46805: RNA-seq Melanocyte BRAF overexpression
- GSE50681: MITF ChIP-seq Melanoma and Melanocyte post-treatment
- GSE82330: ATAC-seq Melanoma A375
- GSE72056: Patient’s Single-cell RNA-seq data
- GSE61966: MITF knock-down RNA-seq in 501 Melanoma and Melanocytes
- GSE50686: COLO829 Melanoma with PLX4032 treatment and Melanocyte
3. Results
3.1. LTR5_Hs Loci form Active Chromatin in Cancer and Pluripotent Cell Lines
3.2. MITF-Regulated LTR5_Hs/HERV-K Expression Is a Hallmark of the ‘Proliferative’ Type of Melanoma
3.3. Inhibition of BRAFV600E Mutant Leads to the MITF Binding over LTR5_Hs/HERV-K in Melanoma
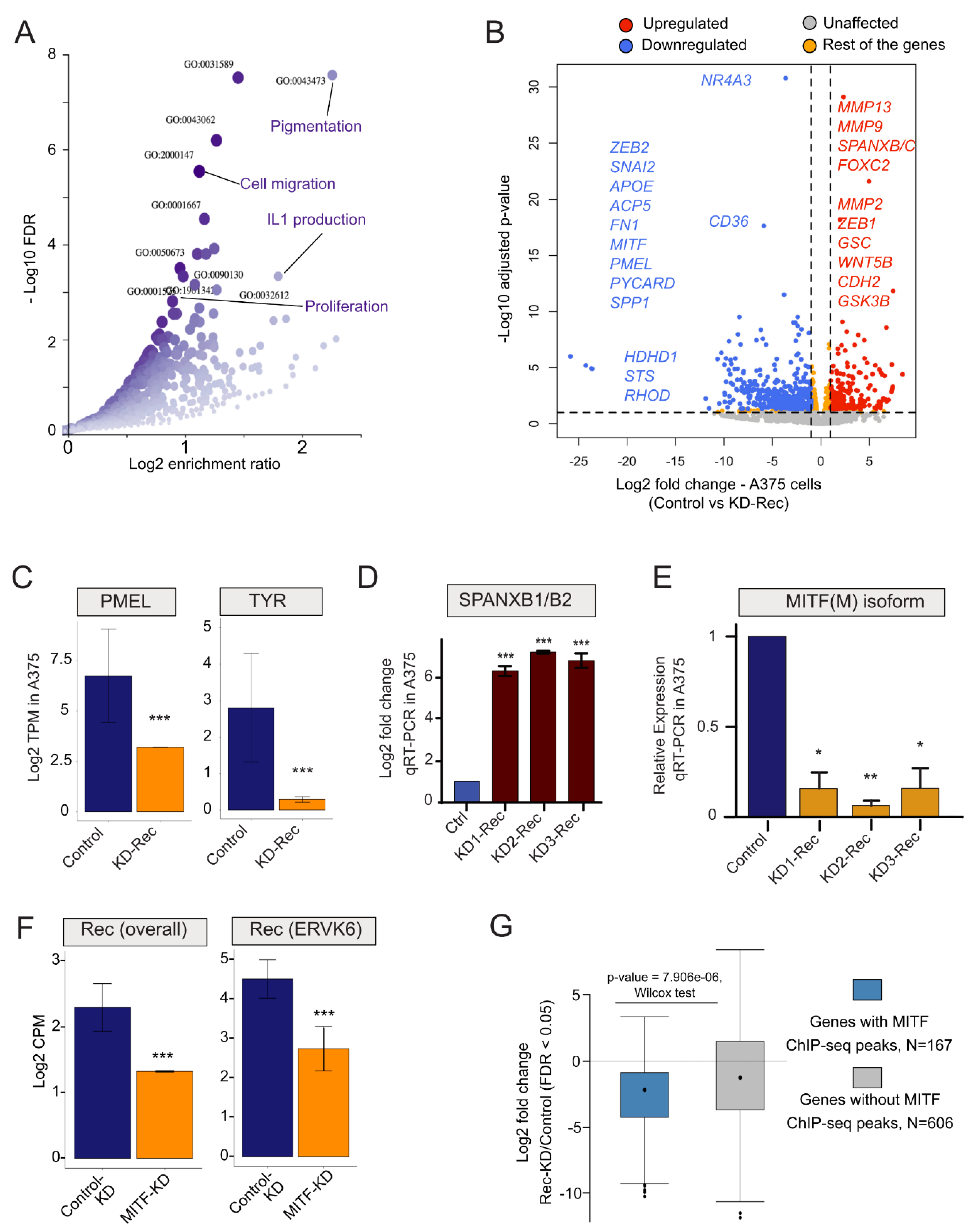
3.4. Depleting HERV-K-Rec May Induce an EMT-Like Process in A375 Melanoma Cells
3.5. Depletion of Rec Results an Enhanced Cell Invasion in A375 Melanoma
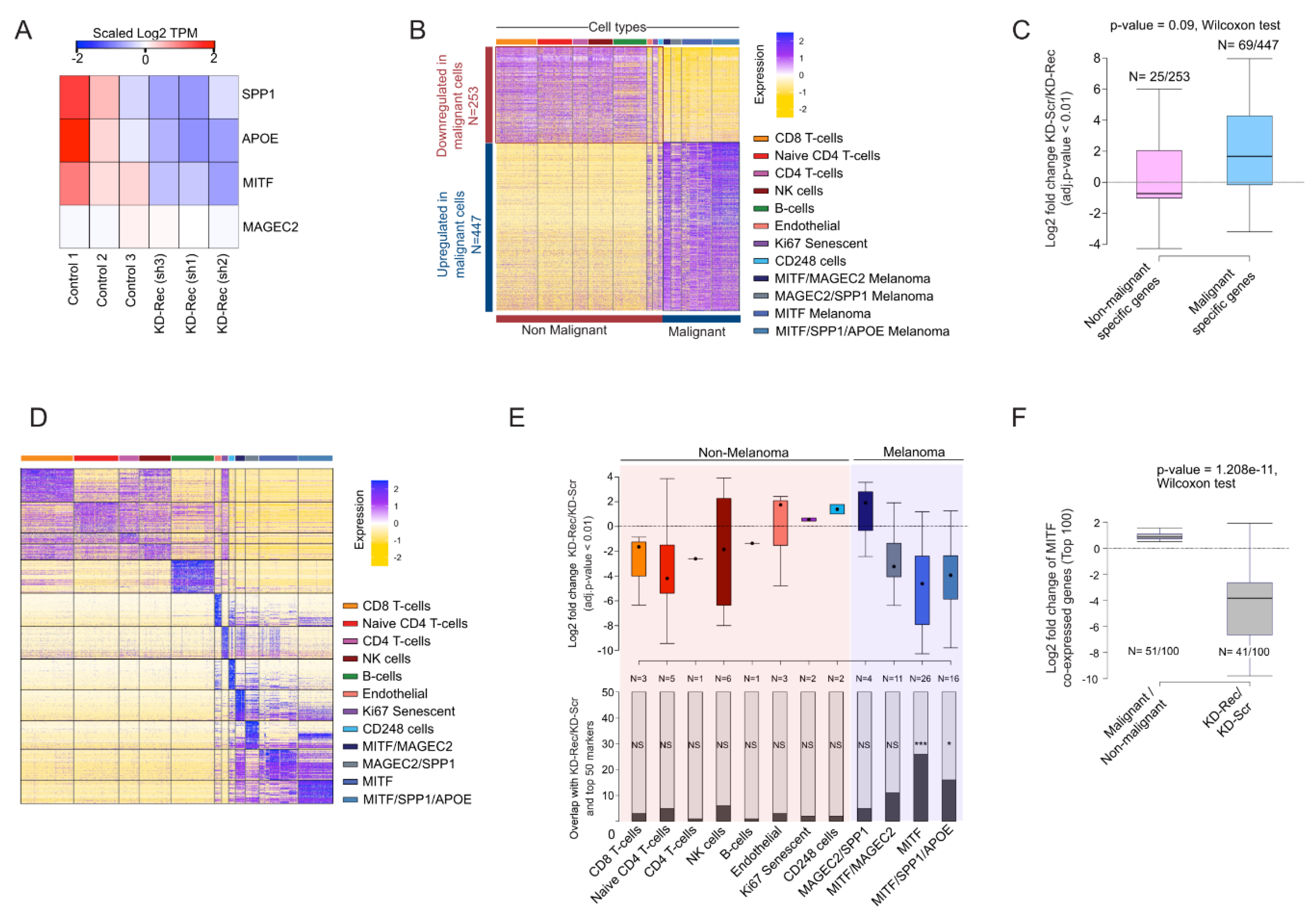
3.6. Comparison of the Rec KD A375 with the Malignant Cells in Melanoma Patients
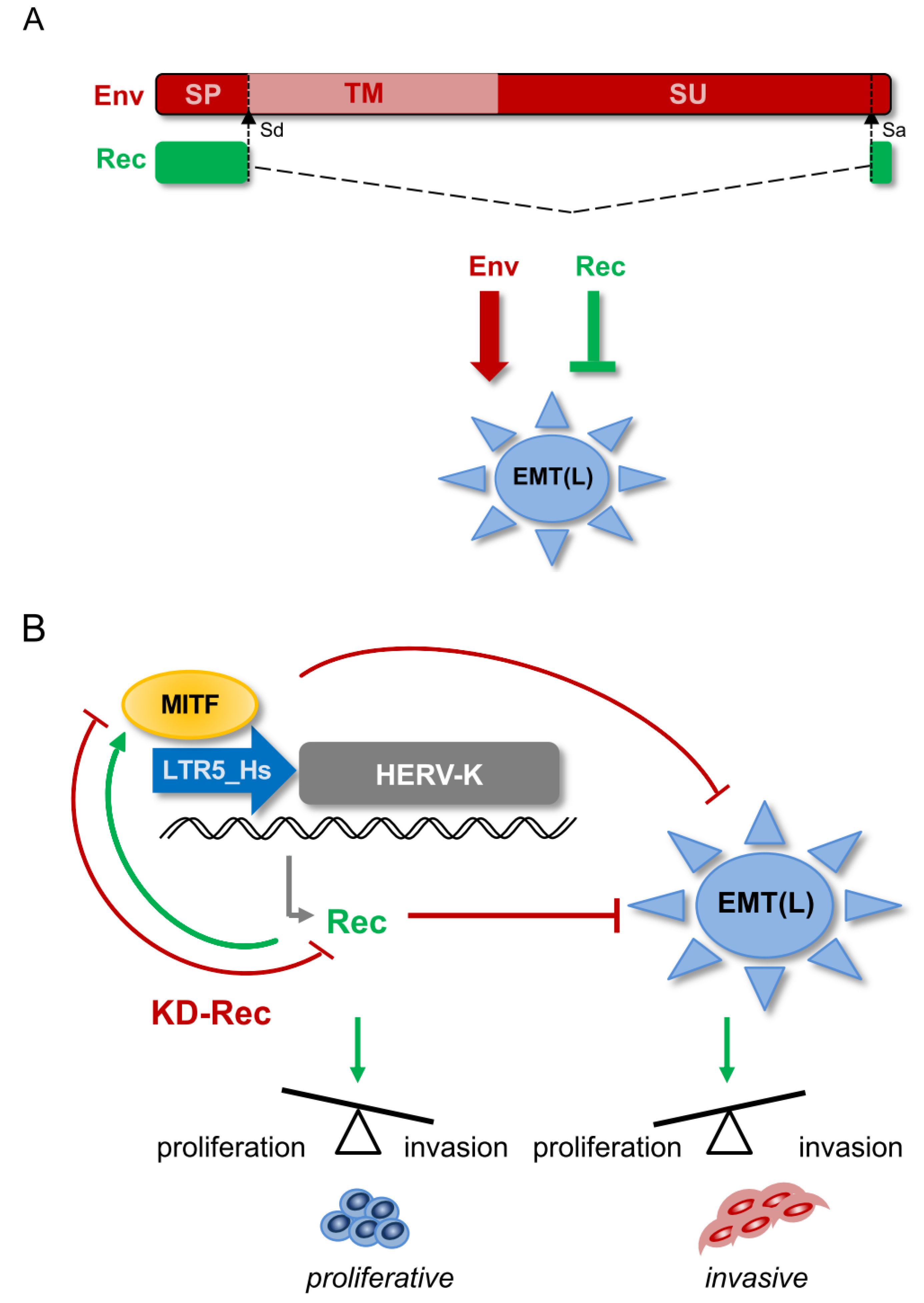
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef]
- Hughes, J.F.; Coffin, J.M. Human endogenous retrovirus K solo-LTR formation and insertional polymorphisms: Implications for human and viral evolution. Proc. Natl. Acad. Sci. USA 2004, 101, 1668–1672. [Google Scholar] [CrossRef] [PubMed]
- Turner, G.; Barbulescu, M.; Su, M.; Jensen-Seaman, M.I.; Kidd, K.K.; Lenz, J. Insertional polymorphisms of full-length endogenous retroviruses in humans. Curr. Biol. 2001, 11, 1531–1535. [Google Scholar] [CrossRef]
- Belshaw, R.; Dawson, A.L.; Woolven-Allen, J.; Redding, J.; Burt, A.; Tristem, M. Genomewide screening reveals high levels of insertional polymorphism in the human endogenous retrovirus family HERV-K(HML2): Implications for present-day activity. J. Virol. 2005, 79, 12507–12514. [Google Scholar] [CrossRef]
- Macfarlane, C.M.; Badge, R.M. Genome-wide amplification of proviral sequences reveals new polymorphic HERV-K(HML-2) proviruses in humans and chimpanzees that are absent from genome assemblies. Retrovirology 2015, 12, 35. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, R.P.; Wildschutte, J.H.; Russo, C.; Coffin, J.M. Identification, characterization, and comparative genomic distribution of the HERV-K (HML-2) group of human endogenous retroviruses. Retrovirology 2011, 8, 90. [Google Scholar] [CrossRef] [PubMed]
- Babaian, A.; Mager, D.L. Endogenous retroviral promoter exaptation in human cancer. Mob. DNA 2016, 7, 24. [Google Scholar] [CrossRef]
- Lower, R.; Tonjes, R.R.; Korbmacher, C.; Kurth, R.; Lower, J. Identification of a Rev-related protein by analysis of spliced transcripts of the human endogenous retroviruses HTDV/HERV-K. J. Virol. 1995, 69, 141–149. [Google Scholar] [CrossRef]
- Dewannieux, M.; Blaise, S.; Heidmann, T. Identification of a functional envelope protein from the HERV-K family of human endogenous retroviruses. J. Virol. 2005, 79, 15573–15577. [Google Scholar] [CrossRef]
- Contreras-Galindo, R.; Kaplan, M.H.; Dube, D.; Gonzalez-Hernandez, M.J.; Chan, S.; Meng, F.; Dai, M.; Omenn, G.S.; Gitlin, S.D.; Markovitz, D.M. Human Endogenous Retrovirus Type K (HERV-K) Particles Package and Transmit HERV-K-Related Sequences. J. Virol. 2015, 89, 7187–7201. [Google Scholar] [CrossRef]
- Lower, R.; Boller, K.; Hasenmaier, B.; Korbmacher, C.; Muller-Lantzsch, N.; Lower, J.; Kurth, R. Identification of human endogenous retroviruses with complex mRNA expression and particle formation. Proc. Natl. Acad. Sci. USA 1993, 90, 4480–4484. [Google Scholar] [CrossRef] [PubMed]
- Armbruester, V.; Sauter, M.; Krautkraemer, E.; Meese, E.; Kleiman, A.; Best, B.; Roemer, K.; Mueller-Lantzsch, N. A novel gene from the human endogenous retrovirus K expressed in transformed cells. Clin. Cancer Res. 2002, 8, 1800–1807. [Google Scholar]
- Magin, C.; Lower, R.; Lower, J. cORF and RcRE, the Rev/Rex and RRE/RxRE homologues of the human endogenous retrovirus family HTDV/HERV-K. J. Virol. 1999, 73, 9496–9507. [Google Scholar] [CrossRef]
