Effects of Soil Microbes on Forest Recovery to Climax Community through the Regulation of Nitrogen Cycling

Abstract
1. Introduction
2. Materials and Methods
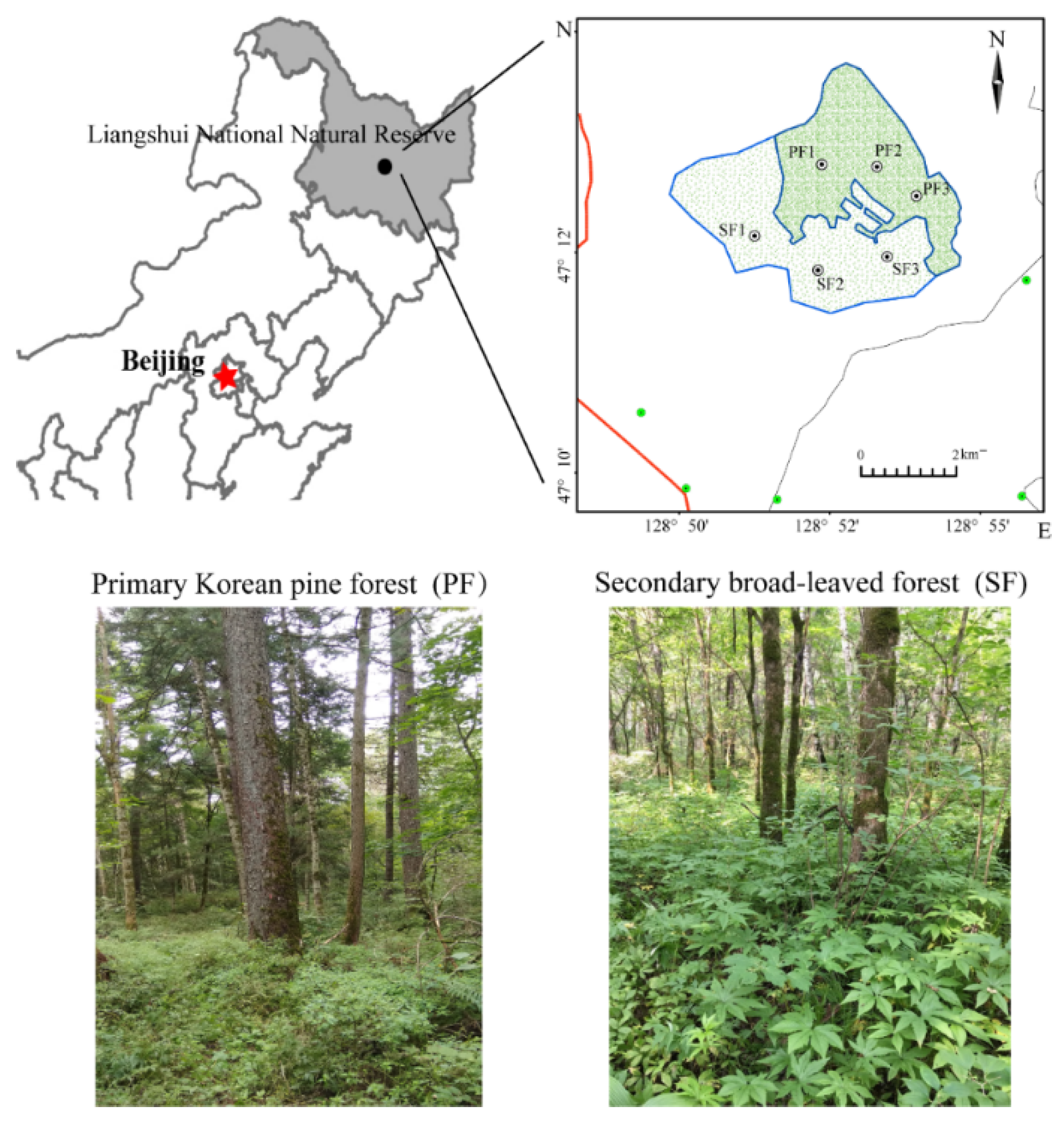
2.1. Experimental Sites
2.2. Sample Collection, Soil Physicochemical Analyses, and In Situ Soil N Mineralization
2.3. Metagenome Sequencing
2.4. Raw Sequences Processing
2.5. Data Analysis
3. Results
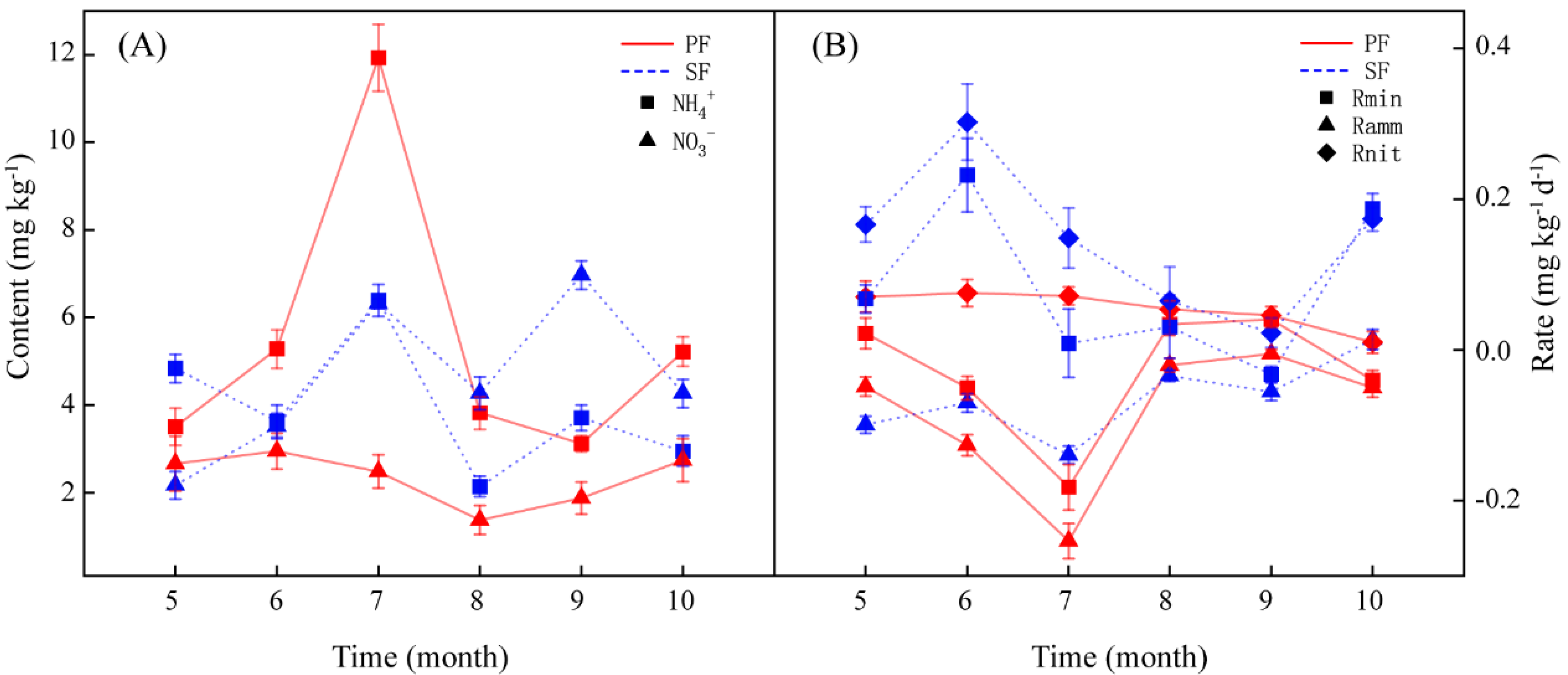
3.1. Seasonal Dynamics of Soil Inorganic N and N Transformation Rates
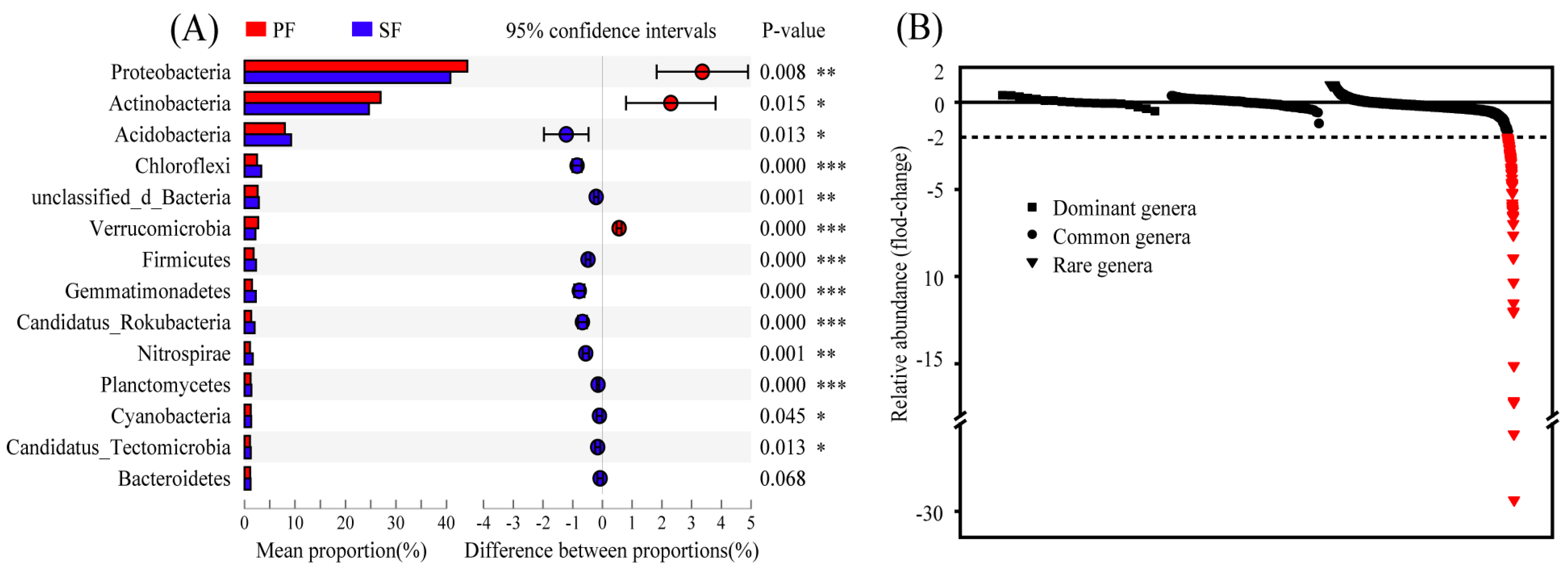
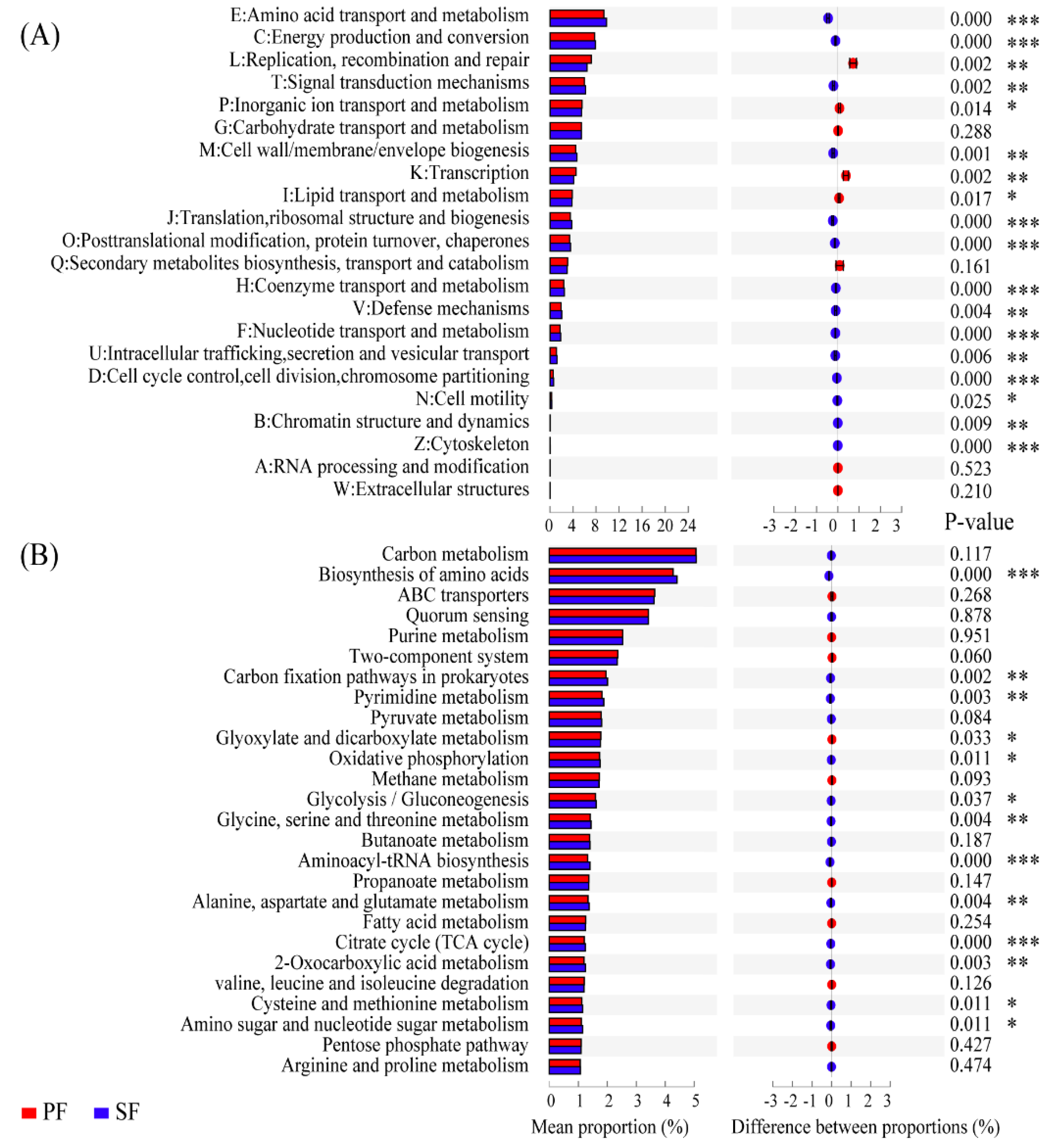
3.2. Variations in Soil Microbial Community Composition and Potential Functions between Two Forest Types
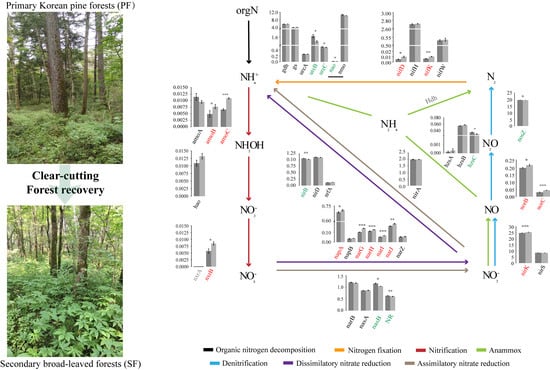
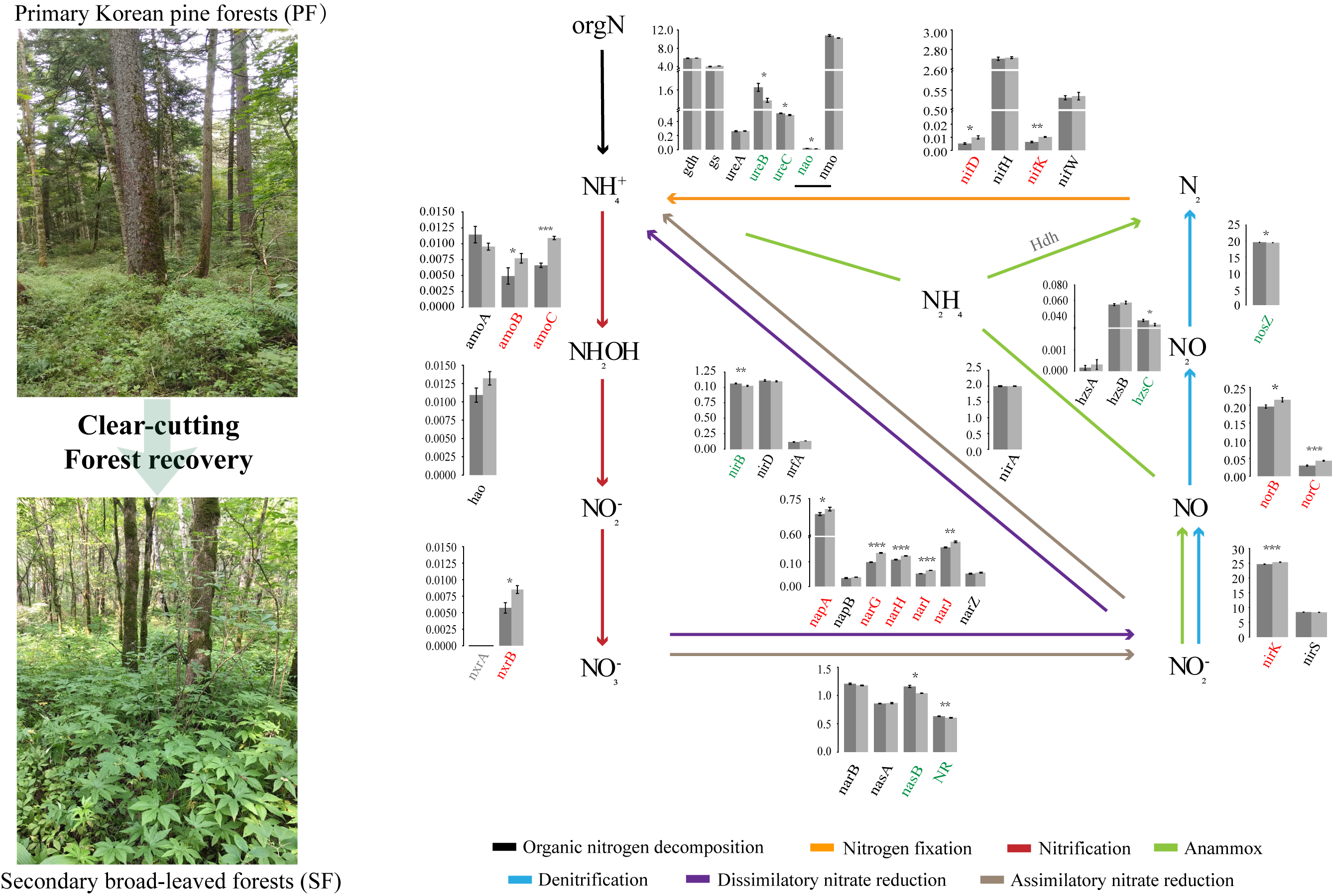
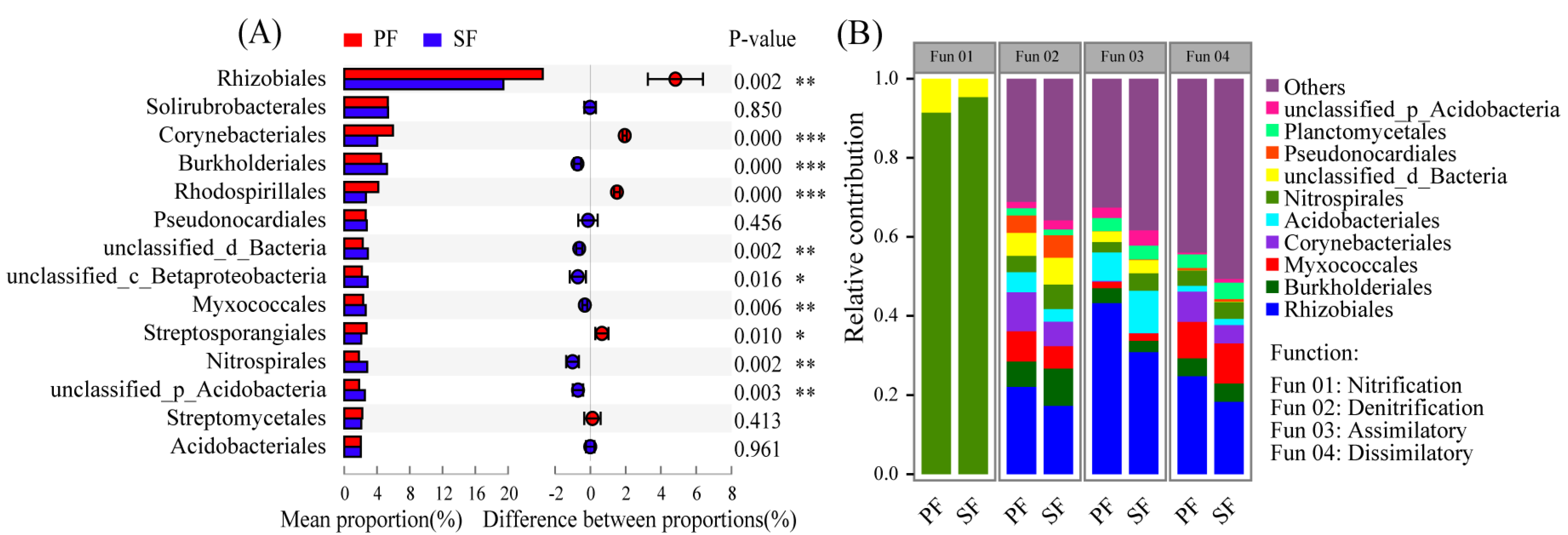
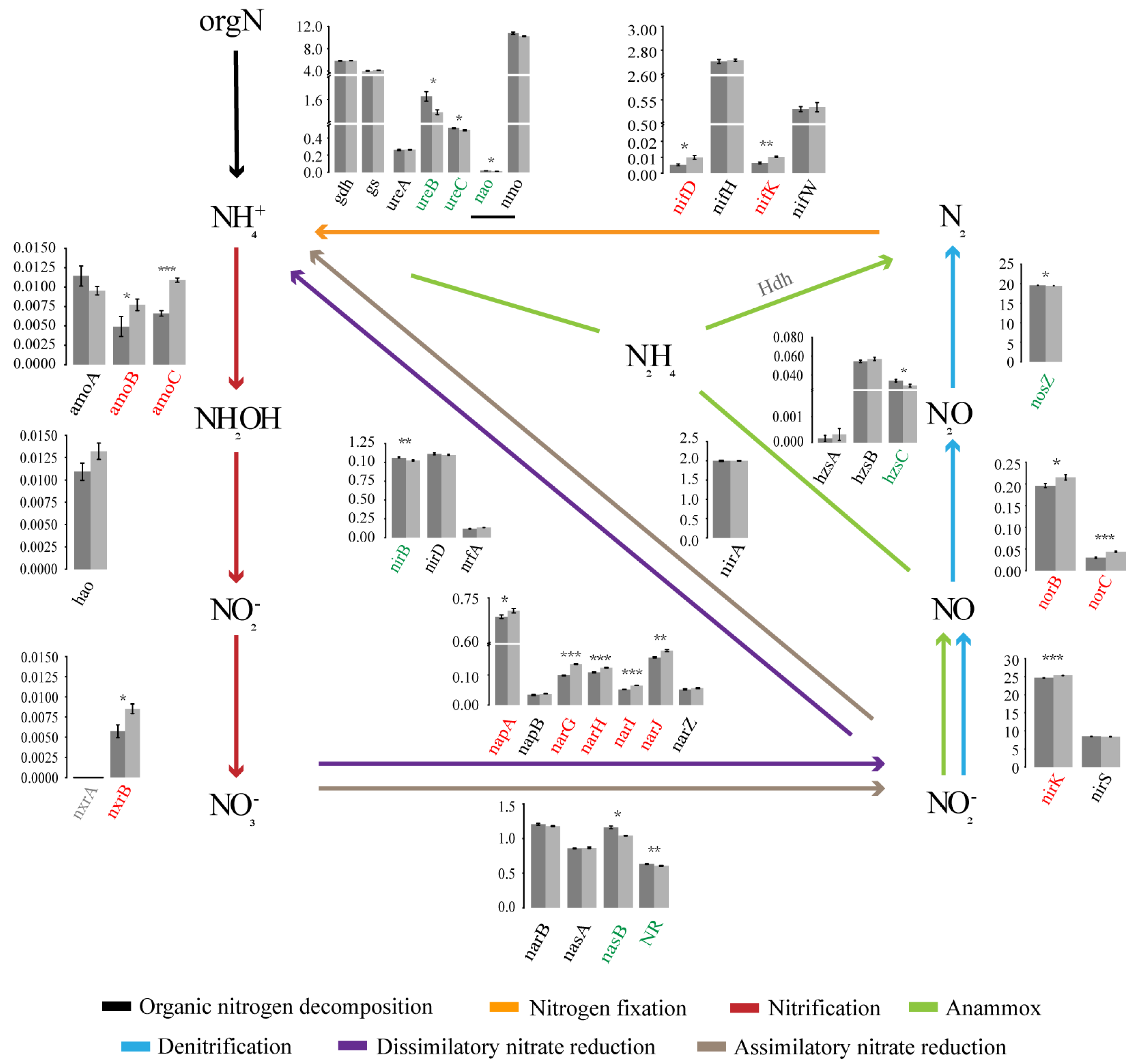
3.3. Variations in Soil Microbial N Cycling between the Two Forest Types
3.4. Relationship between Soil Microbial Community Composition and Functions
4. Discussion
4.1. The Characteristics of Soil Microbial Communities and Functional Changes in the Two Forest Types
4.2. The Characteristics of Soil Microbial N Cycling Processes Changes in the Two Forest Types
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pedrinho, A.; Mendes, L.W.; Merloti, L.F.; Fonseca, M.D.C.D.; Cannavan, F.D.S.; Tsai, S.M. Forest-to-pasture conversion and recovery based on assessment of microbial communities in Eastern Amazon rainforest. FEMS Microbiol. Ecol. 2018, 95. [Google Scholar] [CrossRef]
- Sun, S.; Badgley, B.D. Changes in microbial functional genes within the soil metagenome during forest ecosystem restoration. Soil Boil. Biochem. 2019, 135, 163–172. [Google Scholar] [CrossRef]
- Bardgett, R.D.; De Vries, F.T.; Van Der Putten, W.H.; Tate, K.R. Soil Biodiversity and Ecosystem Functioning. In Microbial Biomass; World Scientific Pub Co Pte Ltd.: London, UK, 2017; pp. 119–140. [Google Scholar]
