In-Vivo Gene Therapy with Foamy Virus Vectors


Abstract
1. Introduction
2. Limitations of Ex-Vivo Gene Therapy
3. Viral and Non-Viral Vectors Platforms for In-Vivo Gene Therapy
4. In-Vivo Gene Therapy for Canine SCID-X1 with FVVs
5. Optimization of FVV In-Vivo Gene Delivery
6. Summary and Future Perspective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mamcarz, E.; Zhou, S.; Lockey, T.; Abdelsamed, H.; Cross, S.J.; Kang, G.; Ma, Z.; Condori, J.; Dowdy, J.; Triplett, B.; et al. Lentiviral gene therapy combined with low-dose busulfan in infants with SCID-X1. N. Engl. J. Med. 2019, 380, 1525–1534. [Google Scholar] [CrossRef]
- Anguela, X.M.; High, K.A. Entering the modern era of gene therapy. Annu. Rev. Med. 2019, 70, 273–288. [Google Scholar] [CrossRef]
- Naldini, L. Genetic engineering of hematopoiesis: Current stage of clinical translation and future perspectives. EMBO Mol. Med. 2019, 11, e9958. [Google Scholar] [CrossRef]
- Cavazzana, M.; Bushman, F.D.; Miccio, A.; Andre-Schmutz, I.; Six, E. Gene therapy targeting haematopoietic stem cells for inherited diseases: Progress and challenges. Nat. Rev. Drug Discov. 2019, 18, 447–462. [Google Scholar] [CrossRef]
- Coquerelle, S.; Ghardallou, M.; Rais, S.; Taupin, P.; Touzot, F.; Boquet, L.; Blanche, S.; Benaouadi, S.; Brice, T.; Tuchmann-Durand, C.; et al. Innovative curative treatment of beta thalassemia: Cost-efficacy analysis of gene therapy versus allogenic hematopoietic stem-cell transplantation. Hum. Gene Ther. 2019, 30, 753–761. [Google Scholar] [CrossRef]
- Touzot, F.; Moshous, D.; Creidy, R.; Neven, B.; Frange, P.; Cros, G.; Caccavelli, L.; Blondeau, J.; Magnani, A.; Luby, J.M.; et al. Faster T-cell development following gene therapy compared with haploidentical HSCT in the treatment of SCID-X1. Blood 2015, 125, 3563–3569. [Google Scholar] [CrossRef]
- Burtner, C.R.; Beard, B.C.; Kennedy, D.R.; Wohlfahrt, M.E.; Adair, J.E.; Trobridge, G.D.; Scharenberg, A.M.; Torgerson, T.R.; Rawlings, D.J.; Felsburg, P.J.; et al. Intravenous injection of a foamy virus vector to correct canine SCID-X1. Blood 2014, 123, 3578–3584. [Google Scholar] [CrossRef]
- Humbert, O.; Chan, F.; Rajawat, Y.S.; Torgerson, T.R.; Burtner, C.R.; Hubbard, N.W.; Humphrys, D.; Norgaard, Z.K.; O’Donnell, P.; Adair, J.E.; et al. Rapid immune reconstitution of SCID-X1 canines after G-CSF/AMD3100 mobilization and in vivo gene therapy. Blood Adv. 2018, 2, 987–999. [Google Scholar] [CrossRef]
- Hacein-Bey-Abina, S.; Pai, S.Y.; Gaspar, H.B.; Armant, M.; Berry, C.C.; Blanche, S.; Bleesing, J.; Blondeau, J.; Buckland, K.F.; Caccavelli, L.; et al. A modified gamma-retrovirus vector for X-linked severe combined immunodeficiency. N. Engl. J. Med. 2014, 371, 1407–1417. [Google Scholar] [CrossRef]
- Cronin, J.; Zhang, X.Y.; Reiser, J. Altering the tropism of lentiviral vectors through pseudotypin. Curr. Gene Ther. 2005, 5, 387–398. [Google Scholar] [CrossRef]