- Magin-Lachmann, C.; Hahn, S.; Strobel, H.; Held, U.; Lower, J.; Lower, R. Rec (formerly Corf) function requires interaction with a complex, folded RNA structure within its responsive element rather than binding to a discrete specific binding site. J. Virol. 2001, 75, 10359–10371. [Google Scholar] [CrossRef]
- Mayer, J.; Ehlhardt, S.; Seifert, M.; Sauter, M.; Muller-Lantzsch, N.; Mehraein, Y.; Zang, K.D.; Meese, E. Human endogenous retrovirus HERV-K(HML-2) proviruses with Rec protein coding capacity and transcriptional activity. Virology 2004, 322, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Buzdin, A.; Ustyugova, S.; Khodosevich, K.; Mamedov, I.; Lebedev, Y.; Hunsmann, G.; Sverdlov, E. Human-specific subfamilies of HERV-K (HML-2) long terminal repeats: Three master genes were active simultaneously during branching of hominoid lineages. Genomics 2003, 81, 149–156. [Google Scholar] [CrossRef]
- Macfarlane, C.; Simmonds, P. Allelic variation of HERV-K(HML-2) endogenous retroviral elements in human populations. J. Mol. Evol. 2004, 59, 642–656. [Google Scholar] [CrossRef] [PubMed]
- Langner, J.S.; Fuchs, N.V.; Hoffmann, J.; Wittmann, A.; Brutschy, B.; Lower, R.; Suess, B. Biochemical analysis of the complex between the tetrameric export adapter protein Rec of HERV-K/HML-2 and the responsive RNA element RcRE pck30. J. Virol. 2012, 86, 9079–9087. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Bogerd, H.P.; Peng, S.; Wiegand, H.; Truant, R.; Cullen, B.R. An ancient family of human endogenous retroviruses encodes a functional homolog of the HIV-1 Rev protein. Proc. Natl. Acad. Sci. USA 1999, 96, 13404–13408. [Google Scholar] [CrossRef]
- Wodrich, H.; Krausslich, H.G. Nucleocytoplasmic RNA transport in retroviral replication. Results Probl. Cell Differ. 2001, 34, 197–217. [Google Scholar] [CrossRef]
- Boese, A.; Sauter, M.; Galli, U.; Best, B.; Herbst, H.; Mayer, J.; Kremmer, E.; Roemer, K.; Mueller-Lantzsch, N. Human endogenous retrovirus protein cORF supports cell transformation and associates with the promyelocytic leukemia zinc finger protein. Oncogene 2000, 19, 4328–4336. [Google Scholar] [CrossRef] [PubMed]
- Hanke, K.; Hohn, O.; Liedgens, L.; Fiddeke, K.; Wamara, J.; Kurth, R.; Bannert, N. Staufen-1 interacts with the human endogenous retrovirus family HERV-K(HML-2) rec and gag proteins and increases virion production. J. Virol. 2013, 87, 11019–11030. [Google Scholar] [CrossRef] [PubMed]
- Denne, M.; Sauter, M.; Armbruester, V.; Licht, J.D.; Roemer, K.; Mueller-Lantzsch, N. Physical and functional interactions of human endogenous retrovirus proteins Np9 and rec with the promyelocytic leukemia zinc finger protein. J. Virol. 2007, 81, 5607–5616. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, S.; Sauter, M.; Schmitt, M.; Baumert, B.; Best, B.; Boese, A.; Roemer, K.; Mueller-Lantzsch, N. Human endogenous retrovirus protein Rec interacts with the testicular zinc-finger protein and androgen receptor. J. Gen. Virol. 2010, 91, 1494–1502. [Google Scholar] [CrossRef] [PubMed]
- Costoya, J.A.; Hobbs, R.M.; Barna, M.; Cattoretti, G.; Manova, K.; Sukhwani, M.; Orwig, K.E.; Wolgemuth, D.J.; Pandolfi, P.P. Essential role of Plzf in maintenance of spermatogonial stem cells. Nat. Genet. 2004, 36, 653–659. [Google Scholar] [CrossRef]
- Grow, E.J.; Flynn, R.A.; Chavez, S.L.; Bayless, N.L.; Wossidlo, M.; Wesche, D.J.; Martin, L.; Ware, C.B.; Blish, C.A.; Chang, H.Y.; et al. Intrinsic retroviral reactivation in human preimplantation embryos and pluripotent cells. Nature 2015, 522, 221–225. [Google Scholar] [CrossRef]
- Chen, T.; Meng, Z.; Gan, Y.; Wang, X.; Xu, F.; Gu, Y.; Xu, X.; Tang, J.; Zhou, H.; Zhang, X.; et al. The viral oncogene Np9 acts as a critical molecular switch for co-activating beta-catenin, ERK, Akt and Notch1 and promoting the growth of human leukemia stem/progenitor cells. Leukemia 2013, 27, 1469–1478. [Google Scholar] [CrossRef]