- Jansson, J.K.; Hofmockel, K.S. Soil microbiomes and climate change. Nat. Rev. Genet. 2019, 18, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Crowther, T.W.; Hoogen, J.V.D.; Wan, J.; Mayes, M.A.; Keiser, A.D.; Mo, L.; Averill, C.; Maynard, D.S. The global soil community and its influence on biogeochemistry. Science 2019, 365, eaav0550. [Google Scholar] [CrossRef] [PubMed]
- Wang, C. Biomass allometric equations for 10 co-occurring tree species in Chinese temperate forests. For. Ecol. Manag. 2006, 222, 9–16. [Google Scholar] [CrossRef]
- Vitousek, P.; Howarth, R.W. Nitrogen limitation on land and in the sea: How can it occur? Biogeochemistry 1991, 13, 87–115. [Google Scholar] [CrossRef]
- Lebauer, D.S.; Treseder, K.K. Nitrogen limitation of net primary productivity in terrestrial ecosystems is globally distributed. Ecology 2008, 89, 371–379. [Google Scholar] [CrossRef]
- Groffman, P.M.; Driscoll, C.T.; Duran, J.; Campbell, J.L.; Christenson, L.M.; Fahey, T.J.; Fisk, M.C.; Fuss, C.; Likens, G.E.; Lovett, G.; et al. Nitrogen oligotrophication in northern hardwood forests. Biogeochemistry 2018, 141, 523–539. [Google Scholar] [CrossRef]
- Du, E.; Terrer, C.; Pellegrini, A.F.A.; Ahlström, A.; Van Lissa, C.J.; Zhao, X.; Xia, N.; Wu, X.; Jackson, R.B. Global patterns of terrestrial nitrogen and phosphorus limitation. Nat. Geosci. 2020, 13, 221–226. [Google Scholar] [CrossRef]
- Mushinski, R.M.; Gentry, T.J.; Dorosky, R.J.; Boutton, T. Forest harvest intensity and soil depth alter inorganic nitrogen pool sizes and ammonia oxidizer community composition. Soil Boil. Biochem. 2017, 112, 216–227. [Google Scholar] [CrossRef]
- Mushinski, R.M.; Zhou, Y.; Gentry, T.J.; Boutton, T. Bacterial metataxonomic profile and putative functional behavior associated with C and N cycle processes remain altered for decades after forest harvest. Soil Boil. Biochem. 2018, 119, 184–193. [Google Scholar] [CrossRef]
- Schimel, J.P.; Schaeffer, S.M. Microbial control over carbon cycling in soil. Front. Microbiol. 2012, 3, 1–11. [Google Scholar] [CrossRef] [PubMed]
