
Boosting Hydroformylation via Reactant Enrichment in Covalent Triazine Frameworks with Atomically Dispersed Rh

Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals
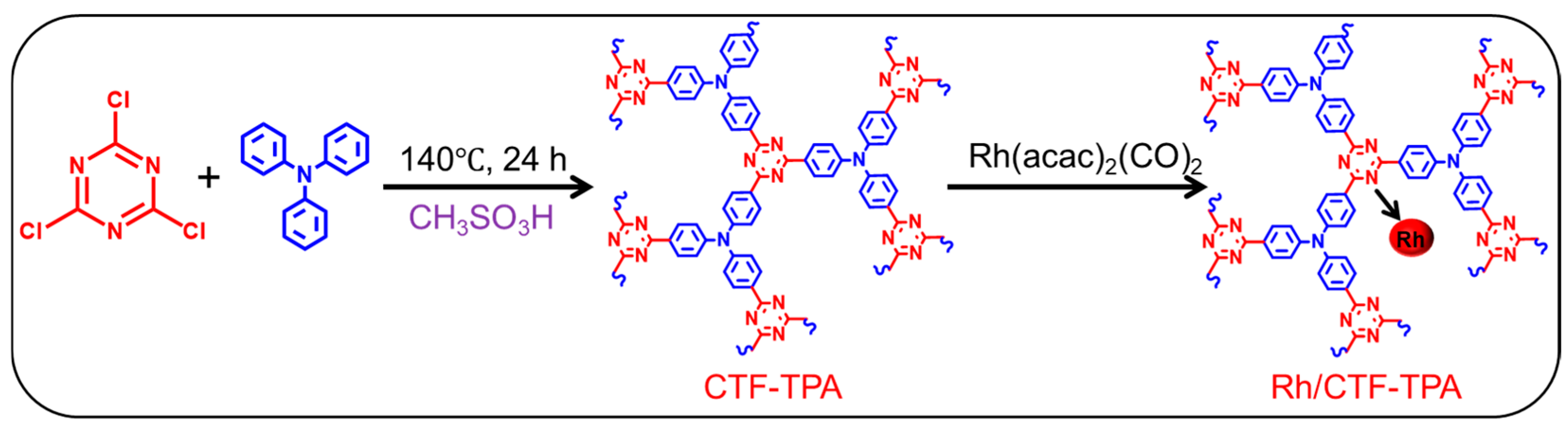
2.2. Synthesis of CTF-TPA and Rh/CTF-TPA