- Schmitt, K.; Heyne, K.; Roemer, K.; Meese, E.; Mayer, J. HERV-K(HML-2) rec and np9 transcripts not restricted to disease but present in many normal human tissues. Mob. DNA 2015, 6, 4. [Google Scholar] [CrossRef]
- Garcia-Montojo, M.; Doucet-O’Hare, T.; Henderson, L.; Nath, A. Human endogenous retrovirus-K (HML-2): A comprehensive review. Crit. Rev. Microbiol. 2018, 44, 715–738. [Google Scholar] [CrossRef]
- Andersson, A.C.; Venables, P.J.; Tonjes, R.R.; Scherer, J.; Eriksson, L.; Larsson, E. Developmental expression of HERV-R (ERV3) and HERV-K in human tissue. Virology 2002, 297, 220–225. [Google Scholar] [CrossRef]
- Izsvak, Z.; Wang, J.; Singh, M.; Mager, D.L.; Hurst, L.D. Pluripotency and the endogenous retrovirus HERVH: Conflict or serendipity? Bioessays 2016, 38, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Kreimer, U.; Schulz, W.A.; Koch, A.; Niegisch, G.; Goering, W. HERV-K and LINE-1 DNA Methylation and Reexpression in Urothelial Carcinoma. Front. Oncol. 2013, 3, 255. [Google Scholar] [CrossRef] [PubMed]
- Serafino, A.; Balestrieri, E.; Pierimarchi, P.; Matteucci, C.; Moroni, G.; Oricchio, E.; Rasi, G.; Mastino, A.; Spadafora, C.; Garaci, E.; et al. The activation of human endogenous retrovirus K (HERV-K) is implicated in melanoma cell malignant transformation. Exp. Cell Res. 2009, 315, 849–862. [Google Scholar] [CrossRef] [PubMed]
- Balestrieri, E.; Argaw-Denboba, A.; Gambacurta, A.; Cipriani, C.; Bei, R.; Serafino, A.; Sinibaldi-Vallebona, P.; Matteucci, C. Human Endogenous Retrovirus K in the Crosstalk Between Cancer Cells Microenvironment and Plasticity: A New Perspective for Combination Therapy. Front. Microbiol. 2018, 9, 1448. [Google Scholar] [CrossRef] [PubMed]
- Reiche, J.; Pauli, G.; Ellerbrok, H. Differential expression of human endogenous retrovirus K transcripts in primary human melanocytes and melanoma cell lines after UV irradiation. Melanoma Res. 2010, 20, 435–440. [Google Scholar] [CrossRef]
- Schanab, O.; Humer, J.; Gleiss, A.; Mikula, M.; Sturlan, S.; Grunt, S.; Okamoto, I.; Muster, T.; Pehamberger, H.; Waltenberger, A. Expression of human endogenous retrovirus K is stimulated by ultraviolet radiation in melanoma. Pigment. Cell Melanoma Res. 2011, 24, 656–665. [Google Scholar] [CrossRef]
- Muster, T.; Waltenberger, A.; Grassauer, A.; Hirschl, S.; Caucig, P.; Romirer, I.; Fodinger, D.; Seppele, H.; Schanab, O.; Magin-Lachmann, C.; et al. An endogenous retrovirus derived from human melanoma cells. Cancer Res. 2003, 63, 8735–8741. [Google Scholar]
- Stengel, S.; Fiebig, U.; Kurth, R.; Denner, J. Regulation of human endogenous retrovirus-K expression in melanomas by CpG methylation. Genes Chromosomes Cancer 2010, 49, 401–411. [Google Scholar] [CrossRef]
- Clausen, J. Endogenous retroviruses and MS: Using ERVs as disease markers. Int. MS J. 2003, 10, 22–28. [Google Scholar]
- Matteucci, C.; Balestrieri, E.; Argaw-Denboba, A.; Sinibaldi-Vallebona, P. Human endogenous retroviruses role in cancer cell stemness. Semin. Cancer Biol. 2018, 53, 17–30. [Google Scholar] [CrossRef]
- Hahn, S.; Ugurel, S.; Hanschmann, K.M.; Strobel, H.; Tondera, C.; Schadendorf, D.; Lower, J.; Lower, R. Serological response to human endogenous retrovirus K in melanoma patients correlates with survival probability. AIDS Res. Hum. Retrovir. 2008, 24, 717–723. [Google Scholar] [CrossRef] [PubMed]
- Buscher, K.; Trefzer, U.; Hofmann, M.; Sterry, W.; Kurth, R.; Denner, J. Expression of human endogenous retrovirus K in melanomas and melanoma cell lines. Cancer Res. 2005, 65, 4172–4180. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, K.; Reichrath, J.; Roesch, A.; Meese, E.; Mayer, J. Transcriptional profiling of human endogenous retrovirus group HERV-K(HML-2) loci in melanoma. Genome Biol. Evol. 2013, 5, 307–328. [Google Scholar] [CrossRef] [PubMed]