- McGuire, K.L.; Treseder, K.K. Microbial communities and their relevance for ecosystem models: Decomposition as a case study. Soil Boil. Biochem. 2010, 42, 529–535. [Google Scholar] [CrossRef]
- Avera, B.N.; Strahm, B.D.; Burger, J.A.; Zipper, C.E. Development of ecosystem structure and function on reforested surface-mined lands in the Central Appalachian Coal Basin of the United States. New Forest 2015, 46, 683–702. [Google Scholar] [CrossRef]
- Tickle, T.L.; Segata, N.; Waldron, L.; Weingart, U.; Huttenhower, C. Two-stage microbial community experimental design. ISME J. 2013, 7, 2330–2339. [Google Scholar] [CrossRef]
- Vazquez, E.; Benito, M.; Espejo, R.; Teutscherova, N. Effects of no-tillage and liming amendment combination on soil carbon and nitrogen mineralization. Eur. J. Soil Boil. 2019, 93, 103090. [Google Scholar] [CrossRef]
- Hart, S.C.; Stark, J.M.; Davidson, E.A.; Firestone, M.K.; Bottomley, P.; Angle, J.; Weaver, R. Nitrogen Mineralization, Immobilization, and Nitrification. In SSSA Book Series; Soil Science Society of America: Madison, WI, USA, 2018; pp. 985–1018. [Google Scholar]
- Lawson, C.E.; Wu, S.; Bhattacharjee, A.S.; Hamilton, J.J.; McMahon, K.D.; Goel, R.; Noguera, D.R. Metabolic network analysis reveals microbial community interactions in anammox granules. Nat. Commun. 2017, 8, 15416. [Google Scholar] [CrossRef]
- Lynch, M.D.J.; Neufeld, J.D. Ecology and exploration of the rare biosphere. Nat. Rev. Genet. 2015, 13, 217–229. [Google Scholar] [CrossRef]
- Tu, Q.; Lin, L.; Cheng, L.; Deng, Y.; He, Z. NCycDB: A curated integrative database for fast and accurate metagenomic profiling of nitrogen cycling genes. Bioinformatics 2018, 35, 1040–1048. [Google Scholar] [CrossRef]
- Shade, A.; Peter, H.; Allison, S.D.; Baho, D.L.; Berga, M.; Bürgmann, H.; Huber, D.H.; Langenheder, S.; Lennon, J.T.; Martiny, J.B.H.; et al. Fundamentals of Microbial Community Resistance and Resilience. Front. Microbiol. 2012, 3, 417. [Google Scholar] [CrossRef]
- Freedman, Z.B.; Zak, D.R. Soil bacterial communities are shaped by temporal and environmental filtering: Evidence from a long-term chronosequence. Environ. Microbiol. 2015, 17, 3208–3218. [Google Scholar] [CrossRef] [PubMed]
