Expanding the Diversity of Myoviridae Phages Infecting Lactobacillus plantarum—A Novel Lineage of Lactobacillus Phages Comprising Five New Members

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Culture Media
2.2. Environmental Sampling, Isolation, Purification, and Enrichment of Phages
2.3. Phage DNA Extraction, Library Preparation, and Sequencing
2.4. Assemblies and Annotations
2.5. Transmission Electron Microscopy
2.6. Identification of Structural Proteins
2.7. Comparative Genomics and Phylogenetics to Distant Relatives
2.8. Phage Genomic Data Availability
3. Results and Discussion
3.1. Isolation of Phages and Basic Features
3.2. Analyses of DNA Sequences and Protein Predictions
3.2.1. Transcription and Translation Takeover
3.2.2. DNA Metabolism, Replication, Recombination, and Repair
3.2.3. Self-Splicing/Selfish Genetic Elements
3.2.4. Morphogenesis, DNA Packaging, and Membrane Transport
3.2.5. Cell Wall and Membrane Degradation
3.2.6. Other Predicted Proteins
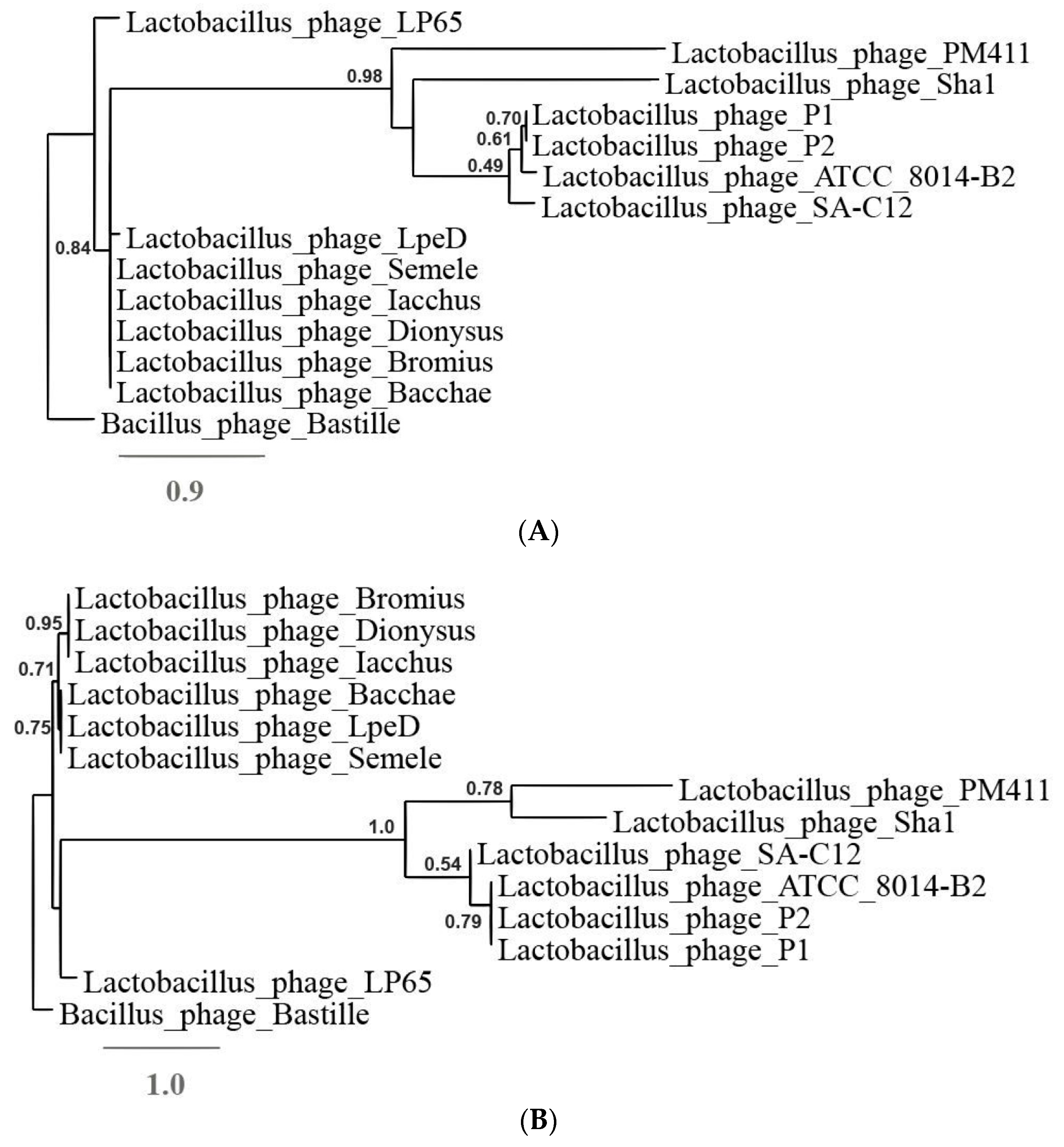
3.3. The Diversity of the Five Phages Supports the Introduction of a New Lactobacillus Phage Genus
4. Outlook
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- König, H.; Fröhlich, J. Lactic Acid Bacteria. In Biology of Microorganisms on Grapes, in Must and in Wine; König, H., Unden, G., Fröhlich, J., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 3–41. [Google Scholar]
- Lamont, J.R.; Wilkins, O.; Bywater-Ekegärd, M.; Smith, D.L. From yogurt to yield: Potential applications of lactic acid bacteria in plant production. Soil Biol. Biochem. 2017, 111, 1–9. [Google Scholar] [CrossRef]
- Du Toit, M.; Engelbrecht, L.; Lerm, E.; Krieger-Weber, S. Lactobacillus: The Next Generation of Malolactic Fermentation Starter Cultures—An Overview. Food Bioprocess Technol. 2011, 4, 876–906. [Google Scholar] [CrossRef]
- Lerm, E.; Engelbrecht, L.; Du Toit, M. Selection and characterisation of Oenococcus oeni and Lactobacillus plantarum South African wine isolates for use as malolactic fermentation starter cultures. S. Afr. J. Enol. Vitic. 2011, 32, 280–295. [Google Scholar] [CrossRef]
- López, I.; López, R.; Santamaría, P.; Torres, C.; Ruiz-Larrea, F. Performance of malolactic fermentation by inoculation of selected Lactobacillus plantarum and Oenococcus oeni strains isolated from Rioja red wines. Vitis 2008, 47, 123–129. [Google Scholar]
- Du Toit, M. Novel lactic acid bacteria for use as MLF starter cultures. Acenología Enoreports 2012, 130, 1–6. [Google Scholar]
- De Vries, M.C.; Vaughan, E.E.; Kleerebezem, M.; de Vos, W.M. Lactobacillus plantarum—Survival, functional and potential probiotic properties in the human intestinal tract. Int. Dairy J. 2006, 16, 1018–1028. [Google Scholar] [CrossRef]