- Buzdin, A.; Kovalskaya-Alexandrova, E.; Gogvadze, E.; Sverdlov, E. At least 50% of human-specific HERV-K (HML-2) long terminal repeats serve in vivo as active promoters for host nonrepetitive DNA transcription. J. Virol. 2006, 80, 10752–10762. [Google Scholar] [CrossRef] [PubMed]
- Tomlins, S.A.; Laxman, B.; Dhanasekaran, S.M.; Helgeson, B.E.; Cao, X.; Morris, D.S.; Menon, A.; Jing, X.; Cao, Q.; Han, B.; et al. Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature 2007, 448, 595–599. [Google Scholar] [CrossRef]
- Oricchio, E.; Sciamanna, I.; Beraldi, R.; Tolstonog, G.V.; Schumann, G.G.; Spadafora, C. Distinct roles for LINE-1 and HERV-K retroelements in cell proliferation, differentiation and tumor progression. Oncogene 2007, 26, 4226–4233. [Google Scholar] [CrossRef]
- Lemaitre, C.; Tsang, J.; Bireau, C.; Heidmann, T.; Dewannieux, M. A human endogenous retrovirus-derived gene that can contribute to oncogenesis by activating the ERK pathway and inducing migration and invasion. PLoS Pathog. 2017, 13, e1006451. [Google Scholar] [CrossRef]
- Li, M.; Radvanyi, L.; Yin, B.; Li, J.; Chivukula, R.; Lin, K.; Lu, Y.; Shen, J.; Chang, D.Z.; Li, D.; et al. Downregulation of Human Endogenous Retrovirus Type K (HERV-K) Viral env RNA in Pancreatic Cancer Cells Decreases Cell Proliferation and Tumor Growth. Clin. Cancer Res. 2017, 23, 5892–5911. [Google Scholar] [CrossRef]
- Zhou, F.; Li, M.; Wei, Y.; Lin, K.; Lu, Y.; Shen, J.; Johanning, G.L.; Wang-Johanning, F. Activation of HERV-K Env protein is essential for tumorigenesis and metastasis of breast cancer cells. Oncotarget 2016, 7, 84093–84117. [Google Scholar] [CrossRef]
- Ishida, T.; Obata, Y.; Ohara, N.; Matsushita, H.; Sato, S.; Uenaka, A.; Saika, T.; Miyamura, T.; Chayama, K.; Nakamura, Y.; et al. Identification of the HERV-K gag antigen in prostate cancer by SEREX using autologous patient serum and its immunogenicity. Cancer Immun. 2008, 8, 15. [Google Scholar]
- Riker, A.I.; Enkemann, S.A.; Fodstad, O.; Liu, S.; Ren, S.; Morris, C.; Xi, Y.; Howell, P.; Metge, B.; Samant, R.S.; et al. The gene expression profiles of primary and metastatic melanoma yields a transition point of tumor progression and metastasis. BMC Med. Genom. 2008, 1, 13. [Google Scholar] [CrossRef] [PubMed]
- Wallace, T.A.; Downey, R.F.; Seufert, C.J.; Schetter, A.; Dorsey, T.H.; Johnson, C.A.; Goldman, R.; Loffredo, C.A.; Yan, P.; Sullivan, F.J.; et al. Elevated HERV-K mRNA expression in PBMC is associated with a prostate cancer diagnosis particularly in older men and smokers. Carcinogenesis 2014, 35, 2074–2083. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Sedrak, M.; Rincon, L.; Sun, H.; Garza, G.; Kelly, B. K-type human endogenous retroviral elements in human melanoma. Adv. Genom. Genet. 2014, 4, 153. [Google Scholar] [CrossRef]
- Chiappinelli, K.B.; Strissel, P.L.; Desrichard, A.; Li, H.; Henke, C.; Akman, B.; Hein, A.; Rote, N.S.; Cope, L.M.; Snyder, A.; et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 2015, 162, 974–986. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.N.; Lever, A.M.; Smith, S.; Pitman, R.; Murray, P.; Perera, S.A.; Westwood, O.M.; Hay, F.C.; Ejtehadi, H.D.; Booth, J.C. Molecular investigations implicate human endogenous retroviruses as mediators of anti-retroviral antibodies in autoimmune rheumatic disease. Immunol. Investig. 1999, 28, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Hirschl, S.; Schanab, O.; Seppele, H.; Waltenberger, A.; Humer, J.; Wolff, K.; Pehamberger, H.; Muster, T. Sequence variability of retroviral particles derived from human melanoma cells melanoma-associated retrovirus. Virus Res. 2007, 123, 211–215. [Google Scholar] [CrossRef]
- Ruprecht, K.; Mayer, J.; Sauter, M.; Roemer, K.; Mueller-Lantzsch, N. Endogenous retroviruses and cancer. Cell. Mol. Life Sci. CMLS 2008, 65, 3366–3382. [Google Scholar] [CrossRef]
- Hohn, O.; Hanke, K.; Bannert, N. HERV-K(HML-2), the Best Preserved Family of HERVs: Endogenization, Expression, and Implications in Health and Disease. Front. Oncol. 2013, 3, 246. [Google Scholar] [CrossRef]