2.3. Characterizations
2.4. Catalytic Reactions
3. Results
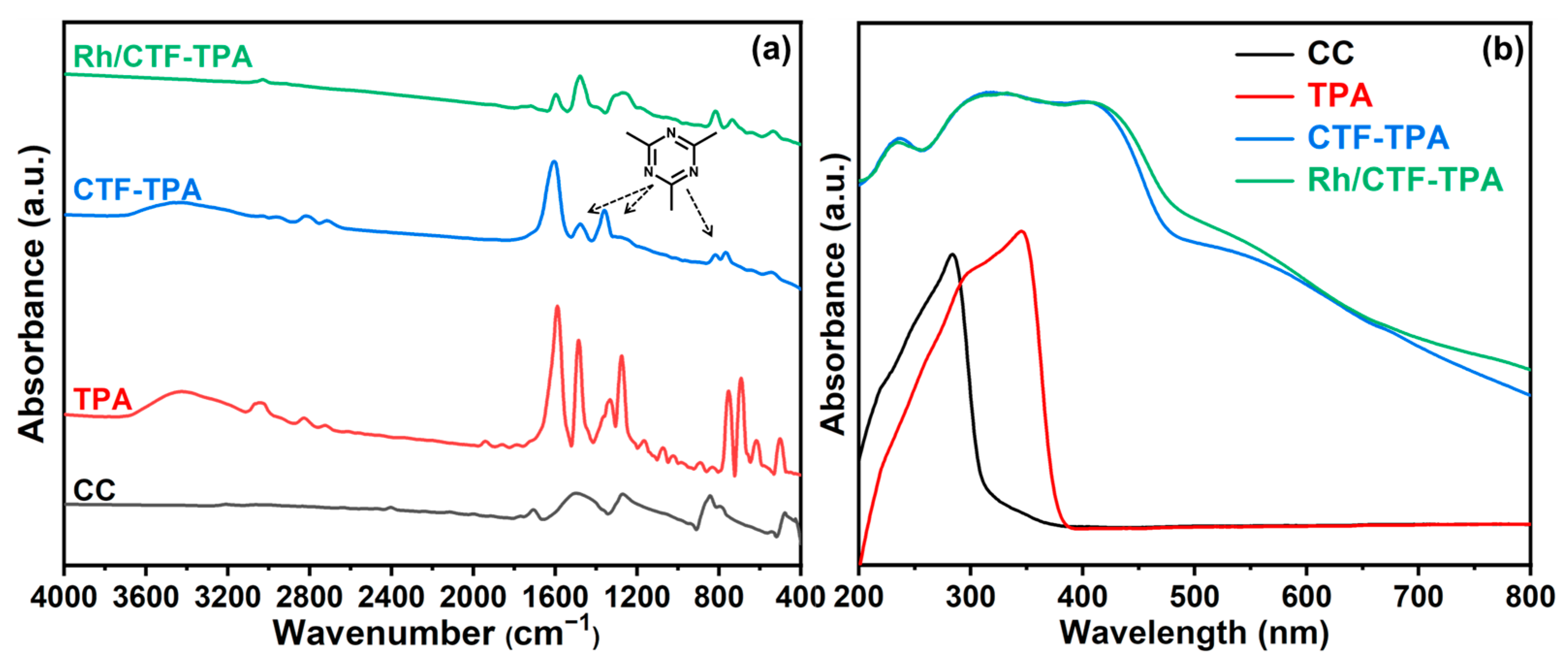