- Samson, J.E.; Moineau, S. Bacteriophages in Food Fermentations: New Frontiers in a Continuous Arms Race. Annu. Rev. Food Sci. Technol. 2013, 4, 347–368. [Google Scholar] [CrossRef] [PubMed]
- Sechaud, L.; Cluzel, P.-J.; Rousseau, M.; Baumgartner, A.; Accolas, J.-P. Bacteriophages of lactobacilli. Biochimie 1988, 70, 401–410. [Google Scholar] [CrossRef]
- Martínez, B.; García, P.; Gonzalez, A.R.; Piuri, M.; Raya, R.R. Bacteriophages of Lactic Acid Bacteria and Biotechnological Tools. In Biotechnology of Lactic Acid Bacteria: Novel Applications; Mozzi, F., Raya, R.R., Vignolo, G.M., Eds.; John Wiley & Sons, Ltd.: Chichester, UK, 2015; pp. 100–119. [Google Scholar]
- Murphy, J.; Mahony, J.; Fitzgerald, G.F.; van Sinderen, D. Bacteriophages infecting lactic acid bacteria. In Cheese; Fox, P.F., McSweeney, P.L.H., Cogan, T.M., Guinee, T.P., Eds.; Academic Press: San Diego, CA, USA, 2017; pp. 249–272. ISBN 9780124170124. [Google Scholar]
- Mahony, J.; van Sinderen, D. Current taxonomy of phages infecting lactic acid bacteria. Front. Microbiol. 2014, 5, 7. [Google Scholar] [CrossRef]
- Villion, M.; Moineau, S. Bacteriophages of Lactobacillus. Front. Biosci. 2009, 14, 1661–1683. [Google Scholar] [CrossRef]
- Sakurai, T.; Takahashi, T.; Kamiyama, K.; Arai, H. Isolation of bacteriophages parasitic on Lactobacillus casei and L. plantarum and their several properties. Virus 1969, 19, 311–324. [Google Scholar] [CrossRef]
- Lu, Z.; Breidt, F.; Fleming, H.P.; Altermann, E.; Klaenhammer, T.R. Isolation and characterization of a Lactobacillus plantarum bacteriophage, φJL-1, from a cucumber fermentation. Int. J. Food Microbiol. 2003, 84, 225–235. [Google Scholar] [CrossRef]
- Trevors, K.E.; Holley, R.A.; Kempton, A.G. Isolation and characterization of a Lactobacillus plantarum bacteriophage isolated from a meat starter culture. J. Appl. Bacteriol. 1983, 54, 281–288. [Google Scholar] [CrossRef]
- Doi, K.; Zhang, Y.; Nishizaki, Y.; Umeda, A.; Ohmomo, S.; Ogata, S. A comparative study and phage typing of silage-making Lactobacillus bacteriophages. J. Biosci. Bioeng. 2003, 95, 518–525. [Google Scholar] [CrossRef]
- Lu, Z.; Breidt, F.; Plengvidhya, V.; Fleming, H.P. Bacteriophage ecology in commercial sauerkraut fermentations. Appl. Environ. Microbiol. 2003, 69, 3192–3202. [Google Scholar] [CrossRef]
- Chibani-Chennoufi, S.; Dillmann, M.-L.; Marvin-Guy, L.; Rami-Shojaei, S.; Brüssow, H. Lactobacillus plantarum bacteriophage LP65: A new member of the SPO1-like genus of the family Myoviridae. J. Bacteriol. 2004, 186, 7069–7083. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.S.; Barrangou-Poueys, R.; Breidt, F.; Klaenhammer, T.R.; Fleming, H.P. Isolation and Characterization of Bacteriophages from Fermenting Sauerkraut. Appl. Environ. Microbiol. 2002, 68, 973–976. [Google Scholar] [CrossRef]