- Eilers, K.G.; Lauber, C.L.; Knight, R.; Fierer, N. Shifts in bacterial community structure associated with inputs of low molecular weight carbon compounds to soil. Soil Boil. Biochem. 2010, 42, 896–903. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, J.; Zhao, B.; Zhou, G.; Ruan, L. Bacterial community structure in maize stubble-amended soils with different moisture levels estimated by bar-coded pyrosequencing. Appl. Soil Ecol. 2015, 86, 62–70. [Google Scholar] [CrossRef]
- Pascault, N.; Ranjard, L.; Kaisermann, A.; Bachar, D.; Christen, R.; Terrat, S.; Mathieu, O.; Levêque, J.; Mougel, C.; Hénault, C.; et al. Stimulation of Different Functional Groups of Bacteria by Various Plant Residues as a Driver of Soil Priming Effect. Ecosystems 2013, 16, 810–822. [Google Scholar] [CrossRef]
- Fierer, N.; Bradford, M.A.; Jackson, R.B. Toward an ecological classification of soil bacteria. Ecology 2007, 88, 1354–1364. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Zhou, Y.; Yao, X.; Cai, J.; Liu, X.; Tang, X.; Zhang, Y.; Jang, K.-S.; Jeppesen, E. Decreasing diversity of rare bacterial subcommunities relates to dissolved organic matter along permafrost thawing gradients. Environ. Int. 2020, 134, 105330. [Google Scholar] [CrossRef]
- Sogin, M.L.; Morrison, H.G.; Huber, J.A.; Welch, D.M.; Huse, S.M.; Neal, P.R.; Arrieta, J.M.; Herndl, G.J. Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proc. Natl. Acad. Sci. USA 2006, 103, 12115–12120. [Google Scholar] [CrossRef]
- Tripathi, B.M.; Edwards, D.P.; Mendes, L.W.; Kim, M.; Dong, K.; Adams, J.M. The impact of tropical forest logging and oil palm agriculture on the soil microbiome. Mol. Ecol. 2016, 25, 2244–2257. [Google Scholar] [CrossRef]
- Fierer, N.; Leff, J.W.; Adams, B.J.; Nielsen, U.N.; Bates, S.T.; Lauber, C.L.; Owens, S.M.; Gilbert, J.A.; Wall, D.H.; Caporaso, J.G. Cross-biome metagenomic analyses of soil microbial communities and their functional attributes. Proc. Natl. Acad. Sci. USA 2012, 109, 21390–21395. [Google Scholar] [CrossRef]
