Distinct Requirements for HIV-1 Accessory Proteins during Cell Coculture and Cell-Free Infection

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Cultures and Reagents
2.2. Plasmids
2.3. Transfections and Infections
2.4. Western Blot
2.5. Flow Cytometry and Cell Sorting
2.6. Fluorescence Microscopy
2.7. ELISA
2.8. Statistical Analysis
3. Results
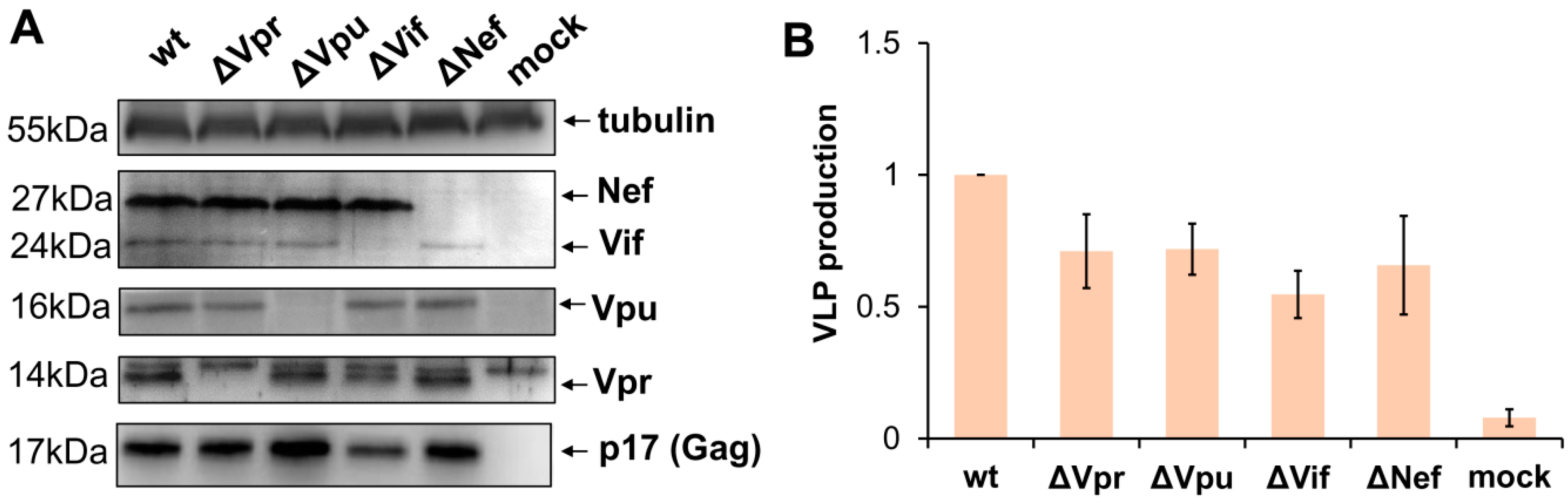
3.1. Generation and Characterization of HIV-1 with Mutations in Accessory Genes
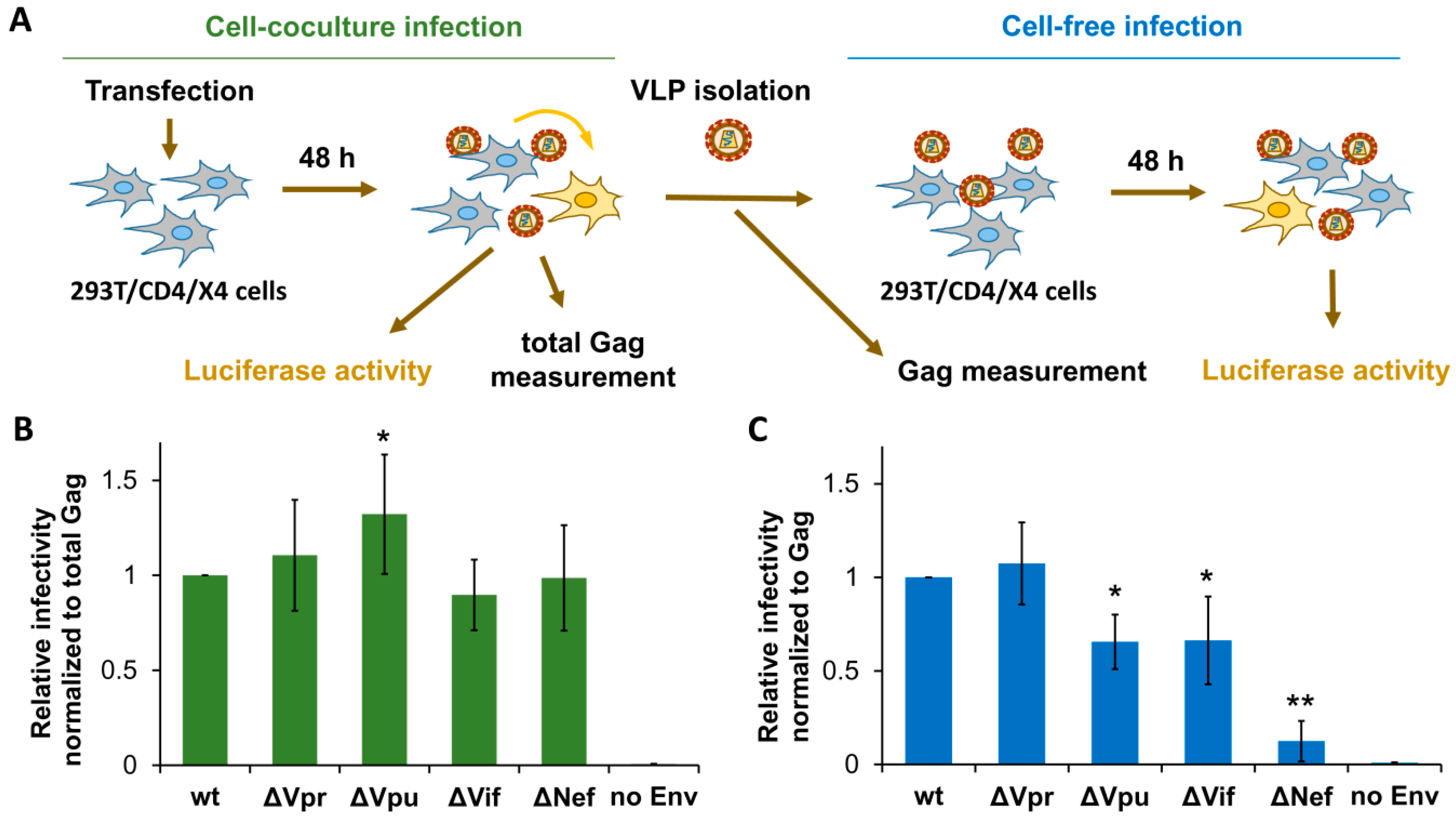
3.2. The Requirements for Accessory Gene Expression during HIV-1 Replication in Nonlymphoid Cells
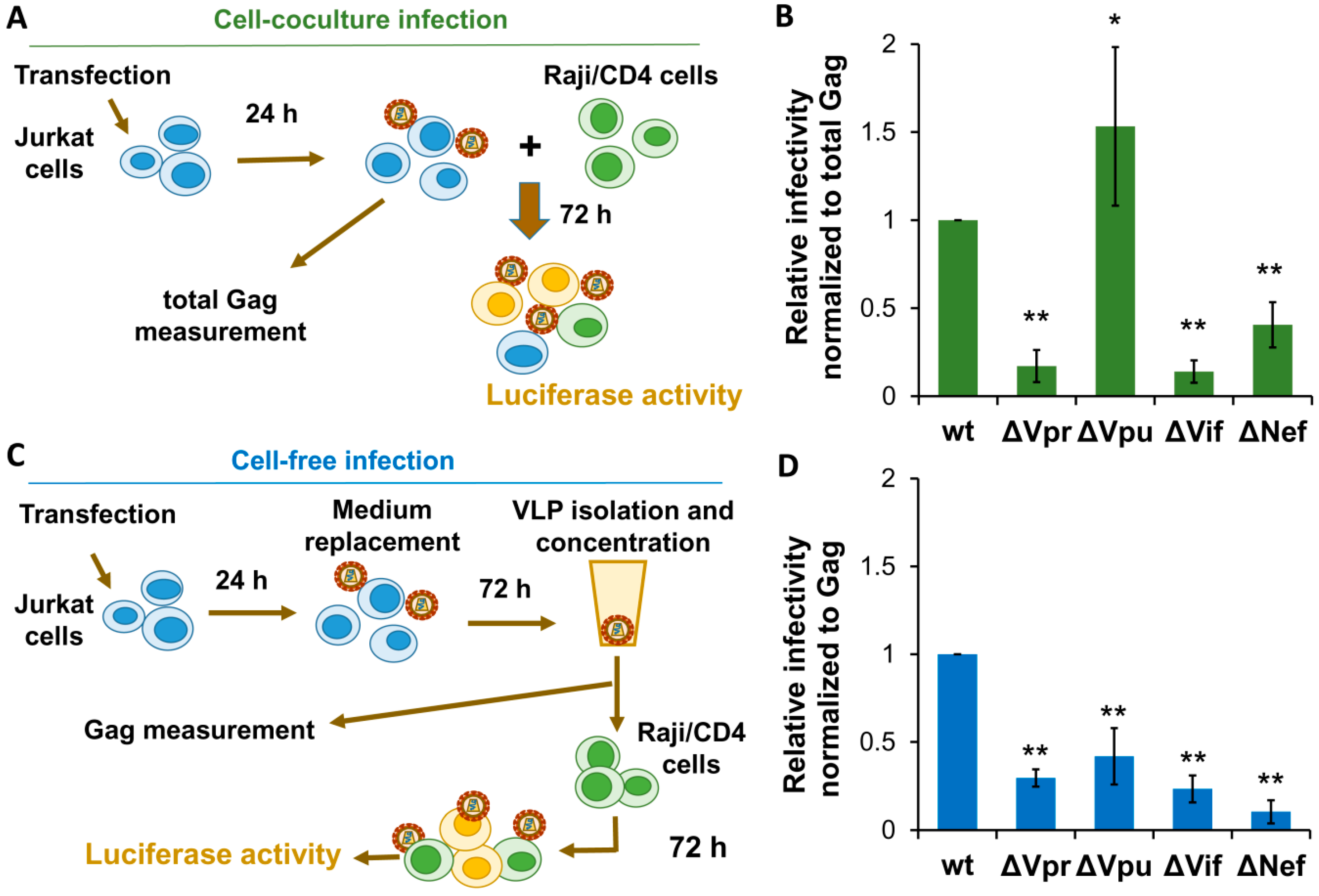
3.3. Effects of Accessory Gene Mutations on HIV-1 Cell Coculture and Cell-Free Infection in Lymphoid Cells
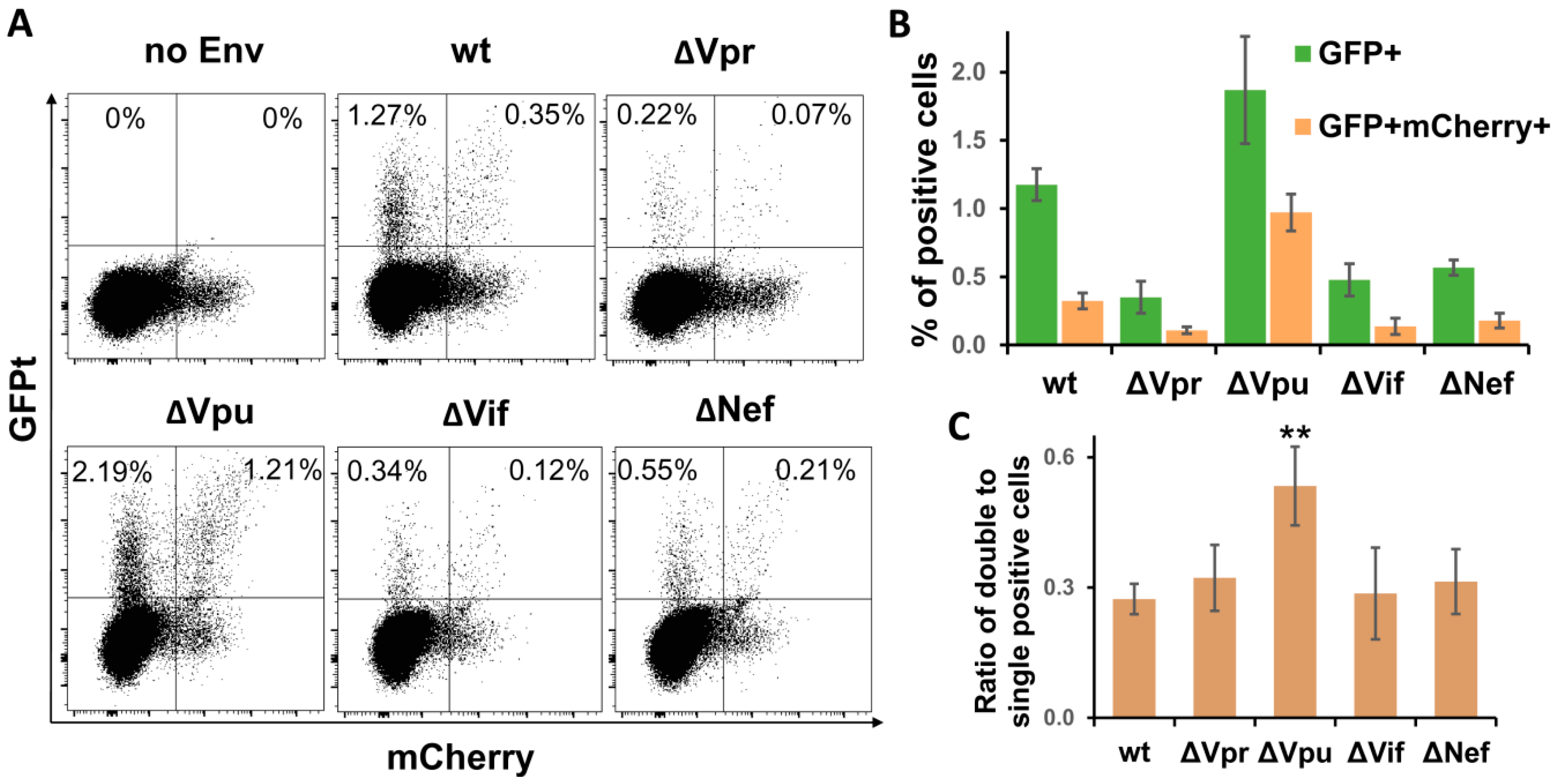
3.4. Vpu Deletion Increases Multiplicity of HIV-1 Infection in Lymphoid Cell Coculture
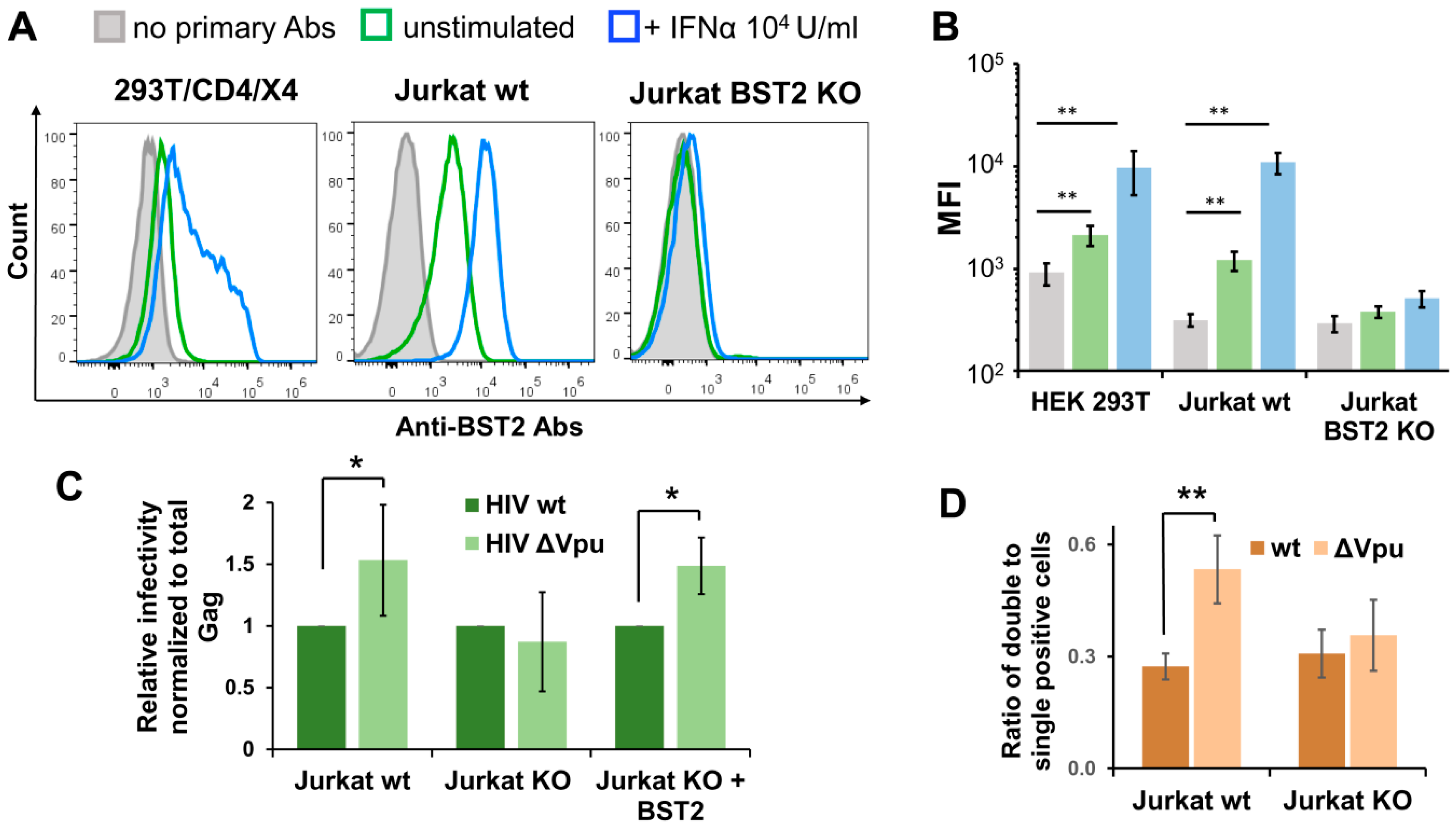
3.5. BST2 Knockout Abolishes Enhanced Replication of HIV-1 ΔVpu in Lymphoid Cell Coculture
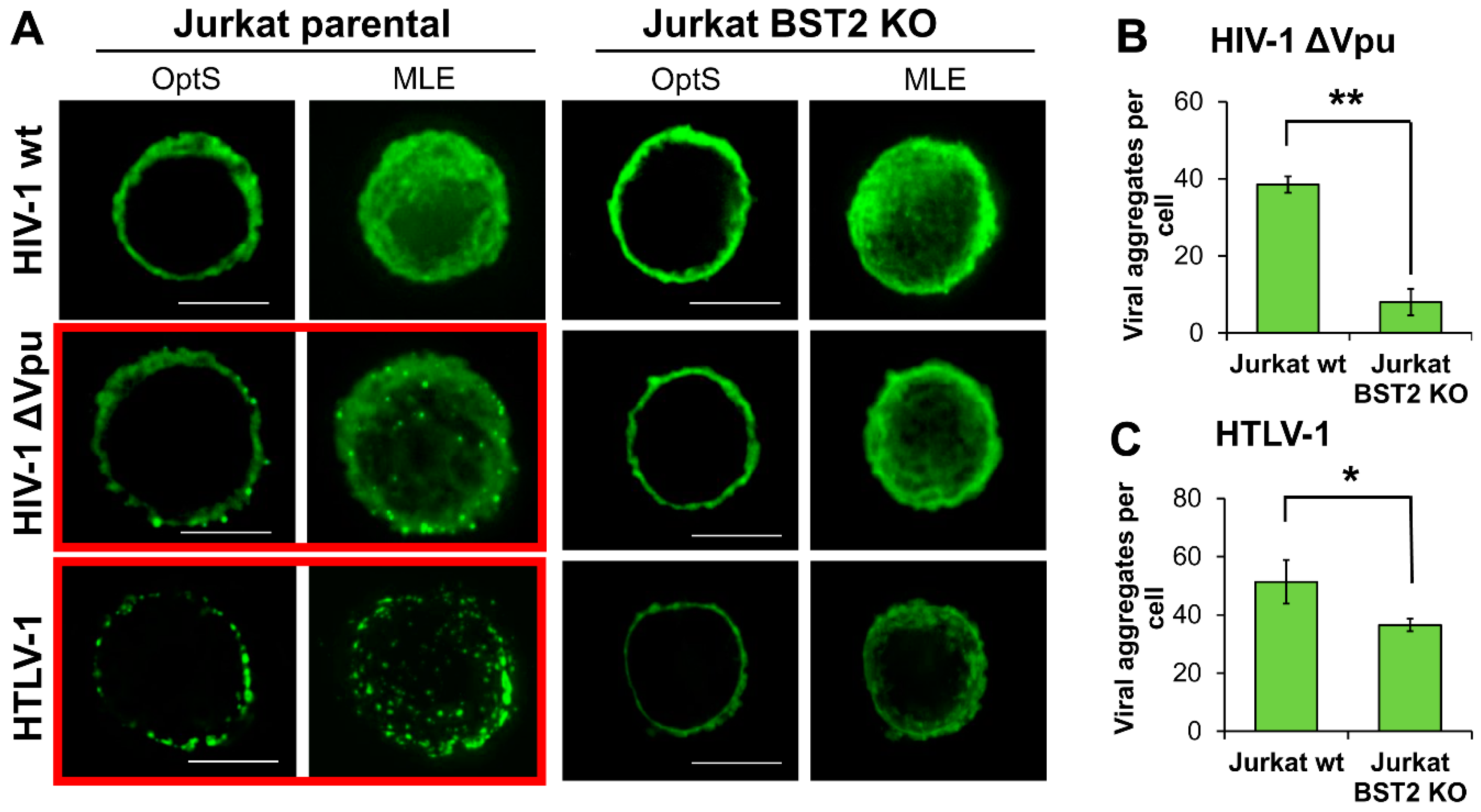
3.6. BST2 Induces Clusterization of HIV-1 ΔVpu and HTLV-1 Viral Particles on the Surface of T Cells
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yan, N.; Chen, Z.J. Intrinsic antiviral immunity. Nat. Immunol. 2012, 13, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Stremlau, M.; Owens, C.M.; Perron, M.J.; Kiessling, M.; Autissier, P.; Sodroski, J. The cytoplasmic body component trim5alpha restricts HIV-1 infection in old world monkeys. Nature 2004, 427, 848–853. [Google Scholar] [CrossRef]
- Stremlau, M.; Perron, M.; Lee, M.; Li, Y.; Song, B.; Javanbakht, H.; Diaz-Griffero, F.; Anderson, D.J.; Sundquist, W.I.; Sodroski, J. Specific recognition and accelerated uncoating of retroviral capsids by the trim5alpha restriction factor. Proc. Nat. Acad. Sci. USA 2006, 103, 5514–5519. [Google Scholar] [CrossRef] [PubMed]