- Daranas, N.; Bonaterra, A.; Francés, J.; Cabrefiga, J.; Montesinos, E.; Badosa, E. Monitoring Viable Cells of the Biological Control Agent Lactobacillus plantarum PM411 in Aerial Plant Surfaces by Means of a Strain-Specific Viability Quantitative PCR Method. Appl. Environ. Microbiol. 2018, 84, e00107-18. [Google Scholar] [CrossRef]
- Kropinski, A.M.; Mazzocco, A.; Waddell, T.E.; Lingohr, E.; Johnson, R.P. Enumeration of Bacteriophages by Double Agar Overlay Plaque Assay. In Bacteriophages: Methods and Protocols, Volume 1: Isolation, Characterization, and Interactions; Clokie, M.R.J., Kropinski, A.M., Eds.; Humana Press: Totowa, NJ, USA, 2009; pp. 69–76. ISBN 978-1-60327-164-6. [Google Scholar]
- Lillehaug, D. An improved plaque assay for poor plaque-producing temperate lactococcal bacteriophages. J. Appl. Microbiol. 1997, 83, 85–90. [Google Scholar] [CrossRef]
- Pires, D.; Sillankorva, S.; Faustino, A.; Azeredo, J. Use of newly isolated phages for control of Pseudomonas aeruginosa PAO1 and ATCC 10145 biofilms. Res. Microbiol. 2011, 162, 798–806. [Google Scholar] [CrossRef]
- Sambrook, J. Molecular Cloning: A Laboratory Manual, 2nd ed.; Fritsch, E.F., Maniatis, T., Eds.; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 1989. [Google Scholar]
- Moineau, S.; Pandian, S.; Klaenhammer, T.R. Evolution of a Lytic Bacteriophage via DNA Acquisition from the Lactococcus lactis Chromosome. Appl. Environ. Microbiol. 1994, 60, 1832–1841. [Google Scholar] [PubMed]
- Kot, W.; Vogensen, F.K.; Sørensen, S.J.; Hansen, L.H. DPS—A rapid method for genome sequencing of DNA-containing bacteriophages directly from a single plaque. J. Virol. Methods 2014, 196, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, T.K.; Carstens, A.B.; Browne, P.; Lametsch, R.; Neve, H.; Kot, W.; Hansen, L.H. The first characterized phage against a member of the ecologically important sphingomonads reveals high dissimilarity against all other known phages. Sci. Rep. 2017, 7, 13566. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [PubMed]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid Annotations using Subsystems Technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef]
- Besemer, J.; Borodovsky, M. GeneMark: Web software for gene finding in prokaryotes, eukaryotes and viruses. Nucleic Acids Res. 2005, 33, W451–W454. [Google Scholar] [CrossRef]
- Altschul, S.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Soding, J.; Biegert, A.; Lupas, A.N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005, 33, W244–W248. [Google Scholar] [CrossRef]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef] [PubMed]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Boratyn, G.M.; Schäffer, A.A.; Agarwala, R.; Altschul, S.F.; Lipman, D.J.; Madden, T.L. Domain enhanced lookup time accelerated BLAST. Biol. Direct 2012, 7, 12. [Google Scholar] [CrossRef] [PubMed]