- Nelson, M.B.; Martiny, A.C.; Martiny, A.C. Global biogeography of microbial nitrogen-cycling traits in soil. Proc. Natl. Acad. Sci. USA 2016, 113, 8033–8040. [Google Scholar] [CrossRef]
- Kuypers, M.M.M.; Marchant, H.K.; Kartal, B. The microbial nitrogen-cycling network. Nat. Rev. Genet. 2018, 16, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; He, H.S.; Dai, L.; Yang, J. Effects of human disturbances on Korean pine coverage and age structure at a landscape scale in Northeast China. Ecol. Eng. 2014, 71, 375–379. [Google Scholar] [CrossRef]
- Hayashida, M. Seed dispersal by red squirrels and subsequent establishment of Korean pine. For. Ecol. Manag. 1989, 28, 115–129. [Google Scholar] [CrossRef]
- Omelko, A.M.; Ukhvatkina, O.N.; Zhmerenetsky, A.; Sibirina, L.A.; Petrenko, T.; Bobrovsky, M. From young to adult trees: How spatial patterns of plants with different life strategies change during age development in an old-growth Korean pine-broadleaved forest. For. Ecol. Manag. 2018, 411, 46–66. [Google Scholar] [CrossRef]
- Zhou, G.; Liu, Q.; Xu, Z.; Du, W.; Yu, J.; Meng, S.; Zhou, H.; Qin, L.; Shah, S. How can the shade intolerant Korean pine survive under dense deciduous canopy? For. Ecol. Manag. 2020, 457, 117735. [Google Scholar] [CrossRef]
- Cui, X.; Song, J. Soil NH4+/NO3—Nitrogen characteristics in primary forests and the adaptability of some coniferous species. Front. For. China 2007, 2, 1–10. [Google Scholar] [CrossRef]
- Axelsson, E.P.; Lundmark, T.; Högberg, P.; Nordin, A. Belowground Competition Directs Spatial Patterns of Seedling Growth in Boreal Pine Forests in Fennoscandia. Forests 2014, 5, 2106–2121. [Google Scholar] [CrossRef]
- Tu, Q.; He, Z.; Wu, L.; Xue, K.; Xie, G.; Chain, P.; Reich, P.B.; Hobbie, S.; Zhou, J. Metagenomic reconstruction of nitrogen cycling pathways in a CO2-enriched grassland ecosystem. Soil Boil. Biochem. 2017, 106, 99–108. [Google Scholar] [CrossRef]