- Sayah, D.M.; Sokolskaja, E.; Berthoux, L.; Luban, J. Cyclophilin a retrotransposition into trim5 explains owl monkey resistance to HIV-1. Nature 2004, 430, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Sheehy, A.M.; Gaddis, N.C.; Choi, J.D.; Malim, M.H. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral vif protein. Nature 2002, 418, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.S.; Bishop, K.N.; Sheehy, A.M.; Craig, H.M.; Petersen-Mahrt, S.K.; Watt, I.N.; Neuberger, M.S.; Malim, M.H. DNA deamination mediates innate immunity to retroviral infection. Cell 2003, 113, 803–809. [Google Scholar] [CrossRef]
- Bishop, K.N.; Verma, M.; Kim, E.-Y.; Wolinsky, S.M.; Malim, M.H. APOBEC3G inhibits elongation of HIV-1 reverse transcripts. PLoS Pathog. 2008, 4, e1000231. [Google Scholar] [CrossRef] [PubMed]
- Hrecka, K.; Hao, C.; Gierszewska, M.; Swanson, S.K.; Kesik-Brodacka, M.; Srivastava, S.; Florens, L.; Washburn, M.P.; Skowronski, J. Vpx relieves inhibition of hiv-1 infection of macrophages mediated by the SAMHD1 protein. Nature 2011, 474, 658–661. [Google Scholar] [CrossRef]
- Berger, A.; Sommer, A.F.R.; Zwarg, J.; Hamdorf, M.; Welzel, K.; Esly, N.; Panitz, S.; Reuter, A.; Ramos, I.; Jatiani, A.; et al. Samhd1-deficient CD14+ cells from individuals with aicardi-goutières syndrome are highly susceptible to HIV-1 infection. PLoS Pathog. 2011, 7, e1002425. [Google Scholar] [CrossRef]
- Goldstone, D.C.; Ennis-Adeniran, V.; Hedden, J.J.; Groom, H.C.T.; Rice, G.I.; Christodoulou, E.; Walker, P.A.; Kelly, G.; Haire, L.F.; Yap, M.W.; et al. HIV-1 restriction factor samhd1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature 2011, 480, 379–382. [Google Scholar] [CrossRef]
- Goujon, C.; Moncorgé, O.; Bauby, H.; Doyle, T.; Ward, C.C.; Schaller, T.; Hué, S.; Barclay, W.S.; Schulz, R.; Malim, M.H. Human MX2 is an interferon-induced post-entry inhibitor of HIV-1 infection. Nature 2013, 502, 559–562. [Google Scholar] [CrossRef] [PubMed]