- Galli, U.M.; Sauter, M.; Lecher, B.; Maurer, S.; Herbst, H.; Roemer, K.; Mueller-Lantzsch, N. Human endogenous retrovirus rec interferes with germ cell development in mice and may cause carcinoma in situ, the predecessor lesion of germ cell tumors. Oncogene 2005, 24, 3223–3228. [Google Scholar] [CrossRef]
- Singh, S.; Kaye, S.; Francis, N.; Peston, D.; Gore, M.; McClure, M.; Bunker, C. Human endogenous retrovirus K (HERV-K) rec mRNA is expressed in primary melanoma but not in benign naevi or normal skin. Pigment. Cell Melanoma Res. 2013, 26, 426–428. [Google Scholar] [CrossRef]
- Lu, X.; Sachs, F.; Ramsay, L.; Jacques, P.E.; Goke, J.; Bourque, G.; Ng, H.H. The retrovirus HERVH is a long noncoding RNA required for human embryonic stem cell identity. Nat. Struct. Mol. Biol. 2014, 21, 423–425. [Google Scholar] [CrossRef] [PubMed]
- Clauss, J.; Obenaus, M.; Miskey, C.; Ivics, Z.; Izsvak, Z.; Uckert, W.; Bunse, M. Efficient Non-Viral T-Cell Engineering by Sleeping Beauty Minicircles Diminishing DNA Toxicity and miRNAs Silencing the Endogenous T-Cell Receptors. Hum. Gene Ther. 2018, 29, 569–584. [Google Scholar] [CrossRef] [PubMed]
- Mates, L.; Chuah, M.K.; Belay, E.; Jerchow, B.; Manoj, N.; Acosta-Sanchez, A.; Grzela, D.P.; Schmitt, A.; Becker, K.; Matrai, J.; et al. Molecular evolution of a novel hyperactive Sleeping Beauty transposase enables robust stable gene transfer in vertebrates. Nat. Genet. 2009, 41, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, D.R.; Swigut, T.; Wysocka, J. Systematic perturbation of retroviral LTRs reveals widespread long-range effects on human gene regulation. eLife 2018, 7, e35989. [Google Scholar] [CrossRef]
- Manghera, M.; Douville, R.N. Endogenous retrovirus-K promoter: A landing strip for inflammatory transcription factors? Retrovirology 2013, 10, 16. [Google Scholar] [CrossRef]
- Sun, Q.; Yang, J.; Xing, G.; Sun, Q.; Zhang, L.; He, F. Expression of GSDML Associates with Tumor Progression in Uterine Cervix Cancer. Transl. Oncol. 2008, 1, 73-IN71. [Google Scholar] [CrossRef]
- Katoh, I.; Mirova, A.; Kurata, S.; Murakami, Y.; Horikawa, K.; Nakakuki, N.; Sakai, T.; Hashimoto, K.; Maruyama, A.; Yonaga, T.; et al. Activation of the long terminal repeat of human endogenous retrovirus K by melanoma-specific transcription factor MITF-M. Neoplasia 2011, 13, 1081–1092. [Google Scholar] [CrossRef]
- Consortium, E.P. The ENCODE (ENCyclopedia Of DNA Elements) Project. Science 2004, 306, 636–640. [Google Scholar] [CrossRef]
- Chan, Y.S.; Goke, J.; Ng, J.H.; Lu, X.; Gonzales, K.A.; Tan, C.P.; Tng, W.Q.; Hong, Z.Z.; Lim, Y.S.; Ng, H.H. Induction of a human pluripotent state with distinct regulatory circuitry that resembles preimplantation epiblast. Cell Stem Cell 2013, 13, 663–675. [Google Scholar] [CrossRef]
- Laurette, P.; Strub, T.; Koludrovic, D.; Keime, C.; Le Gras, S.; Seberg, H.; Van Otterloo, E.; Imrichova, H.; Siddaway, R.; Aerts, S.; et al. Transcription factor MITF and remodeller BRG1 define chromatin organisation at regulatory elements in melanoma cells. eLife 2015, 4, e06857. [Google Scholar] [CrossRef]
- Levy, C.; Khaled, M.; Fisher, D.E. MITF: Master regulator of melanocyte development and melanoma oncogene. Trends Mol. Med. 2006, 12, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Steingrimsson, E.; Copeland, N.G.; Jenkins, N.A. Melanocytes and the microphthalmia transcription factor network. Annu. Rev. Genet. 2004, 38, 365–411. [Google Scholar] [CrossRef] [PubMed]
- Verfaillie, A.; Imrichova, H.; Atak, Z.K.; Dewaele, M.; Rambow, F.; Hulselmans, G.; Christiaens, V.; Svetlichnyy, D.; Luciani, F.; Van den Mooter, L.; et al. Decoding the regulatory landscape of melanoma reveals TEADS as regulators of the invasive cell state. Nat. Commun. 2015, 6, 6683. [Google Scholar] [CrossRef] [PubMed]