- Moreno-Vivián, C.; Cabello, P.; Martiínez-Luque, M.; Blasco, R.; Castillo, F. Prokaryotic Nitrate Reduction: Molecular Properties and Functional Distinction among Bacterial Nitrate Reductases. J. Bacteriol. 1999, 181, 6573–6584. [Google Scholar] [CrossRef]
- Fang, Y.; Gundersen, P.; Mo, J.M.; Zhu, W.X. Input and output of dissolved organic and inorganic nitrogen in subtropical forests of South China under high air pollution. Biogeosciences 2008, 5, 339–352. [Google Scholar] [CrossRef]
- Davidson, E.A.; De Carvalho, C.J.R.; Figueira, A.M.; Ishida, F.Y.; Ometto, J.P.H.B.; Nardoto, G.B.; Sabá, R.T.; Hayashi, S.N.; Leal, E.C.; Vieira, I.C.G.; et al. Recuperation of nitrogen cycling in Amazonian forests following agricultural abandonment. Nature 2007, 447, 995–998. [Google Scholar] [CrossRef] [PubMed]
- Scott, C.E.; Rap, A.; Spracklen, D.V.; Forster, P.M.D.F.; Carslaw, K.; Mann, G.; Pringle, K.J.; Kivekäs, N.; Kulmala, M.; Lihavainen, H.; et al. The direct and indirect radiative effects of biogenic secondary organic aerosol. Atmospheric Chem. Phys. Discuss. 2014, 14, 447–470. [Google Scholar] [CrossRef]
- Tang, Y.; Yu, G.-R.; Zhang, X.; Wang, Q.; Ge, J.; Liu, S. Changes in nitrogen-cycling microbial communities with depth in temperate and subtropical forest soils. Appl. Soil Ecol. 2018, 124, 218–228. [Google Scholar] [CrossRef]
- Thébault, A.; Clement, J.-C.; Ibanez, S.; Roy, J.; Geremia, R.A.; Pérez, C.A.; Buttler, A.; Estienne, Y.; Lavorel, S. Nitrogen limitation and microbial diversity at the treeline. Oikos 2014, 123, 729–740. [Google Scholar] [CrossRef]
- Rennenberg, H.; Dannenmann, M.; Gessler, A.; Kreuzwieser, J.; Simon, J.; Papen, H. Nitrogen balance in forest soils: Nutritional limitation of plants under climate change stresses. Plant Boil. 2009, 11, 4–23. [Google Scholar] [CrossRef] [PubMed]
- Kaye, J.P.; Hart, S.C. Competition for nitrogen between plants and soil microorganisms. Trends Ecol. Evol. 1997, 12, 139–143. [Google Scholar] [CrossRef]
- Moreau, D.; Bardgett, R.D.; Finlay, R.D.; Jones, D.L.; Philippot, L. A plant perspective on nitrogen cycling in the rhizosphere. Funct. Ecol. 2019, 33, 540–552. [Google Scholar] [CrossRef]