- Kane, M.; Yadav, S.S.; Bitzegeio, J.; Kutluay, S.B.; Zang, T.; Wilson, S.J.; Schoggins, J.W.; Rice, C.M.; Yamashita, M.; Hatziioannou, T.; et al. MX2 is an interferon-induced inhibitor of HIV-1 infection. Nature 2013, 502, 563–566. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Pan, Q.; Ding, S.; Qian, J.; Xu, F.; Zhou, J.; Cen, S.; Guo, F.; Liang, C. The interferon-inducible MXB protein inhibits HIV-1 infection. Cell Host Microbe 2013, 14, 398–410. [Google Scholar]
- Li, M.; Kao, E.; Gao, X.; Sandig, H.; Limmer, K.; Pavon-Eternod, M.; Jones, T.E.; Landry, S.; Pan, T.; Weitzman, M.D.; et al. Codon-usage-based inhibition of hiv protein synthesis by human schlafen 11. Nature 2012, 491, 125–128. [Google Scholar] [CrossRef]
- Jakobsen, M.R.; Mogensen, T.H.; Paludan, S.R. Caught in translation: Innate restriction of HIV mRNA translation by a schlafen family protein. Cell. Res. 2013, 23, 320–322. [Google Scholar] [CrossRef] [PubMed]
- Neil, S.J.D.; Zang, T.; Bieniasz, P.D. Tetherin inhibits retrovirus release and is antagonized by HIV-1 vpu. Nature 2008, 451, 425–430. [Google Scholar] [CrossRef]
- Usami, Y.; Wu, Y.; Gottlinger, H.G. Serinc3 and serinc5 restrict HIV-1 infectivity and are counteracted by nef. Nature 2015, 526, 218–223. [Google Scholar] [CrossRef]
- Sood, C.; Marin, M.; Chande, A.; Pizzato, M.; Melikyan, G.B. Serinc5 protein inhibits HIV-1 fusion pore formation by promoting functional inactivation of envelope glycoproteins. J. Biol. Chem. 2017, 292, 6014–6026. [Google Scholar] [CrossRef]
- Sauter, D.; Schindler, M.; Specht, A.; Landford, W.N.; Münch, J.; Kim, K.-A.; Votteler, J.; Schubert, U.; Bibollet-Ruche, F.; Keele, B.F.; et al. Tetherin-driven adaptation of vpu and nef function and the evolution of pandemic and nonpandemic hiv-1 strains. Cell Host Microbe 2009, 6, 409–421. [Google Scholar]
- Agosto, L.M.; Uchil, P.D.; Mothes, W. HIV cell-to-cell transmission: Effects on pathogenesis and antiretroviral therapy. Trends Microbiol. 2015, 23, 289–295. [Google Scholar] [CrossRef]
- Zhong, P.; Agosto, L.M.; Ilinskaya, A.; Dorjbal, B.; Truong, R.; Derse, D.; Uchil, P.D.; Heidecker, G.; Mothes, W. Cell-to-cell transmission can overcome multiple donor and target cell barriers imposed on cell-free HIV. PLoS ONE 2013, 8, e53138. [Google Scholar] [CrossRef]