- Schattner, P.; Brooks, A.N.; Lowe, T.M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005, 33, W686–W689. [Google Scholar] [CrossRef] [PubMed]
- Siguier, P.; Perochon, J.; Lestrade, L.; Mahillon, J.; Chandler, M. ISfinder: The reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006, 34, D32–D36. [Google Scholar] [CrossRef] [PubMed]
- Carstens, A.B.; Kot, W.; Lametsch, R.; Neve, H.; Hansen, L.H. Characterisation of a novel enterobacteria phage, CAjan, isolated from rat faeces. Arch. Virol. 2016, 161, 2219–2226. [Google Scholar] [CrossRef]
- Tolstoy, I.; Kropinski, A.M.; Brister, J.R. Bacteriophage Taxonomy: An Evolving Discipline. In Bacteriophage Therapy; Azeredo, J., Sillankorva, S., Eds.; Humana Press: New York, NY, USA, 2018; Volume 1693, pp. 57–71. [Google Scholar]
- Ågren, J.; Sundström, A.; Håfström, T.; Segerman, B. Gegenees: Fragmented Alignment of Multiple Genomes for Determining Phylogenomic Distances and Genetic Signatures Unique for Specified Target Groups. PLoS ONE 2012, 7, e39107. [Google Scholar] [CrossRef]
- Dereeper, A.; Guignon, V.; Blanc, G.; Audic, S.; Buffet, S.; Chevenet, F.; Dufayard, J.-F.; Guindon, S.; Lefort, V.; Lescot, M.; et al. Phylogeny.fr: Robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008, 36, W465–W469. [Google Scholar] [CrossRef]
- Vesth, T.; Lagesen, K.; Acar, Ö.; Ussery, D. CMG-Biotools, a Free Workbench for Basic Comparative Microbial Genomics. PLoS ONE 2013, 8, e60120. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.S.; Kutter, E.; Mosig, G.; Arisaka, F.; Kunisawa, T.; Rüger, W. Bacteriophage T4 genome. Microbiol. Mol. Biol. Rev. 2003, 67, 86–156. [Google Scholar] [CrossRef] [PubMed]
- Travers, A.A. Bacteriophage Sigma Factor for RNA Polymerase. Nature 1969, 223, 1107–1110. [Google Scholar] [CrossRef] [PubMed]
- Malys, N.; Chang, D.-Y.; Baumann, R.G.; Xie, D.; Black, L.W. A Bipartite Bacteriophage T4 SOC and HOC Randomized Peptide Display Library: Detection and Analysis of Phage T4 Terminase (gp17) and Late σ Factor (gp55) Interaction. J. Mol. Biol. 2002, 319, 289–304. [Google Scholar] [CrossRef]
- Mikhailopulo, I. Biotechnology of Nucleic Acid Constituents—State of the Art and Perspectives. Curr. Org. Chem. 2007, 11, 317–335. [Google Scholar] [CrossRef]
- Lee, J.Y.; Li, Z.; Miller, E.S. Vibrio Phage KVP40 Encodes a Functional NAD+ Salvage Pathway. J. Bacteriol. 2017, 199. [Google Scholar] [CrossRef]
- Greiner, T.; Moroni, A.; van Etten, J.; Thiel, G. Genes for Membrane Transport Proteins: Not So Rare in Viruses. Viruses 2018, 10, 456. [Google Scholar] [CrossRef]
- Arias-Palomo, E.; Berger, J.M. An Atypical AAA+ ATPase Assembly Controls Efficient Transposition through DNA Remodeling and Transposase Recruitment. Cell 2015, 162, 860–871. [Google Scholar] [CrossRef]