- Kafri, T.; Blomer, U.; Peterson, D.A.; Gage, F.H.; Verma, I.M. Sustained expression of genes delivered directly into liver and muscle by lentiviral vectors. Nat. Genet. 1997, 17, 314–317. [Google Scholar] [CrossRef]
- Zufferey, R.; Dull, T.; Mandel, R.J.; Bukovsky, A.; Quiroz, D.; Naldini, L.; Trono, D. Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. J. Virol. 1998, 72, 9873–9880. [Google Scholar]
- Richter, M.; Saydaminova, K.; Yumul, R.; Krishnan, R.; Liu, J.; Nagy, E.E.; Singh, M.; Izsvak, Z.; Cattaneo, R.; Uckert, W.; et al. In vivo transduction of primitive mobilized hematopoietic stem cells after intravenous injection of integrating adenovirus vectors. Blood 2016, 128, 2206–2217. [Google Scholar] [CrossRef]
- Schmid, M.; Ernst, P.; Honegger, A.; Suomalainen, M.; Zimmermann, M.; Braun, L.; Stauffer, S.; Thom, C.; Dreier, B.; Eibauer, M.; et al. Adenoviral vector with shield and adapter increases tumor specificity and escapes liver and immune control. Nat. Commun. 2018, 9, 450. [Google Scholar] [CrossRef]
- Wold, W.S.; Toth, K. Adenovirus vectors for gene therapy, vaccination and cancer gene therapy. Curr. Gene Ther. 2013, 13, 421–433. [Google Scholar] [CrossRef]
- Wang, H.; Georgakopoulou, A.; Psatha, N.; Li, C.; Capsali, C.; Samal, H.B.; Anagnostopoulos, A.; Ehrhardt, A.; Izsvak, Z.; Papayannopoulou, T.; et al. In vivo hematopoietic stem cell gene therapy ameliorates murine thalassemia intermedia. J. Clin. Investig. 2019, 129, 598–615. [Google Scholar] [CrossRef]
- Zhong, L.; Zhao, W.; Wu, J.; Maina, N.; Han, Z.; Srivastava, A. Adeno-associated virus-mediated gene transfer in hematopoietic stem/ progenitor cells as a therapeutic tool (Review). Curr. Gene Ther. 2006, 6, 683–698. [Google Scholar] [CrossRef]
- Srivastava, A. In vivo tissue-tropism of adeno-associated viral vectors. Curr. Opin. Virol. 2016, 21, 75–80. [Google Scholar] [CrossRef]
- Colella, P.; Ronzitti, G.; Mingozzi, F. Emerging issues in AAV-mediated in vivo gene therapy. Mol. Ther. Methods Clin. Dev. 2018, 8, 87–104. [Google Scholar] [CrossRef]
- Manno, C.S.; Pierce, G.F.; Arruda, V.R.; Glader, B.; Ragni, M.; Rasko, J.J.; Ozelo, M.C.; Hoots, K.; Blatt, P.; Konkle, B.; et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat. Med. 2006, 12, 342–347. [Google Scholar] [CrossRef]
- Marshall, E. Gene therapy death prompts review of adenovirus vector. Science 1999, 286, 2244–2245. [Google Scholar] [CrossRef]
- Raper, S.E.; Chirmule, N.; Lee, F.S.; Wivel, N.A.; Bagg, A.; Gao, G.P.; Wilson, J.M.; Batshaw, M.L. Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer. Mol. Genet. Metab. 2003, 80, 148–158. [Google Scholar] [CrossRef]
- Van der Loo, J.C.; Wright, J.F. Progress and challenges in viral vector manufacturing. Hum. Mol. Genet. 2016, 25, R42–R52. [Google Scholar] [CrossRef]
- Milone, M.C.; O’Doherty, U. Clinical use of lentiviral vectors. Leukemia 2018, 32, 1529–1541. [Google Scholar] [CrossRef]
- Follenzi, A.; Santambrogio, L.; Annoni, A. Immune responses to lentiviral vectors. Curr. Gene Ther. 2007, 7, 306–315. [Google Scholar] [CrossRef]