- Galloway, N.L.; Doitsh, G.; Monroe, K.M.; Yang, Z.; Munoz-Arias, I.; Levy, D.N.; Greene, W.C. Cell-to-cell transmission of HIV-1 is required to trigger pyroptotic death of lymphoid-tissue-derived cd4 t cells. Cell. Rep. 2015, 12, 1555–1563. [Google Scholar] [CrossRef]
- Sewald, X.; Motamedi, N.; Mothes, W. Viruses exploit the tissue physiology of the host to spread in vivo. Curr. Opin. Cell. Biol. 2016, 41, 81–90. [Google Scholar] [CrossRef]
- Blanchet, F.P.; Stalder, R.; Czubala, M.; Lehmann, M.; Rio, L.; Mangeat, B.; Piguet, V. TLR-4 engagement of dendritic cells confers a BST-2/tetherin-mediated restriction of HIV-1 infection to CD4+ T cells across the virological synapse. Retrovirology 2013, 10, 6. [Google Scholar] [CrossRef] [PubMed]
- Casartelli, N.; Sourisseau, M.; Feldmann, J.; Guivel-Benhassine, F.; Mallet, A.; Marcelin, A.-G.; Guatelli, J.; Schwartz, O. Tetherin restricts productive HIV-1 cell-to-cell transmission. PLoS Pathog. 2010, 6, e1000955. [Google Scholar] [CrossRef]
- Chu, H.; Wang, J.-J.; Qi, M.; Yoon, J.-J.; Chen, X.; Wen, X.; Hammonds, J.; Ding, L.; Spearman, P. Tetherin/BST-2 is essential for the formation of the intracellular virus-containing compartment in HIV-infected macrophages. Cell Host Microbe 2012, 12, 360–372. [Google Scholar]
- Giese, S.; Marsh, M. Tetherin can restrict cell-free and cell-cell transmission of HIV from primary macrophages to t cells. PLoS Pathog. 2014, 10, e1004189. [Google Scholar] [CrossRef]
- Kuhl, B.D.; Sloan, R.D.; Donahue, D.A.; Bar-Magen, T.; Liang, C.; Wainberg, M.A. Tetherin restricts direct cell-to-cell infection of HIV-1. Retrovirology 2010, 7, 115. [Google Scholar] [CrossRef] [PubMed]
- Jolly, C.; Booth, N.J.; Neil, S.J.D. Cell-cell spread of human immunodeficiency virus type 1 overcomes tetherin/BST-2-mediated restriction in t cells. J. Virol. 2010, 84, 12185–12199. [Google Scholar] [CrossRef]
- Coleman, C.M.; Spearman, P.; Wu, L. Tetherin does not significantly restrict dendritic cell-mediated HIV-1 transmission and its expression is upregulated by newly synthesized HIV-1 Nef. Retrovirology 2011, 8, 26. [Google Scholar] [CrossRef]