- Che, R.; Wang, Y.; Li, K.; Xu, Z.; Hu, J.; Wang, F.; Rui, Y.; Li, L.; Pang, Z.; Cui, X. Degraded patch formation significantly changed microbial community composition in alpine meadow soils. Soil Tillage Res. 2019, 195, 104426. [Google Scholar] [CrossRef]
- Fang, Y.; Koba, K.; Makabe, A.; Takahashi, C.; Zhu, W.; Hayashi, T.; Hokari, A.A.; Urakawa, R.; Bai, E.; Houlton, B.Z.; et al. Microbial denitrification dominates nitrate losses from forest ecosystems. Proc. Natl. Acad. Sci. USA 2015, 112, 1470–1474. [Google Scholar] [CrossRef]
- Wieder, W.R.; Cleveland, C.C.; Smith, W.K.; Todd-Brown, K. Future productivity and carbon storage limited by terrestrial nutrient availability. Nat. Geosci. 2015, 8, 441–444. [Google Scholar] [CrossRef]
- Banning, N.C.; Gleeson, D.B.; Grigg, A.H.; Grant, C.D.; Andersen, G.L.; Brodie, E.L.; Murphy, D.V. Soil Microbial Community Successional Patterns during Forest Ecosystem Restoration. Appl. Environ. Microbiol. 2011, 77, 6158–6164. [Google Scholar] [CrossRef] [PubMed]
- Veen, G.F.; Fry, E.L.; Hooven, F.C.T.; Kardol, P.; Morriën, E.; De Long, J.R. The Role of Plant Litter in Driving Plant-Soil Feedbacks. Front. Environ. Sci. 2019, 7, 168. [Google Scholar] [CrossRef]
- Köster, K.; Berninger, F.; Lindén, A.; Köster, E.; Pumpanen, J. Recovery in fungal biomass is related to decrease in soil organic matter turnover time in a boreal fire chronosequence. Geoderma 2014, 235, 74–82. [Google Scholar] [CrossRef]
- Jin, X.; Liu, Y.; Hu, W.; Wang, G.; Kong, Z.; Wu, L.; Ge, G. Soil bacterial and fungal communities and the associated nutrient cycling responses to forest conversion after selective logging in a subtropical forest of China. For. Ecol. Manag. 2019, 444, 308–317. [Google Scholar] [CrossRef]
- Allison, S.D. Cheaters, diffusion and nutrients constrain decomposition by microbial enzymes in spatially structured environments. Ecol. Lett. 2005, 8, 626–635. [Google Scholar] [CrossRef]