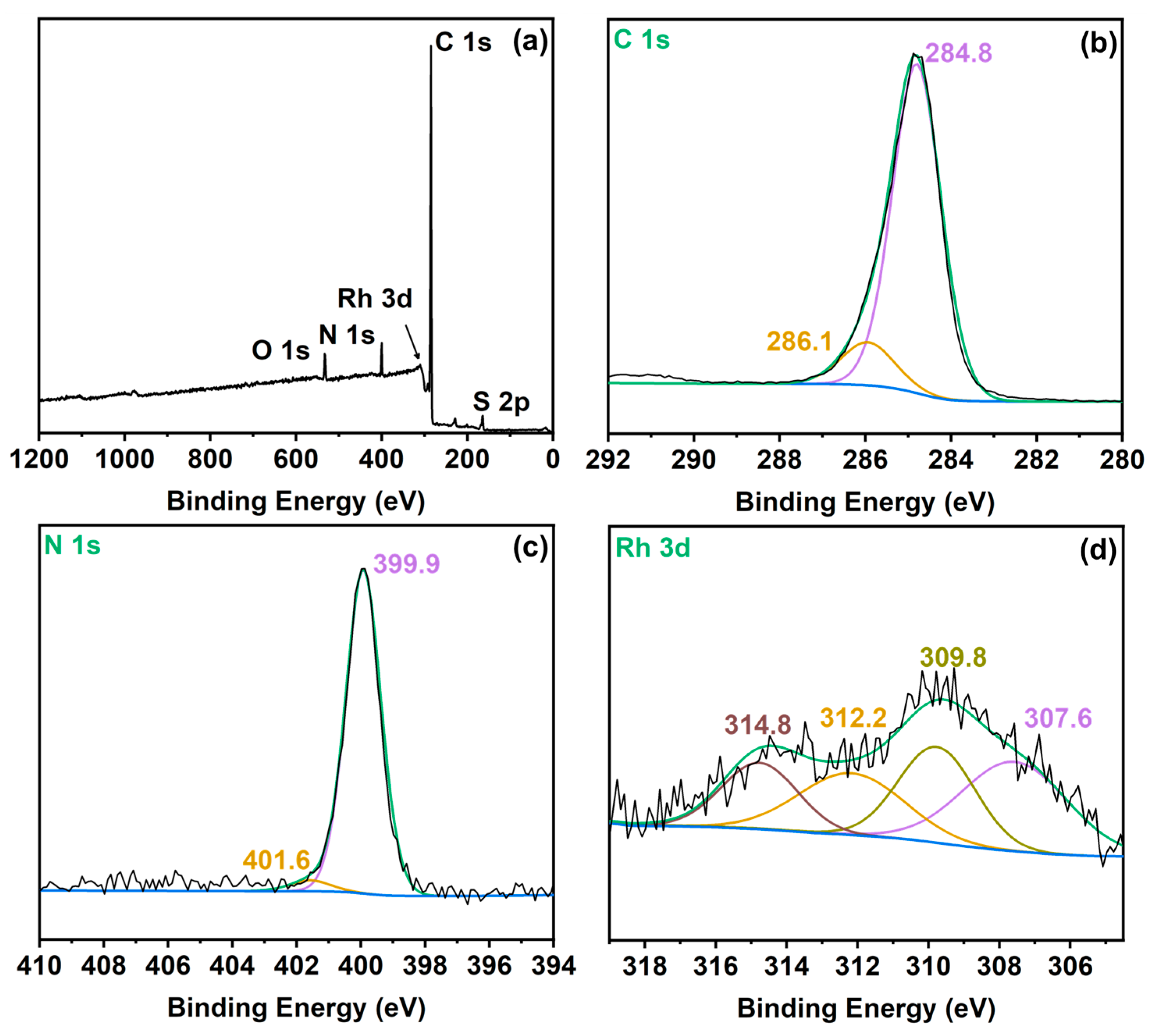
3.1. Catalysts Characterizations
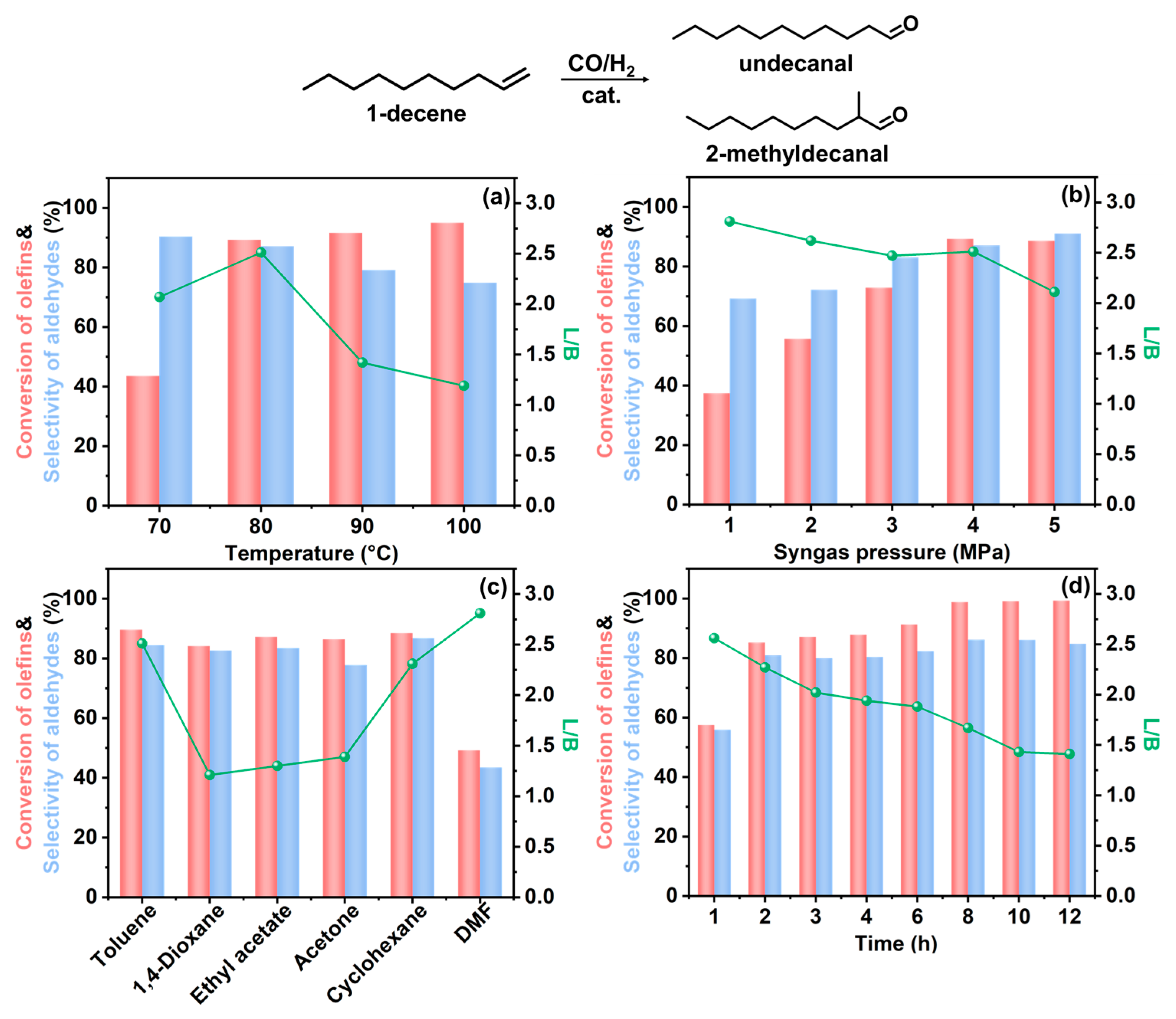