- Shunaeva, A.; Potashnikova, D.; Pichugin, A.; Mishina, A.; Filatov, A.; Nikolaitchik, O.; Hu, W.-S.; Mazurov, D. Improvement of HIV-1 and human T cell lymphotropic virus type 1 replication-dependent vectors via optimization of reporter gene reconstitution and modification with intronic short hairpin RNA. J. Virol. 2015, 89, 10591–10601. [Google Scholar] [CrossRef] [PubMed]
- Mazurov, D.; Ilinskaya, A.; Heidecker, G.; Lloyd, P.; Derse, D. Quantitative comparison of HTLV-1 and HIV-1 cell-to-cell infection with new replication dependent vectors. PLoS Pathog. 2010, 6, e1000788. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Freed, E.O. The long cytoplasmic tail of gp41 is required in a cell type-dependent manner for HIV-1 envelope glycoprotein incorporation into virions. Proc. Nat. Acad. Sci. USA 2000, 97, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Tarasevich, A.; Filatov, A.; Pichugin, A.; Mazurov, D. Monoclonal antibody profiling of cell surface proteins associated with the viral biofilms on HTLV-1 transformed cells. Acta Virolog. 2015, 59, 247–256. [Google Scholar] [CrossRef]
- Mazurov, D.; Ilinskaya, A.; Heidecker, G.; Filatov, A. Role of O-glycosylation and expression of CD43 and CD45 on the surfaces of effector t cells in human T cell leukemia virus type 1 cell-to-cell infection. J. Virol. 2012, 86, 2447–2458. [Google Scholar] [CrossRef]
- Zotova, A.; Lopatukhina, E.; Filatov, A.; Khaitov, M.; Mazurov, D. Gene editing in human lymphoid cells: Role for donor DNA, type of genomic nuclease and cell selection method. Viruses 2017, 9, 325. [Google Scholar] [CrossRef]
- Zotova, A.; Pichugin, A.; Atemasova, A.; Knyazhanskaya, E.; Lopatukhina, E.; Mitkin, N.; Holmuhamedov, E.; Gottikh, M.; Kuprash, D.; Filatov, A.; et al. Isolation of gene-edited cells via knock-in of short glycophosphatidylinositol-anchored epitope tags. Sci. Rep. 2019, 9, 3132. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Lin, C.Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.E.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef]