- Blackwood, E.M.; Eisenman, R.N. Max: A helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science 1991, 251, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Lopez-Beltran, A.; Massari, F.; MacLennan, G.T.; Montironi, R. Molecular testing for BRAF mutations to inform melanoma treatment decisions: A move toward precision medicine. Mod. Pathol. 2018, 31, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Halaban, R.; Zhang, W.; Bacchiocchi, A.; Cheng, E.; Parisi, F.; Ariyan, S.; Krauthammer, M.; McCusker, J.P.; Kluger, Y.; Sznol, M. PLX4032, a selective BRAF(V600E) kinase inhibitor, activates the ERK pathway and enhances cell migration and proliferation of BRAF melanoma cells. Pigment. Cell Melanoma Res. 2010, 23, 190–200. [Google Scholar] [CrossRef] [PubMed]
- Hatzivassiliou, G.; Song, K.; Yen, I.; Brandhuber, B.J.; Anderson, D.J.; Alvarado, R.; Ludlam, M.J.; Stokoe, D.; Gloor, S.L.; Vigers, G.; et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 2010, 464, 431–435. [Google Scholar] [CrossRef]
- George, A.L.; Suriano, R.; Rajoria, S.; Osso, M.C.; Tuli, N.; Hanly, E.; Geliebter, J.; Arnold, A.N.; Wallack, M.; Tiwari, R.K. PLX4032 Mediated Melanoma Associated Antigen Potentiation in Patient Derived Primary Melanoma Cells. J. Cancer 2015, 6, 1320–1330. [Google Scholar] [CrossRef]
- Webster, D.E.; Barajas, B.; Bussat, R.T.; Yan, K.J.; Neela, P.H.; Flockhart, R.J.; Kovalski, J.; Zehnder, A.; Khavari, P.A. Enhancer-targeted genome editing selectively blocks innate resistance to oncokinase inhibition. Genome Res. 2014, 24, 751–760. [Google Scholar] [CrossRef]
- Elia, I.; Broekaert, D.; Christen, S.; Boon, R.; Radaelli, E.; Orth, M.F.; Verfaillie, C.; Grunewald, T.G.P.; Fendt, S.M. Proline metabolism supports metastasis formation and could be inhibited to selectively target metastasizing cancer cells. Nat. Commun. 2017, 8, 15267. [Google Scholar] [CrossRef]
- Tang, L.; Zeng, J.; Geng, P.; Fang, C.; Wang, Y.; Sun, M.; Wang, C.; Wang, J.; Yin, P.; Hu, C.; et al. Global Metabolic Profiling Identifies a Pivotal Role of Proline and Hydroxyproline Metabolism in Supporting Hypoxic Response in Hepatocellular Carcinoma. Clin. Cancer Res. 2018, 24, 474–485. [Google Scholar] [CrossRef] [PubMed]
- Suntsova, M.; Gogvadze, E.V.; Salozhin, S.; Gaifullin, N.; Eroshkin, F.; Dmitriev, S.E.; Martynova, N.; Kulikov, K.; Malakhova, G.; Tukhbatova, G.; et al. Human-specific endogenous retroviral insert serves as an enhancer for the schizophrenia-linked gene PRODH. Proc. Natl. Acad. Sci. USA 2013, 110, 19472–19477. [Google Scholar] [CrossRef] [PubMed]
- Sultana, T.; Zamborlini, A.; Cristofari, G.; Lesage, P. Integration site selection by retroviruses and transposable elements in eukaryotes. Nat. Rev. Genet. 2017, 18, 292–308. [Google Scholar] [CrossRef] [PubMed]
- Reus, K.; Mayer, J.; Sauter, M.; Scherer, D.; Muller-Lantzsch, N.; Meese, E. Genomic organization of the human endogenous retrovirus HERV-K(HML-2.HOM) (ERVK6) on chromosome 7. Genomics 2001, 72, 314–320. [Google Scholar] [CrossRef]
- Kim, H.Y.; Lee, H.; Kim, S.H.; Jin, H.; Bae, J.; Choi, H.K. Discovery of potential biomarkers in human melanoma cells with different metastatic potential by metabolic and lipidomic profiling. Sci. Rep. 2017, 7, 8864. [Google Scholar] [CrossRef]
- Hoek, K.S.; Eichhoff, O.M.; Schlegel, N.C.; Dobbeling, U.; Kobert, N.; Schaerer, L.; Hemmi, S.; Dummer, R. In vivo switching of human melanoma cells between proliferative and invasive states. Cancer Res. 2008, 68, 650–656. [Google Scholar] [CrossRef]
- Li, F.Z.; Dhillon, A.S.; Anderson, R.L.; McArthur, G.; Ferrao, P.T. Phenotype switching in melanoma: Implications for progression and therapy. Front. Oncol. 2015, 5, 31. [Google Scholar] [CrossRef]
- Alonso, S.R.; Tracey, L.; Ortiz, P.; Perez-Gomez, B.; Palacios, J.; Pollan, M.; Linares, J.; Serrano, S.; Saez-Castillo, A.I.; Sanchez, L.; et al. A high-throughput study in melanoma identifies epithelial-mesenchymal transition as a major determinant of metastasis. Cancer Res. 2007, 67, 3450–3460. [Google Scholar] [CrossRef]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. Emt: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef]