- Venugopal, P.; Junninen, K.; Linnakoski, R.; Edman, M.; Kouki, J. Climate and wood quality have decayer-specific effects on fungal wood decomposition. For. Ecol. Manag. 2016, 360, 341–351. [Google Scholar] [CrossRef]
- Bani, A.; Pioli, S.; Ventura, M.; Panzacchi, P.; Borruso, L.; Tognetti, R.; Tonon, G.; Brusetti, L. The role of microbial community in the decomposition of leaf litter and deadwood. Appl. Soil Ecol. 2018, 126, 75–84. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Forest Type | Location | Altitude (m) | Area (hm2) | Main Tree Species |
|---|---|---|---|---|
| PF | 47°12′57″ N, 128°52′17″ E | 402 | 11.7 | Pinus koraiensis Siebold & Zucc., Fraxinus mandschurica Rupr., Tilia mandschurica Rupr. & Maxim., Tilia amurensis Rupr., Acer mono (Maxim.) H.Ohashi, Acer ukurunduense Trautv. & Mey., Acer tegmentosum Maxim., Ulmus laciniata (Trautv.) Mayr, Syringa reticulata (Blume) H.Hara, Padus racemosa L., Betula costata Trautv., Abies nephrolepis (Trautv.) Maxim. |
| SF | 47°12′49″ N, 128°52′12″ E | 390 | 9.3 | Fraxinus mandschurica, Tilia amurensis, Phellodendron amurense Rupr., Acer mono (Maxim.) H.Ohashi, Padus racemosa L., Syringa reticulata (Blume) H.Hara, Ulmus laciniata (Trautv.) Mayr |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qi, D.; Feng, F.; Fu, Y.; Ji, X.; Liu, X. Effects of Soil Microbes on Forest Recovery to Climax Community through the Regulation of Nitrogen Cycling. Forests 2020, 11, 1027. https://doi.org/10.3390/f11101027
Qi D, Feng F, Fu Y, Ji X, Liu X. Effects of Soil Microbes on Forest Recovery to Climax Community through the Regulation of Nitrogen Cycling. Forests. 2020; 11(10):1027. https://doi.org/10.3390/f11101027
Chicago/Turabian StyleQi, Dandan, Fujuan Feng, Yanmei Fu, Ximei Ji, and Xianfa Liu. 2020. "Effects of Soil Microbes on Forest Recovery to Climax Community through the Regulation of Nitrogen Cycling" Forests 11, no. 10: 1027. https://doi.org/10.3390/f11101027
APA StyleQi, D., Feng, F., Fu, Y., Ji, X., & Liu, X. (2020). Effects of Soil Microbes on Forest Recovery to Climax Community through the Regulation of Nitrogen Cycling. Forests, 11(10), 1027. https://doi.org/10.3390/f11101027