- Pais-Correia, A.M.; Sachse, M.; Guadagnini, S.; Robbiati, V.; Lasserre, R.; Gessain, A.; Gout, O.; Alcover, A.; Thoulouze, M.I. Biofilm-like extracellular viral assemblies mediate HTLV-1 cell-to-cell transmission at virological synapses. Nat. Med. 2010, 16, 83–89. [Google Scholar] [CrossRef]
- Jolly, C. Cell-to-cell transmission of retroviruses: Innate immunity and interferon-induced restriction factors. Virology 2011, 411, 251–259. [Google Scholar] [CrossRef]
- Andrew, A.; Strebel, K. The interferon-inducible host factor bone marrow stromal antigen 2/tetherin restricts virion release, but is it actually a viral restriction factor? J. Interferon Cytokine Res. 2011, 31, 137–144. [Google Scholar] [CrossRef]
- Rosa, A.; Chande, A.; Ziglio, S.; De Sanctis, V.; Bertorelli, R.; Goh, S.L.; McCauley, S.M.; Nowosielska, A.; Antonarakis, S.E.; Luban, J.; et al. HIV-1 Nef promotes infection by excluding serinc5 from virion incorporation. Nature 2015, 526, 212–217. [Google Scholar] [CrossRef]
- Aiken, C. Pseudotyping human immunodeficiency virus type 1 (HIV-1) by the glycoprotein of vesicular stomatitis virus targets HIV-1 entry to an endocytic pathway and suppresses both the requirement for Nef and the sensitivity to cyclosporin A. J. Virol. 1997, 71, 5871–5877. [Google Scholar]
- Dale, B.M.; McNerney, G.P.; Thompson, D.L.; Hubner, W.; de Los Reyes, K.; Chuang, F.Y.; Huser, T.; Chen, B.K. Cell-to-cell transfer of HIV-1 via virological synapses leads to endosomal virion maturation that activates viral membrane fusion. Cell Host Microbe 2011, 10, 551–562. [Google Scholar] [CrossRef]
- Lama, J.; Mangasarian, A.; Trono, D. Cell-surface expression of CD4 reduces HIV-1 infectivity by blocking Env incorporation in a Nef- and Vpu-inhibitable manner. Curr. Biol. 1999, 9, 622–631. [Google Scholar] [CrossRef]
- Hammonds, J.; Wang, J.-J.; Yi, H.; Spearman, P. Immunoelectron microscopic evidence for tetherin/BST2 as the physical bridge between HIV-1 virions and the plasma membrane. PLoS Pathog. 2010, 6, e1000749. [Google Scholar] [CrossRef]