- Annoni, A.; Gregori, S.; Naldini, L.; Cantore, A. Modulation of immune responses in lentiviral vector-mediated gene transfer. Cell. Immunol. 2019, 342, 103802. [Google Scholar] [CrossRef]
- Russell, D.W.; Miller, A.D. Foamy virus vectors. J. Virol. 1996, 70, 217–222. [Google Scholar]
- Vassilopoulos, G.; Trobridge, G.; Josephson, N.C.; Russell, D.W. Gene transfer into murine hematopoietic stem cells with helper-free foamy virus vectors. Blood 2001, 98, 604–609. [Google Scholar] [CrossRef]
- Trobridge, G.D.; Russell, D.W. Helper-free foamy virus vectors. Hum. Gene Ther. 1998, 9, 2517–2525. [Google Scholar] [CrossRef]
- Hirata, R.K.; Miller, A.D.; Andrews, R.G.; Russell, D.W. Transduction of hematopoietic cells by foamy virus vectors. Blood 1996, 88, 3654–3661. [Google Scholar] [CrossRef]
- Trobridge, G.D.; Miller, D.G.; Jacobs, M.A.; Allen, J.M.; Kiem, H.P.; Kaul, R.; Russell, D.W. Foamy virus vector integration sites in normal human cells. Proc. Natl. Acad. Sci. USA 2006, 103, 1498–1503. [Google Scholar] [CrossRef]
- Kiem, H.P.; Allen, J.; Trobridge, G.; Olson, E.; Keyser, K.; Peterson, L.; Russell, D.W. Foamy virus-mediated gene transfer to canine repopulating cells. Blood 2007, 109, 65–70. [Google Scholar] [CrossRef]
- Trobridge, G.D.; Allen, J.M.; Peterson, L.; Ironside, C.G.; Russell, D.W.; Kiem, H.P. Foamy and lentiviral vectors transduce canine long-term repopulating cells at similar efficiency. Hum. Gene Ther. 2009, 20, 519–523. [Google Scholar] [CrossRef]
- Trobridge, G.D.; Beard, B.C.; Wu, R.A.; Ironside, C.; Malik, P.; Kiem, H.P. Stem cell selection in vivo using foamy vectors cures canine pyruvate kinase deficiency. PLoS ONE 2012, 7, e45173. [Google Scholar] [CrossRef]
- Trobridge, G.D.; Horn, P.A.; Beard, B.C.; Kiem, H.P. Large animal models for foamy virus vector gene therapy. Viruses 2012, 4, 3572–3588. [Google Scholar] [CrossRef]
- Thompson, A.A.; Walters, M.C.; Kwiatkowski, J.; Rasko, J.E.J.; Ribeil, J.A.; Hongeng, S.; Magrin, E.; Schiller, G.J.; Payen, E.; Semeraro, M.; et al. Gene therapy in patients with transfusion-dependent beta-thalassemia. N. Engl. J. Med. 2018, 378, 1479–1493. [Google Scholar] [CrossRef]
- Ribeil, J.A.; Hacein-Bey-Abina, S.; Payen, E.; Magnani, A.; Semeraro, M.; Magrin, E.; Caccavelli, L.; Neven, B.; Bourget, P.; El Nemer, W.; et al. Gene therapy in a patient with sickle cell disease. N. Engl. J. Med. 2017, 376, 848–855. [Google Scholar] [CrossRef]
- Hacein-Bey-Abina, S.; Hauer, J.; Lim, A.; Picard, C.; Wang, G.P.; Berry, C.C.; Martinache, C.; Rieux-Laucat, F.; Latour, S.; Belohradsky, B.H.; et al. Efficacy of gene therapy for X-linked severe combined immunodeficiency. N. Engl. J. Med. 2010, 363, 355–364. [Google Scholar] [CrossRef]
- Aiuti, A.; Biasco, L.; Scaramuzza, S.; Ferrua, F.; Cicalese, M.P.; Baricordi, C.; Dionisio, F.; Calabria, A.; Giannelli, S.; Castiello, M.C.; et al. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science 2013, 341, 1233151. [Google Scholar] [CrossRef]