- Stetter, K.O. Evidence for frequent lysogeny in lactobacilli: Temperate bacteriophages within the subgenus Streptobacterium. J. Virol. 1977, 24, 685–689. [Google Scholar]
- Hausner, G.; Hafez, M.; Edgell, D.R. Bacterial group I introns: Mobile RNA catalysts. Mob. DNA 2014, 5, 8. [Google Scholar] [CrossRef]
- Petrov, V.M.; Ratnayaka, S.; Nolan, J.M.; Miller, E.S.; Karam, J.D. Genomes of the T4-related bacteriophages as windows on microbial genome evolution. Virol. J. 2010, 7, 292–311. [Google Scholar] [CrossRef] [PubMed]
- Mills, K.V. Self-Splicing Proteins. In Handbook of Proteolytic Enzymes; Rawlings, N.D., Salvesen, G., Eds.; Academic Press: Cambridge, MA, USA, 2013; pp. 315–321. ISBN 9780123822192. [Google Scholar]
- Elleuche, S.; Pöggeler, S. Inteins, valuable genetic elements in molecular biology and biotechnology. Appl. Microbiol. Biotechnol. 2010, 87, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Kot, W.; Hansen, L.H.; Neve, H.; Hammer, K.; Jacobsen, S.; Pedersen, P.D.; Sørensen, S.J.; Heller, K.J.; Vogensen, F.K. Sequence and comparative analysis of Leuconostoc dairy bacteriophages. Int. J. Food Microbiol. 2014, 176, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Goodrich-Blair, H.; Shub, D.A. Beyond homing: Competition between intron endonucleases confers a selective advantage on flanking genetic markers. Cell 1996, 84, 211–221. [Google Scholar] [CrossRef]
- Perler, F.B. InBase: The Intein Database. Nucleic Acids Res. 2002, 30, 383–384. [Google Scholar] [CrossRef] [PubMed]
- Landthaler, M.; Shub, D.A. Unexpected abundance of self-splicing introns in the genome of bacteriophage Twort: Introns in multiple genes, a single gene with three introns, and exon skipping by group I ribozymes. Proc. Natl. Acad. Sci. USA 1999, 96, 7005–7010. [Google Scholar] [CrossRef] [PubMed]
- Riipinen, K.A.; Alatossava, T. Two self-splicing group I introns interrupt two late transcribed genes of prolate-headed Lactobacillus delbrueckii phage JCL1032. Arch. Virol. 2004, 149, 2013–2024. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, M.S.; Rao, V.B. Functional analysis of the bacteriophage T4 DNA-packaging ATPase motor. J. Biol. Chem. 2006, 281, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Ugorčáková, J.; Bukovská, G. Lysins and holins: Tools of phage-induced lysis. Biologia 2003, 58, 327–334. [Google Scholar]
- Josephsen, J.Y.; Neve, H.O. Bacteriophage and antiphage mechanisms of lactic acid bacteria. In Lactic Acid Bacteria: Microbiological and Functional Aspects; Salminen, S., von Wright, A., Ouwehand, A., Eds.; CRC Press: New York, NY, USA, 2004; pp. 295–350. [Google Scholar]
- Wang, I.-N.; Smith, D.L.; Young, R. Holins: The Protein Clocks of Bacteriophage Infections. Annu. Rev. Microbiol. 2000, 54, 799–825. [Google Scholar] [CrossRef]