- Perez-Caballero, D.; Zang, T.; Ebrahimi, A.; McNatt, M.W.; Gregory, D.A.; Johnson, M.C.; Bieniasz, P.D. Tetherin inhibits HIV-1 release by directly tethering virions to cells. Cell 2009, 139, 499–511. [Google Scholar] [CrossRef]
- Van Damme, N.; Goff, D.; Katsura, C.; Jorgenson, R.L.; Mitchell, R.; Johnson, M.C.; Stephens, E.B.; Guatelli, J. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral vpu protein. Cell Host Microbe 2008, 3, 245–252. [Google Scholar] [CrossRef]
- Douglas, J.L.; Viswanathan, K.; McCarroll, M.N.; Gustin, J.K.; Fruh, K.; Moses, A.V. Vpu directs the degradation of the human immunodeficiency virus restriction factor BST-2/tetherin via a trcp-dependent mechanism. J. Virol 2009, 83, 7931–7947. [Google Scholar] [CrossRef]
- Goffinet, C.; Allespach, I.; Homann, S.; Tervo, H.-M.; Habermann, A.; Rupp, D.; Oberbremer, L.; Kern, C.; Tibroni, N.; Welsch, S.; et al. HIV-1 antagonism of CD317 is species specific and involves Vpu-mediated proteasomal degradation of the restriction factor. Cell Host Microbe 2009, 5, 285–297. [Google Scholar]
- Mangeat, B.; Gers-Huber, G.; Lehmann, M.; Zufferey, M.; Luban, J.; Piguet, V. HIV-1 vpu neutralizes the antiviral factor tetherin/BST-2 by binding it and directing its beta-trcp2-dependent degradation. PLoS Pathog. 2009, 5, e1000574. [Google Scholar] [CrossRef]
- Madjo, U.; Leymarie, O.; Fremont, S.; Kuster, A.; Nehlich, M.; Gallois-Montbrun, S.; Janvier, K.; Berlioz-Torrent, C. Lc3c contributes to vpu-mediated antagonism of BST2/tetherin restriction on HIV-1 release through a non-canonical autophagy pathway. Cell. Rep. 2016, 17, 2221–2233. [Google Scholar] [CrossRef]
- Klimkait, T.; Strebel, K.; Hoggan, M.D.; Martin, M.A.; Orenstein, J.M. The human immunodeficiency virus type 1-specific protein vpu is required for efficient virus maturation and release. J. Virol. 1990, 64, 621–629. [Google Scholar]