- Biffi, A.; Montini, E.; Lorioli, L.; Cesani, M.; Fumagalli, F.; Plati, T.; Baldoli, C.; Martino, S.; Calabria, A.; Canale, S.; et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science 2013, 341, 1233158. [Google Scholar] [CrossRef]
- Sessa, M.; Lorioli, L.; Fumagalli, F.; Acquati, S.; Redaelli, D.; Baldoli, C.; Canale, S.; Lopez, I.D.; Morena, F.; Calabria, A.; et al. Lentiviral haemopoietic stem-cell gene therapy in early-onset metachromatic leukodystrophy: An ad-hoc analysis of a non-randomised, open-label, phase 1/2 trial. Lancet 2016, 388, 476–487. [Google Scholar] [CrossRef]
- De Ravin, S.S.; Wu, X.; Moir, S.; Anaya-O’Brien, S.; Kwatemaa, N.; Littel, P.; Theobald, N.; Choi, U.; Su, L.; Marquesen, M.; et al. Lentiviral hematopoietic stem cell gene therapy for X-linked severe combined immunodeficiency. Sci. Transl. Med. 2016, 8, 335ra57. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, D.R.; McLellan, K.; Moore, P.F.; Henthorn, P.S.; Felsburg, P.J. Effect of ex vivo culture of CD34+ bone marrow cells on immune reconstitution of XSCID dogs following allogeneic bone marrow transplantation. Biol. Blood Marrow Transplant. 2009, 15, 662–670. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cavazzana, M. Innovations needed for effective implementation of ex vivo gene therapies. Front. Med. 2017, 4, 29. [Google Scholar] [CrossRef]
- Yin, H.; Kanasty, R.L.; Eltoukhy, A.A.; Vegas, A.J.; Dorkin, J.R.; Anderson, D.G. Non-viral vectors for gene-based therapy. Nat. Rev. Genet. 2014, 15, 541–555. [Google Scholar] [CrossRef]
- Lundstrom, K. Viral vectors in gene therapy. Diseases 2018, 6, 42. [Google Scholar] [CrossRef]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef]
- Pardi, N.; Secreto, A.J.; Shan, X.; Debonera, F.; Glover, J.; Yi, Y.; Muramatsu, H.; Ni, H.; Mui, B.L.; Tam, Y.K.; et al. Administration of nucleoside-modified mRNA encoding broadly neutralizing antibody protects humanized mice from HIV-1 challenge. Nat. Commun. 2017, 8, 14630. [Google Scholar] [CrossRef]
- Naldini, L.; Blömer, U.; Gallay, P.; Ory, D.; Mulligan, R.; Gage, F.H.; Verma, I.M.; Trono, D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 1996, 272, 263–267. [Google Scholar] [CrossRef]
- Raper, S.E.; Yudkoff, M.; Chirmule, N.; Gao, G.P.; Nunes, F.; Haskal, Z.J.; Furth, E.E.; Propert, K.J.; Robinson, M.B.; Magosin, S.; et al. A pilot study of in vivo liver-directed gene transfer with an adenoviral vector in partial ornithine transcarbamylase deficiency. Hum. Gene Ther. 2002, 13, 163–175. [Google Scholar] [CrossRef]
- Hacein-Bey-Abina, S.; von Kalle, C.; Schmidt, M.; Le Deist, F.; Wulffraat, N.; McIntyre, E.; Radford, I.; Villeval, J.L.; Fraser, C.C.; Cavazzana-Calvo, M.; et al. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N. Eng. J. Med. 2003, 348, 255–256. [Google Scholar] [CrossRef]
- Hacein-Bey-Abina, S.; von Kalle, C.; Schmidt, M.; McCormack, M.P.; Wulffraat, N.; Leboulch, P.; Lim, A.; Osborne, C.S.; Pawliuk, R.; Morillon, E.; et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 2003, 302, 415–419. [Google Scholar] [CrossRef]