- Henrich, B.; Binishofer, B.; Bläsi, U. Primary structure and functional analysis of the lysis genes of Lactobacillus gasseri bacteriophage phi adh. J. Bacteriol. 1995, 177, 723–732. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Oki, M.; Kakikawa, M.; Nakamura, S.; Yamamura, E.-T.; Watanabe, K.; Sasamoto, M.; Taketo, A.; Kodaira, K.-I. Functional and structural features of the holin HOL protein of the Lactobacillus plantarum phage φg1e: Analysis in Escherichia coli system. Gene 1997, 197, 137–145. [Google Scholar] [CrossRef]
- Fogg, P.C.M.; Rigden, D.J.; Saunders, J.R.; McCarthy, A.J.; Allison, H.E. Characterization of the relationship between integrase, excisionase and antirepressor activities associated with a superinfecting Shiga toxin encoding bacteriophage. Nucleic Acids Res. 2011, 39, 2116–2129. [Google Scholar] [CrossRef]
- Casey, E.; Mahony, J.; O’Connell-Motherway, M.; Bottacini, F.; Cornelissen, A.; Neve, H.; Heller, K.J.; Noben, J.-P.; Dal Bello, F.; van Sinderen, D. Molecular characterization of three Lactobacillus delbrueckii subsp. bulgaricus phages. Appl. Environ. Microbiol. 2014, 80, 5623–5635. [Google Scholar] [CrossRef] [PubMed]
- Cornelissen, A.; Sadovskaya, I.; Vinogradov, E. The baseplate of Lactobacillus delbrueckii bacteriophage Ld17 harbours a glycerophosphodiesterase. J. Biol. Chem. 2016, 291, 16816–16827. [Google Scholar] [CrossRef]
- Samson, J.E.; Magadán, A.H.; Sabri, M.; Moineau, S. Revenge of the phages: Defeating bacterial defences. Nat. Rev. Microbiol. 2013, 11, 675–687. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Rubio, L.; Martínez, B.; Donovan, D.M.; Rodríguez, A.; García, P. Bacteriophage virion-associated peptidoglycan hydrolases: Potential new enzybiotics. Crit. Rev. Microbiol. 2013, 39, 427–434. [Google Scholar] [CrossRef]
- Pulliainen, A.T.; Kauko, A.; Haataja, S.; Papageorgiou, A.C.; Finne, J. Dps/Dpr ferritin-like protein: Insights into the mechanism of iron incorporation and evidence for a central role in cellular iron homeostasis in Streptococcus suis. Mol. Microbiol. 2005, 57, 1086–1100. [Google Scholar] [CrossRef] [PubMed]
- Guidone, A.; Ianniello, R.G.; Ricciardi, A.; Zotta, T.; Parente, E. Aerobic metabolism and oxidative stress tolerance in the Lactobacillus plantarum group. World J. Microbiol. Biotechnol. 2013, 29, 1713–1722. [Google Scholar] [CrossRef]
- Archibald, F.S.; Duong, M.N. Manganese acquisition by Lactobacillus plantarum. J. Bacteriol. 1984, 158, 1–8. [Google Scholar]
- Dai, G.; Li, R.; Chen, H.; Jiang, C.; You, X.; Wu, Y. A ferritin-like protein with antioxidant activity in Ureaplasma urealyticum. BMC Microbiol. 2015, 15, 145. [Google Scholar] [CrossRef] [PubMed]