- Zhang, Y.; Ozono, S.; Yao, W.; Tobiume, M.; Yamaoka, S.; Kishigami, S.; Fujita, H.; Tokunaga, K. CRISPR-mediated activation of endogenous BST-2/tetherin expression inhibits wild-type HIV-1 production. Sci. Rep. 2019, 9, 3134. [Google Scholar] [CrossRef]
- Jager, S.; Kim, D.Y.; Hultquist, J.F.; Shindo, K.; LaRue, R.S.; Kwon, E.; Li, M.; Anderson, B.D.; Yen, L.; Stanley, D.; et al. Vif hijacks cbf-beta to degrade APOBEC3G and promote HIV-1 infection. Nature 2011, 481, 371–375. [Google Scholar] [CrossRef]
- Chaipan, C.; Smith, J.L.; Hu, W.S.; Pathak, V.K. APOBEC3G restricts HIV-1 to a greater extent than APOBEC3F and APOBEC3DE in human primary CD4+ T cells and macrophages. J. Virol. 2013, 87, 444–453. [Google Scholar] [CrossRef]
- Stopak, K.; de Noronha, C.; Yonemoto, W.; Greene, W.C. HIV-1 Vif blocks the antiviral activity of APOBEC3G by impairing both its translation and intracellular stability. Mol. Cell. 2003, 12, 591–601. [Google Scholar] [CrossRef]
- Yuan, T.; Yao, W.; Huang, F.; Sun, B.; Yang, R. The human antiviral factor trim11 is under the regulation of HIV-1 Vpr. PLoS ONE 2014, 9, e104269. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zotova, A.; Atemasova, A.; Pichugin, A.; Filatov, A.; Mazurov, D. Distinct Requirements for HIV-1 Accessory Proteins during Cell Coculture and Cell-Free Infection. Viruses 2019, 11, 390. https://doi.org/10.3390/v11050390
Zotova A, Atemasova A, Pichugin A, Filatov A, Mazurov D. Distinct Requirements for HIV-1 Accessory Proteins during Cell Coculture and Cell-Free Infection. Viruses. 2019; 11(5):390. https://doi.org/10.3390/v11050390
Chicago/Turabian StyleZotova, Anastasia, Anastasia Atemasova, Alexey Pichugin, Alexander Filatov, and Dmitriy Mazurov. 2019. "Distinct Requirements for HIV-1 Accessory Proteins during Cell Coculture and Cell-Free Infection" Viruses 11, no. 5: 390. https://doi.org/10.3390/v11050390
APA StyleZotova, A., Atemasova, A., Pichugin, A., Filatov, A., & Mazurov, D. (2019). Distinct Requirements for HIV-1 Accessory Proteins during Cell Coculture and Cell-Free Infection. Viruses, 11(5), 390. https://doi.org/10.3390/v11050390