- Hacein-Bey-Abina, S.; Garrigue, A.; Wang, G.P.; Soulier, J.; Lim, A.; Morillon, E.; Clappier, E.; Caccavelli, L.; Delabesse, E.; Beldjord, K.; et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Investig. 2008, 118, 3132–3142. [Google Scholar] [CrossRef] [PubMed]
- Alba, R.; Bosch, A.; Chillon, M. Gutless adenovirus: Last-generation adenovirus for gene therapy. Gene Ther. 2005, 12 (Suppl. S1), S18–S27. [Google Scholar] [CrossRef]
- Li, C.; Psatha, N.; Wang, H.; Singh, M.; Samal, H.B.; Zhang, W.; Ehrhardt, A.; Izsvak, Z.; Papayannopoulou, T.; Lieber, A. Integrating HDAd5/35++ vectors as a new platform for HSC gene therapy of hemoglobinopathies. Mol. Ther. Methods Clin. Dev. 2018, 9, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.S.; Bishop, E.S.; Zhang, R.; Yu, X.; Farina, E.M.; Yan, S.; Zhao, C.; Zheng, Z.; Shu, Y.; Wu, X.; et al. Adenovirus-mediated gene delivery: Potential applications for gene and cell-based therapies in the new era of personalized medicine. Genes Dis. 2017, 4, 43–63. [Google Scholar] [CrossRef] [PubMed]
- Thornhill, S.I.; Schambach, A.; Howe, S.J.; Ulaganathan, M.; Grassman, E.; Williams, D.; Schiedlmeier, B.; Sebire, N.J.; Gaspar, H.B.; Kinnon, C.; et al. Self-inactivating gammaretroviral vectors for gene therapy of X-linked severe combined immunodeficiency. Mol. Ther. 2008, 16, 590–598. [Google Scholar] [CrossRef]
- Miyoshi, H.; Blomer, U.; Takahashi, M.; Gage, F.H.; Verma, I.M. Development of a self-inactivating lentivirus vector. J. Virol. 1998, 72, 8150–8157. [Google Scholar]
- Rio, P.; Navarro, S.; Wang, W.; Sanchez-Dominguez, R.; Pujol, R.M.; Segovia, J.C.; Bogliolo, M.; Merino, E.; Wu, N.; Salgado, R.; et al. Successful engraftment of gene-corrected hematopoietic stem cells in non-conditioned patients with Fanconi anemia. Nat. Med. 2019, 25, 1396–1401. [Google Scholar] [CrossRef]
- High, K.A.; Roncarolo, M.G. Gene Therapy. N. Engl. J. Med. 2019, 381, 455–464. [Google Scholar] [CrossRef]
- Kotterman, M.A.; Chalberg, T.W.; Schaffer, D.V. Viral vectors for gene therapy: Translational and clinical outlook. Annu. Rev. Biomed. Eng. 2015, 17, 63–89. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.T.; Parab, S.; Burnett, R.; Diago, O.; Ostertag, D.; Hofman, F.M.; Espinoza, F.L.; Martin, B.; Ibanez, C.E.; Kasahara, N.; et al. Intravenous administration of retroviral replicating vector, Toca 511, demonstrates therapeutic efficacy in orthotopic immune-competent mouse glioma model. Hum. Gene Ther. 2015, 26, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Palfi, S.; Gurruchaga, J.M.; Ralph, G.S.; Lepetit, H.; Lavisse, S.; Buttery, P.C.; Watts, C.; Miskin, J.; Kelleher, M.; Deeley, S.; et al. Long-term safety and tolerability of ProSavin, a lentiviral vector-based gene therapy for Parkinson’s disease: A dose escalation, open-label, phase 1/2 trial. Lancet 2014, 383, 1138–1146. [Google Scholar] [CrossRef]