- Kumaran, D.; Bonanno, J.B.; Burley, S.K.; Swaminathan, S. Crystal structure of phosphatidylglycerophosphatase (PGPase), a putative membrane-bound lipid phosphatase, reveals a novel binuclear metal binding site and two “proton wires”. Proteins 2006, 64, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, D.; Martínez, B.; Rodríguez, A.; García, P. Genomic characterization of two Staphylococcus epidermidis bacteriophages with anti-biofilm potential. BMC Genom. 2012, 13, 228. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, D.M.; Cai, X.; Mushegian, A. Evolutionarily conserved orthologous families in phages are relatively rare in their prokaryotic hosts. J. Bacteriol. 2011, 193, 1806–1814. [Google Scholar] [CrossRef] [PubMed]
- Schumann, W. Dynamics of the Bacterial Chromosome: Structure and Function; Wiley-VCH: Weinheim, Germany, 2006; ISBN 3527304967. [Google Scholar]
- Ignacio-Espinoza, J.C.; Sullivan, M.B. Phylogenomics of T4 cyanophages: Lateral gene transfer in the ‘core’ and origins of host genes. Environ. Microbiol. 2012, 14, 2113–2126. [Google Scholar] [CrossRef] [PubMed]
- Millard, A.D.; Zwirglmaier, K.; Downey, M.J.; Mann, N.H.; Scanlan, D.J. Comparative genomics of marine cyanomyoviruses reveals the widespread occurrence of Synechococcus host genes localized to a hyperplastic region: Implications for mechanisms of cyanophage evolution. Environ. Microbiol. 2009, 11, 2370–2387. [Google Scholar] [CrossRef] [PubMed]
- Vigil-Stenman, T.; Ininbergs, K.; Bergman, B.; Ekman, M. High abundance and expression of transposases in bacteria from the Baltic Sea. ISME J. 2017, 11, 2611–2623. [Google Scholar] [CrossRef] [PubMed]
- De Melo, A.G.; Levesque, S.; Moineau, S. Phages as friends and enemies in food processing. Curr. Opin. Biotechnol. 2018, 49, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.; Neve, H.; McAuliffe, O.; Ross, R.P.; Arendt, E.K.; Coffey, A. Isolation and Characterization of Bacteriophages That Inhibit Strains of Pediococcus Damnosus, Lactobacillus Brevis, and Lactobacillus paraplantarum That Cause Beer Spoilage. J. Am. Soc. Brew. Chem. 2011, 69, 8–12. [Google Scholar] [CrossRef]
- Deasy, T.; Mahony, J.; Neve, H.; Heller, K.J.; van Sinderen, D. Isolation of a Virulent Lactobacillus brevis Phage and Its Application in the Control of Beer Spoilage. J. Food Prot. 2011, 74, 2157–2161. [Google Scholar] [CrossRef]
- Kelly, D.; O’Sullivan, O.; Mills, S.; McAuliffe, O.; Ross, R.P.; Neve, H.; Coffey, A. Genome sequence of the phage clP1, which infects the beer spoilage bacterium Pediococcus damnosus. Gene 2012, 504, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Kim, Y.; Ma, Q.; Hong, S.H.; Pokusaeva, K.; Sturino, J.M.; Wood, T.K. Cryptic prophages help bacteria cope with adverse environments. Nat. Commun. 2010, 1, 147. [Google Scholar] [CrossRef] [PubMed]
- Lepeuple, A.-S.; Van Gemert, E.; Chapot-Chartier, M.-P. Analysis of the bacteriolytic enzymes of the autolytic lactococcus lactis subsp. cremoris strain AM2 by renaturing polyacrylamide gel electrophoresis: Identification of a prophage-encoded enzyme. Appl. Environ. Microbiol. 1998, 64, 4142–4148. [Google Scholar] [PubMed]