- Chae, Y.K.; Chang, S.; Ko, T.; Anker, J.; Agte, S.; Iams, W.; Choi, W.M.; Lee, K.; Cruz, M. Epithelial-mesenchymal transition (EMT) signature is inversely associated with T-cell infiltration in non-small cell lung cancer (NSCLC). Sci. Rep. 2018, 8, 2918. [Google Scholar] [CrossRef]
- Dissanayake, S.K.; Wade, M.; Johnson, C.E.; O’Connell, M.P.; Leotlela, P.D.; French, A.D.; Shah, K.V.; Hewitt, K.J.; Rosenthal, D.T.; Indig, F.E.; et al. The Wnt5A/protein kinase C pathway mediates motility in melanoma cells via the inhibition of metastasis suppressors and initiation of an epithelial to mesenchymal transition. J. Biol. Chem. 2007, 282, 17259–17271. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, M.P.; Marchbank, K.; Webster, M.R.; Valiga, A.A.; Kaur, A.; Vultur, A.; Li, L.; Herlyn, M.; Villanueva, J.; Liu, Q.; et al. Hypoxia induces phenotypic plasticity and therapy resistance in melanoma via the tyrosine kinase receptors ROR1 and ROR2. Cancer Discov. 2013, 3, 1378–1393. [Google Scholar] [CrossRef] [PubMed]
- Caramel, J.; Papadogeorgakis, E.; Hill, L.; Browne, G.J.; Richard, G.; Wierinckx, A.; Saldanha, G.; Osborne, J.; Hutchinson, P.; Tse, G.; et al. A switch in the expression of embryonic EMT-inducers drives the development of malignant melanoma. Cancer Cell 2013, 24, 466–480. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.; Baritaki, S.; Militello, L.; Malaponte, G.; Bevelacqua, Y.; Bonavida, B. The Role of B-RAF Mutations in Melanoma and the Induction of EMT via Dysregulation of the NF-kappaB/Snail/RKIP/PTEN Circuit. Genes Cancer 2010, 1, 409–420. [Google Scholar] [CrossRef] [PubMed]
- Weiss, M.B.; Abel, E.V.; Mayberry, M.M.; Basile, K.J.; Berger, A.C.; Aplin, A.E. TWIST1 is an ERK1/2 effector that promotes invasion and regulates MMP-1 expression in human melanoma cells. Cancer Res. 2012, 72, 6382–6392. [Google Scholar] [CrossRef] [PubMed]
- Almanzar, G.; Olkhanud, P.B.; Bodogai, M.; Dell’agnola, C.; Baatar, D.; Hewitt, S.M.; Ghimenton, C.; Tummala, M.K.; Weeraratna, A.T.; Hoek, K.S.; et al. Sperm-derived SPANX-B is a clinically relevant tumor antigen that is expressed in human tumors and readily recognized by human CD4+ and CD8+ T cells. Clin. Cancer Res. 2009, 15, 1954–1963. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Goldstein, N.B.; Koster, M.I.; Hoaglin, L.G.; Wright, M.J.; Robinson, S.E.; Robinson, W.A.; Roop, D.R.; Norris, D.A.; Birlea, S.A. Isolating RNA from precursor and mature melanocytes from human vitiligo and normal skin using laser capture microdissection. Exp. Dermatol. 2016, 25, 805–811. [Google Scholar] [CrossRef]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H., 2nd; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef]
- Pencheva, N.; Tran, H.; Buss, C.; Huh, D.; Drobnjak, M.; Busam, K.; Tavazoie, S.F. Convergent multi-miRNA targeting of ApoE drives LRP1/LRP8-dependent melanoma metastasis and angiogenesis. Cell 2012, 151, 1068–1082. [Google Scholar] [CrossRef]
- Castello, L.M.; Raineri, D.; Salmi, L.; Clemente, N.; Vaschetto, R.; Quaglia, M.; Garzaro, M.; Gentilli, S.; Navalesi, P.; Cantaluppi, V.; et al. Osteopontin at the Crossroads of Inflammation and Tumor Progression. Mediat. Inflamm. 2017, 2017, 4049098. [Google Scholar] [CrossRef]
- MacFadyen, J.R.; Haworth, O.; Roberston, D.; Hardie, D.; Webster, M.T.; Morris, H.R.; Panico, M.; Sutton-Smith, M.; Dell, A.; van der Geer, P.; et al. Endosialin (TEM1, CD248) is a marker of stromal fibroblasts and is not selectively expressed on tumour endothelium. FEBS Lett. 2005, 579, 2569–2575. [Google Scholar] [CrossRef] [PubMed]
- Hoek, K.S.; Schlegel, N.C.; Brafford, P.; Sucker, A.; Ugurel, S.; Kumar, R.; Weber, B.L.; Nathanson, K.L.; Phillips, D.J.; Herlyn, M.; et al. Metastatic potential of melanomas defined by specific gene expression profiles with no BRAF signature. Pigment. Cell Res. 2006, 19, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Goding, C.R. Commentary. A picture of Mitf in melanoma immortality. Oncogene 2011, 30, 2304–2306. [Google Scholar] [CrossRef] [PubMed]