- Campochiaro, P.A.; Lauer, A.K.; Sohn, E.H.; Mir, T.A.; Naylor, S.; Anderton, M.C.; Kelleher, M.; Harrop, R.; Ellis, S.; Mitrophanous, K.A. Lentiviral vector gene transfer of endostatin/angiostatin for macular degeneration (GEM) study. Hum. Gene Ther. 2017, 28, 99–111. [Google Scholar] [CrossRef]
- Cantore, A.; Ranzani, M.; Bartholomae, C.C.; Volpin, M.; Valle, P.D.; Sanvito, F.; Sergi, L.S.; Gallina, P.; Benedicenti, F.; Bellinger, D.; et al. Liver-directed lentiviral gene therapy in a dog model of hemophilia B. Sci. Transl. Med. 2015, 7, 277ra28. [Google Scholar] [CrossRef]
- Milani, M.; Annoni, A.; Bartolaccini, S.; Biffi, M.; Russo, F.; Di Tomaso, T.; Raimondi, A.; Lengler, J.; Holmes, M.C.; Scheiflinger, F.; et al. Genome editing for scalable production of alloantigen-free lentiviral vectors for in vivo gene therapy. EMBO Mol. Med. 2017, 9, 1558–1573. [Google Scholar] [CrossRef]
- Bernadin, O.; Amirache, F.; Girard-Gagnepain, A.; Moirangthem, R.D.; Levy, C.; Ma, K.; Costa, C.; Negre, D.; Reimann, C.; Fenard, D.; et al. Baboon envelope LVs efficiently transduced human adult, fetal, and progenitor T cells and corrected SCID-X1 T-cell deficiency. Blood Adv. 2019, 3, 461–475. [Google Scholar] [CrossRef]
- Munis, A.M.; Mattiuzzo, G.; Bentley, E.M.; Collins, M.K.; Eyles, J.E.; Takeuchi, Y. Use of heterologous vesiculovirus G proteins circumvents the humoral anti-envelope immunity in lentivector-based in vivo gene delivery. Mol. Ther. Nucleic Acids 2019, 17, 126–137. [Google Scholar] [CrossRef]
- Donsante, A.; Miller, D.G.; Li, Y.; Vogler, C.; Brunt, E.M.; Russell, D.W.; Sands, M.S. AAV vector integration sites in mouse hepatocellular carcinoma. Science 2007, 317, 477. [Google Scholar] [CrossRef]
- Xiong, W.; Wu, D.M.; Xue, Y.; Wang, S.K.; Chung, M.J.; Ji, X.; Rana, P.; Zhao, S.R.; Mai, S.; Cepko, C.L. AAV cis-regulatory sequences are correlated with ocular toxicity. Proc. Natl. Acad. Sci. USA 2019, 116, 5785–5794. [Google Scholar] [CrossRef]
- Hinderer, C.; Katz, N.; Buza, E.L.; Dyer, C.; Goode, T.; Bell, P.; Richman, L.K.; Wilson, J.M. Severe toxicity in nonhuman primates and piglets following high-dose intravenous administration of an adeno-associated virus vector expressing human SMN. Hum. Gene Ther. 2018, 29, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Lindemann, D.; Rethwilm, A. Foamy virus biology and its application for vector development. Viruses 2011, 3, 561–585. [Google Scholar] [CrossRef] [PubMed]
- Meiering, C.D.; Linial, M.L. Historical perspective of foamy virus epidemiology and infection. Clin. Microbiol. Rev. 2001, 14, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Nasimuzzaman, M.; Persons, D.A. Cell membrane-associated heparan sulfate is a receptor for prototype foamy virus in human, monkey, and rodent cells. Mol. Ther. 2012, 20, 1158–1166. [Google Scholar] [CrossRef] [PubMed]
- Linial, M. Why aren’t foamy viruses pathogenic? Trends Microbiol. 2000, 8, 284–289. [Google Scholar] [CrossRef]