- SnapGene Software. From GSL Biotech. Available online: https://www.snapgene.com/ (accessed on 1 October 2018).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structural Components | Dimensions (nm) | Counted Phage Particles |
|---|---|---|
| Head Diameter | 92.7 ± 3.5 | 17 |
| Tail Length (with Baseplate) | 222.4 ± 9.0 | 17 |
| Tail Width | 20.9 ± 0.6 | 17 |
| Baseplate Width | 44.1 ± 3.9 | 16 |
| Baseplate Length | 25.0 ± 2.4 | 16 |
| Baseplate Appendages Length | 21.8 ± 1.8 | 10 |
| Globular Structure Diameter | 9.2 ± 1.3 | 12 |
| Tail Fiber Length | 30.4 ± 2.2 | 13 |
| Phage Isolate | Open Reading Frames with Assigned Function | Genome Size (bp) | G/C Content (%) | tRNA Genes |
|---|---|---|---|---|
| Semele | 40/177 | 139,450 | 36.3 | 11 |
| Bacchae | 38/180 | 141,124 | 36.3 | 15 |
| Iacchus | 44/170 | 137,973 | 36.5 | 7 |
| Dionysus | 42/172 | 141,344 | 36.6 | 7 |
| Bromius | 39/172 | 140,527 | 36.5 | 7 |
| Description | FDR | Coverage (%) | No of Peptides | Molecular Mass (kDa) | Gene |
|---|---|---|---|---|---|
| Major capsid protein | High | 2 | 1 | 53.5 | peg. 170 |
| Putative portal protein | High | 2 | 1 | 60.9 | peg. 2 |
| Hypothetical protein | Low | 3 | 1 | 101.9 | peg. 151 |
| Hydrolase | Low | 5 | 1 | 35.9 | peg. 5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kyrkou, I.; Byth Carstens, A.; Ellegaard-Jensen, L.; Kot, W.; Zervas, A.; Djurhuus, A.M.; Neve, H.; Hansen, M.; Hestbjerg Hansen, L. Expanding the Diversity of Myoviridae Phages Infecting Lactobacillus plantarum—A Novel Lineage of Lactobacillus Phages Comprising Five New Members. Viruses 2019, 11, 611. https://doi.org/10.3390/v11070611
Kyrkou I, Byth Carstens A, Ellegaard-Jensen L, Kot W, Zervas A, Djurhuus AM, Neve H, Hansen M, Hestbjerg Hansen L. Expanding the Diversity of Myoviridae Phages Infecting Lactobacillus plantarum—A Novel Lineage of Lactobacillus Phages Comprising Five New Members. Viruses. 2019; 11(7):611. https://doi.org/10.3390/v11070611
Chicago/Turabian StyleKyrkou, Ifigeneia, Alexander Byth Carstens, Lea Ellegaard-Jensen, Witold Kot, Athanasios Zervas, Amaru Miranda Djurhuus, Horst Neve, Martin Hansen, and Lars Hestbjerg Hansen. 2019. "Expanding the Diversity of Myoviridae Phages Infecting Lactobacillus plantarum—A Novel Lineage of Lactobacillus Phages Comprising Five New Members" Viruses 11, no. 7: 611. https://doi.org/10.3390/v11070611
APA StyleKyrkou, I., Byth Carstens, A., Ellegaard-Jensen, L., Kot, W., Zervas, A., Djurhuus, A. M., Neve, H., Hansen, M., & Hestbjerg Hansen, L. (2019). Expanding the Diversity of Myoviridae Phages Infecting Lactobacillus plantarum—A Novel Lineage of Lactobacillus Phages Comprising Five New Members. Viruses, 11(7), 611. https://doi.org/10.3390/v11070611