- Arozarena, I.; Wellbrock, C. Phenotype plasticity as enabler of melanoma progression and therapy resistance. Nat. Rev. Cancer 2019, 19, 377–391. [Google Scholar] [CrossRef] [PubMed]
- Hoek, K.S.; Goding, C.R. Cancer stem cells versus phenotype-switching in melanoma. Pigment. Cell Melanoma Res. 2010, 23, 746–759. [Google Scholar] [CrossRef] [PubMed]
- Vachtenheim, J.; Ondrusova, L. Microphthalmia-associated transcription factor expression levels in melanoma cells contribute to cell invasion and proliferation. Exp. Dermatol. 2015, 24, 481–484. [Google Scholar] [CrossRef]
- Carreira, S.; Goodall, J.; Denat, L.; Rodriguez, M.; Nuciforo, P.; Hoek, K.S.; Testori, A.; Larue, L.; Goding, C.R. Mitf regulation of Dia1 controls melanoma proliferation and invasiveness. Genes Dev. 2006, 20, 3426–3439. [Google Scholar] [CrossRef]
- Lauss, M.; Haq, R.; Cirenajwis, H.; Phung, B.; Harbst, K.; Staaf, J.; Rosengren, F.; Holm, K.; Aine, M.; Jirstrom, K.; et al. Genome-Wide DNA Methylation Analysis in Melanoma Reveals the Importance of CpG Methylation in MITF Regulation. J. Investig. Dermatol. 2015, 135, 1820–1828. [Google Scholar] [CrossRef]
- Litvin, O.; Schwartz, S.; Wan, Z.; Schild, T.; Rocco, M.; Oh, N.L.; Chen, B.J.; Goddard, N.; Pratilas, C.; Pe’er, D. Interferon alpha/beta Enhances the Cytotoxic Response of MEK Inhibition in Melanoma. Mol. Cell 2015, 57, 784–796. [Google Scholar] [CrossRef]
- Perez-Guijarro, E.; Day, C.P.; Merlino, G.; Zaidi, M.R. Genetically engineered mouse models of melanoma. Cancer 2017, 123, 2089–2103. [Google Scholar] [CrossRef]
- Tuncer, E.; Calcada, R.R.; Zingg, D.; Varum, S.; Cheng, P.; Freiberger, S.N.; Deng, C.X.; Kleiter, I.; Levesque, M.P.; Dummer, R.; et al. SMAD signaling promotes melanoma metastasis independently of phenotype switching. J. Clin. Investig. 2019, 129, 2702–2716. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Application | Sequence (5′–3′) | Tm (°C) |
|---|---|---|---|
| HML-2-Env | Real-Time qPCR | F: GCTGCCCTGCCAAACCTGAG R: CCTGAGTGACATCCCGCTTACC | 60 |
| HML-2-Gag | Real-Time qPCR | F: AGCAGGTCAGGTGCCTGTAACATT R: TGGTGCCGTAGGATTAAGTCTCCT | 60 |
| HML-2-Np9 | Real-Time qPCR | F: AGATGTCTGCAGGTGTACCCA R: CTCTTGCTTTTCCCCACATTTC | 60 |
| HML-2-Rec | Real-Time qPCR | F: ATCGAGCACCGTTGACTCACAAGA R: GGTACACCTGCAGACACCATTGAT | 60 |
| MITF(M) | Real-Time qPCR | F:ATGCTGGAAATGCTAGAATATAATCACT R: GAATGTGTGTTCATGCCTGG | 60 |
| SPANXB1/B2 | Real-Time qPCR | F: AGGCCAATGAGGCCAACAAGAC R: TCCTCCTGTAGCGAACCACTAG | 60 |
| 18S RNA | Real-Time qPCR | F: GTAACCCGTTGAACCCCATT R: CCATCCAATCGGTAGTAGCG | 60 |
| GAPDH | Real-Time qPCR | F: CAATGACCCCTTCATTGACCTC R: AGCATCGCCCCACTTGATT | 60 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, M.; Cai, H.; Bunse, M.; Feschotte, C.; Izsvák, Z. Human Endogenous Retrovirus K Rec Forms a Regulatory Loop with MITF that Opposes the Progression of Melanoma to an Invasive Stage. Viruses 2020, 12, 1303. https://doi.org/10.3390/v12111303
Singh M, Cai H, Bunse M, Feschotte C, Izsvák Z. Human Endogenous Retrovirus K Rec Forms a Regulatory Loop with MITF that Opposes the Progression of Melanoma to an Invasive Stage. Viruses. 2020; 12(11):1303. https://doi.org/10.3390/v12111303
Chicago/Turabian StyleSingh, Manvendra, Huiqiang Cai, Mario Bunse, Cédric Feschotte, and Zsuzsanna Izsvák. 2020. "Human Endogenous Retrovirus K Rec Forms a Regulatory Loop with MITF that Opposes the Progression of Melanoma to an Invasive Stage" Viruses 12, no. 11: 1303. https://doi.org/10.3390/v12111303
APA StyleSingh, M., Cai, H., Bunse, M., Feschotte, C., & Izsvák, Z. (2020). Human Endogenous Retrovirus K Rec Forms a Regulatory Loop with MITF that Opposes the Progression of Melanoma to an Invasive Stage. Viruses, 12(11), 1303. https://doi.org/10.3390/v12111303