- Everson, E.M.; Olzsko, M.E.; Leap, D.J.; Hocum, J.D.; Trobridge, G.D. A comparison of foamy and lentiviral vector genotoxicity in SCID-repopulating cells shows foamy vectors are less prone to clonal dominance. Mol. Ther. Methods Clin. Dev. 2016, 3, 16048. [Google Scholar] [CrossRef]
- Beard, B.C.; Keyser, K.A.; Trobridge, G.D.; Peterson, L.J.; Miller, D.G.; Jacobs, M.; Kaul, R.; Kiem, H.P. Unique integration profiles in a canine model of long-term repopulating cells transduced with gammaretrovirus, lentivirus, and foamy virus. Hum. Gene Ther. 2007, 18, 423–434. [Google Scholar] [CrossRef]
- Takeuchi, Y.; Liong, S.H.; Bieniasz, P.D.; Jager, U.; Porter, C.D.; Friedman, T.; McClure, M.O.; Weiss, R.A. Sensitization of rhabdo-, lenti-, and spumaviruses to human serum by galactosyl(alpha1-3)galactosylation. J. Virol. 1997, 71, 6174–6178. [Google Scholar]
- Trobridge, G.D. Foamy virus vectors for gene transfer (Review). Expert Opin. Biol. Ther. 2009, 9, 1427–1436. [Google Scholar] [CrossRef]
- Jezyk, P.F.; Felsburg, P.J.; Haskins, M.E.; Patterson, D.F. X-linked severe combined immunodeficiency in the dog. Clin. Immunol. Immunopathol. 1989, 52, 173–189. [Google Scholar] [CrossRef]
- Felsburg, P.J.; Somberg, R.L.; Hartnett, B.J.; Henthorn, P.S.; Carding, S.R. Canine X-linked severe combined immunodeficiency. A model for investigating the requirement for the common gamma chain (gamma c) in human lymphocyte development and function (Review). Immunol. Res. 1998, 17, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Henthorn, P.S.; Somberg, R.L.; Fimiani, V.M.; Puck, J.M.; Patterson, D.F.; Felsburg, P.J. IL-2R gamma gene microdeletion demonstrates that canine X-linked severe combined immunodeficiency is a homologue of the human disease. Genomics 1994, 23, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Ting-De Ravin, S.S.; Kennedy, D.R.; Naumann, N.; Kennedy, J.S.; Choi, U.; Hartnett, B.J.; Linton, G.F.; Whiting-Theobald, N.L.; Moore, P.F.; Vernau, W.; et al. Correction of canine X-linked severe combined immunodeficiency by in vivo retroviral gene therapy. Blood 2006, 107, 3091–3097. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hopman, R.K.; DiPersio, J.F. Advances in stem cell mobilization. Blood Rev. 2014, 28, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Liles, W.C.; Broxmeyer, H.E.; Rodger, E.; Wood, B.; Hubel, K.; Cooper, S.; Hangoc, G.; Bridger, G.J.; Henson, G.W.; Calandra, G.; et al. Mobilization of hematopoietic progenitor cells in healthy volunteers by AMD3100, a CXCR4 antagonist. Blood 2003, 102, 2728–2730. [Google Scholar] [CrossRef]
- Broxmeyer, H.E.; Orschell, C.M.; Clapp, D.W.; Hangoc, G.; Cooper, S.; Plett, P.A.; Liles, W.C.; Li, X.; Graham-Evans, B.; Campbell, T.B.; et al. Rapid mobilization of murine and human hematopoietic stem and progenitor cells with AMD3100, a CXCR4 antagonist. J. Exp. Med. 2005, 201, 1307–1318. [Google Scholar] [CrossRef]
- DiPersio, J.F.; Stadtmauer, E.A.; Nademanee, A.; Micallef, I.N.; Stiff, P.J.; Kaufman, J.L.; Maziarz, R.T.; Hosing, C.; Fruehauf, S.; Horwitz, M.; et al. Plerixafor and G-CSF versus placebo and G-CSF to mobilize hematopoietic stem cells for autologous stem cell transplantation in patients with multiple myeloma. Blood 2009, 113, 5720–5726. [Google Scholar] [CrossRef]
- Flomenberg, N.; Devine, S.M.; DiPersio, J.F.; Liesveld, J.L.; McCarty, J.M.; Rowley, S.D.; Vesole, D.H.; Badel, K.; Calandra, G. The use of AMD3100 plus G-CSF for autologous hematopoietic progenitor cell mobilization is superior to G-CSF alone. Blood 2005, 106, 1867–1874. [Google Scholar] [CrossRef]
- Burroughs, L.; Mielcarek, M.; Little, M.T.; Bridger, G.; MacFarland, R.; Fricker, S.; LaBrecque, J.; Sandmaier, B.M.; Storb, R. Durable engraftment of AMD3100-mobilized autologous and allogeneic peripheral blood mononuclear cells in a canine transplantation model. Blood 2005, 106, 4002–4008. [Google Scholar] [CrossRef]
- Wang, X.; Shin, S.C.; Chiang, A.F.; Khan, I.; Pan, D.; Rawlings, D.J.; Miao, C.H. Intraosseous delivery of lentiviral vectors targeting factor VIII expression in platelets corrects murine hemophilia A. Mol. Ther. 2015, 23, 617–626. [Google Scholar] [CrossRef]
- Wang, H.; Richter, M.; Psatha, N.; Li, C.; Kim, J.; Liu, J.; Ehrhardt, A.; Nilsson, S.K.; Cao, B.; Palmer, D.; et al. A combined in vivo HSC transduction/selection approach results in efficient and stable gene expression in peripheral blood cells in mice. Mol. Ther. Methods Clin. Dev. 2018, 8, 52–64. [Google Scholar] [CrossRef] [PubMed]


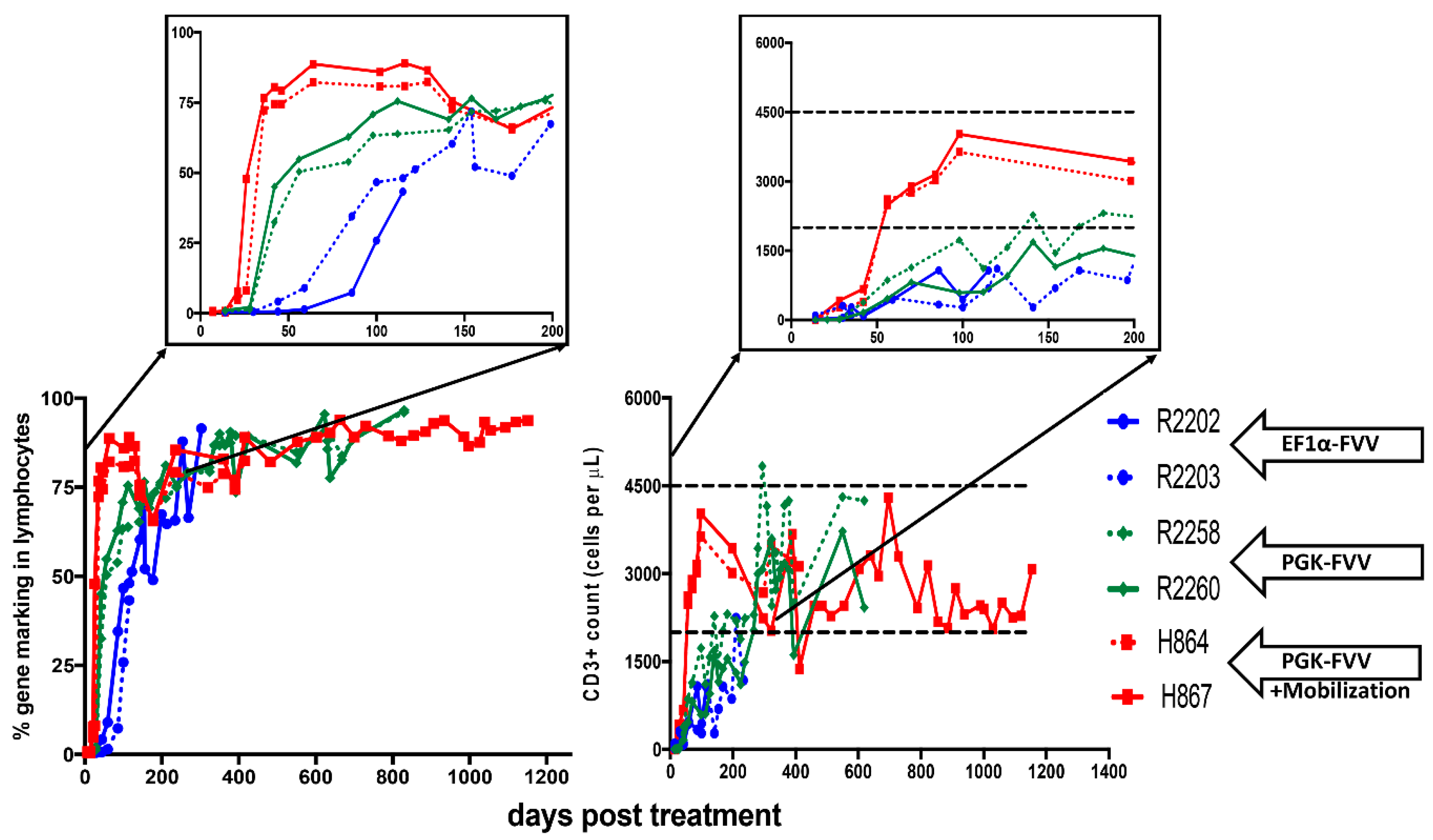{kind=link}
| ID | Age at Injection (Days Old.) | Foamy Viral Vector | Dose of Vector (Infectious Units) | Mobilization | Survival of Dogs (Days Post Treatment) | Health Status or Infectious Complications |
|---|---|---|---|---|---|---|
| H867 | 16 | PGK.mCherry.2A.γC | 4.0 × 108 | G-CSF/AMD3100 | 1260 | Healthy and Alive |
| H864 | 16 | PGK.mCherry.2A.γC | 4.0 × 108 | G-CSF/AMD3100 | ~486 | Bordetella bronchiseptica |
| R2258 | 18 | EF1⍺.EGFP.2A.γC | 4.0 × 108 | NO | ~820 | Papillomavirus |
| PGK.mCherry.2A.γC | 4.0 × 108 | |||||
| R2260 | 18 | EF1⍺.mCherry.2A.γC | 4.0 × 108 | NO | ~820 | Papillomavirus |
| PGK.EGFP.2A.γC | 4.0 × 108 | |||||
| R2202 | 1 | EF1α-EGFP-2A-γC | 4.2 × 108 | NO | ~334 | Coccidiosis; Canine Distemper virus |
| R2203 | 1 | EF1α-EGFP-2A-γC | 4.2 × 108 | NO | ~120 | Canine Parainfluenza virus |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rajawat, Y.S.; Humbert, O.; Kiem, H.-P. In-Vivo Gene Therapy with Foamy Virus Vectors. Viruses 2019, 11, 1091. https://doi.org/10.3390/v11121091
Rajawat YS, Humbert O, Kiem H-P. In-Vivo Gene Therapy with Foamy Virus Vectors. Viruses. 2019; 11(12):1091. https://doi.org/10.3390/v11121091
Chicago/Turabian StyleRajawat, Yogendra Singh, Olivier Humbert, and Hans-Peter Kiem. 2019. "In-Vivo Gene Therapy with Foamy Virus Vectors" Viruses 11, no. 12: 1091. https://doi.org/10.3390/v11121091
APA StyleRajawat, Y. S., Humbert, O., & Kiem, H.-P. (2019). In-Vivo Gene Therapy with Foamy Virus Vectors. Viruses, 11(12), 1091. https://doi.org/10.3390/v11121091