3.2. Catalytic Performance
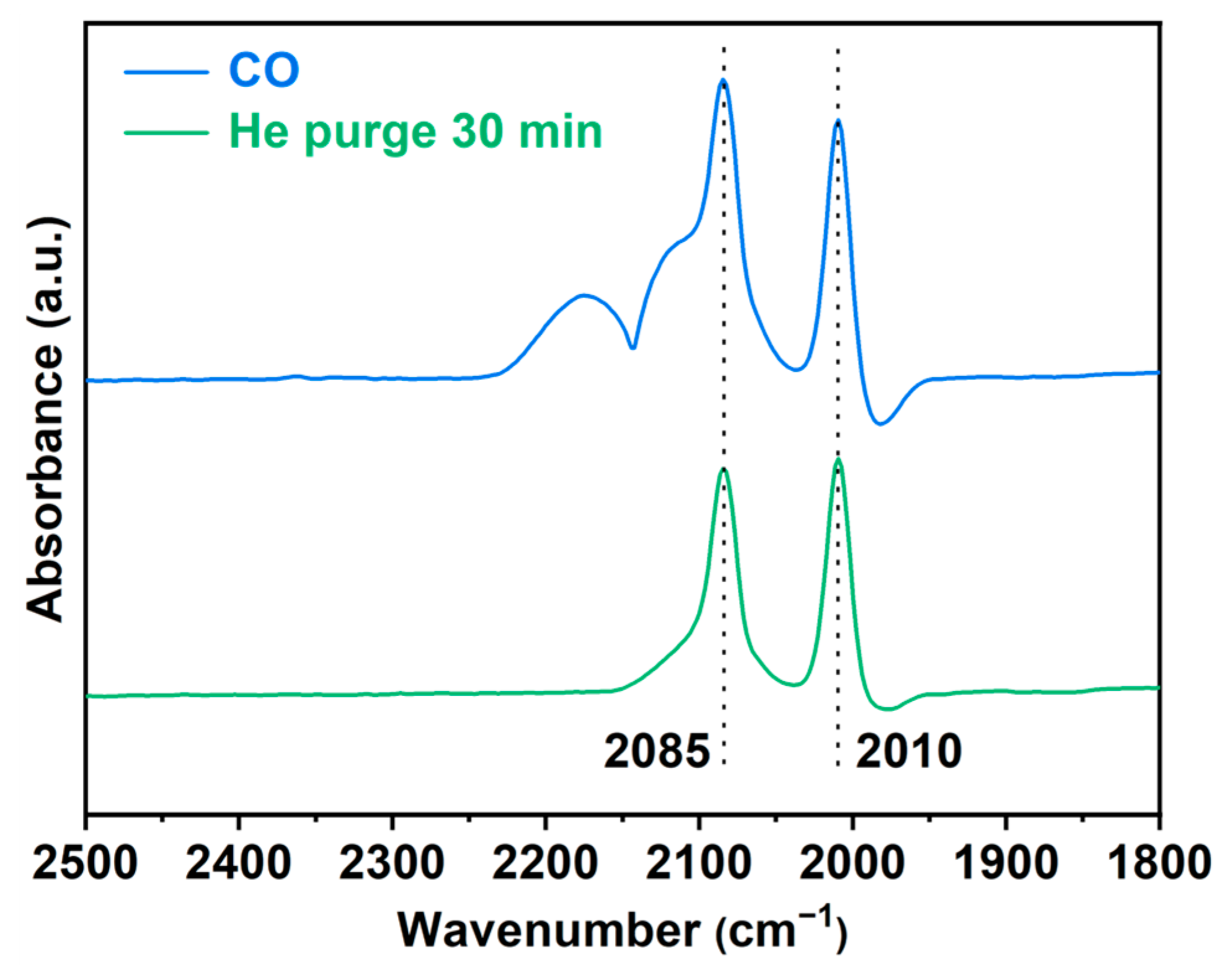
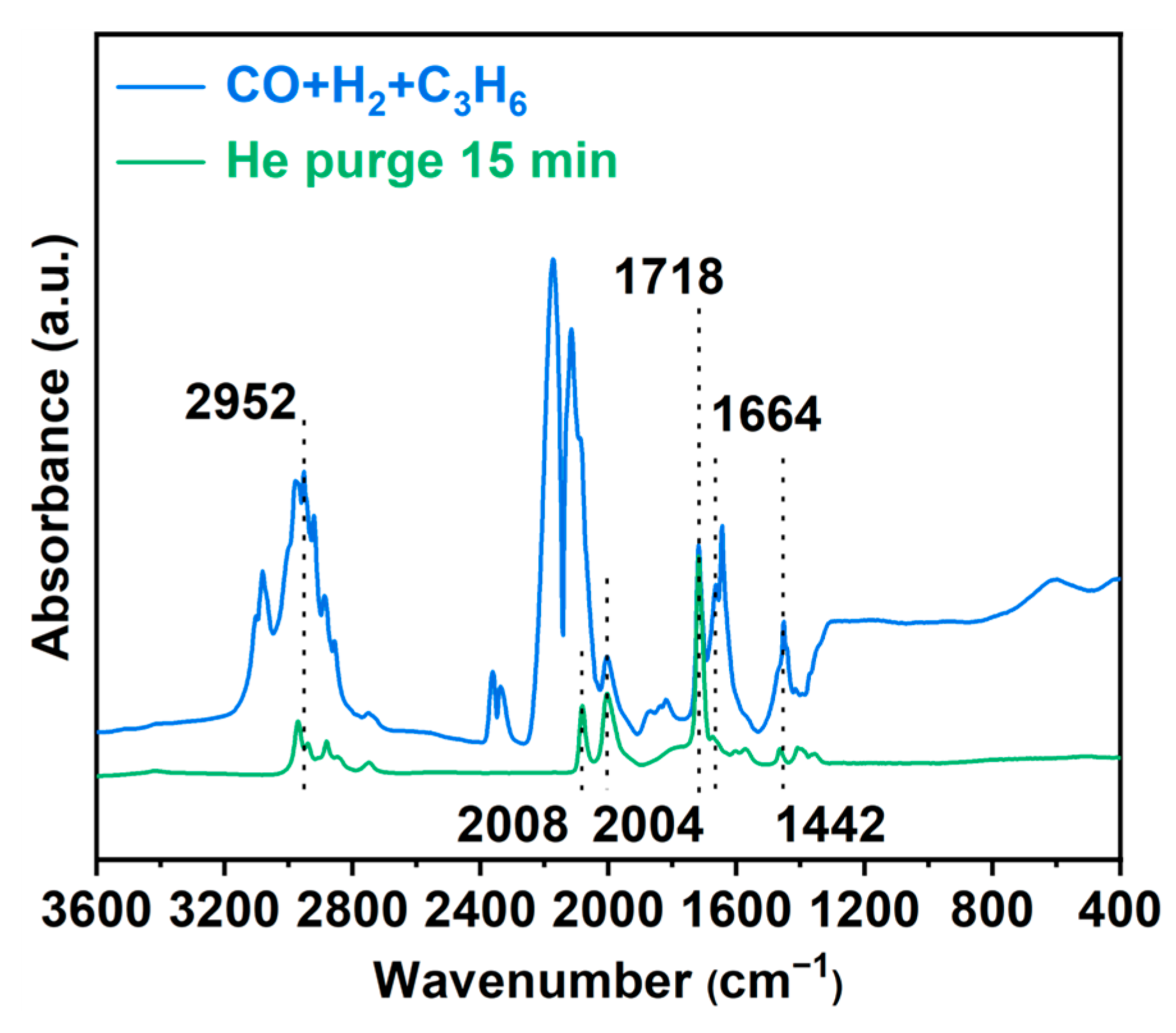
3.3. In Situ FT-IR
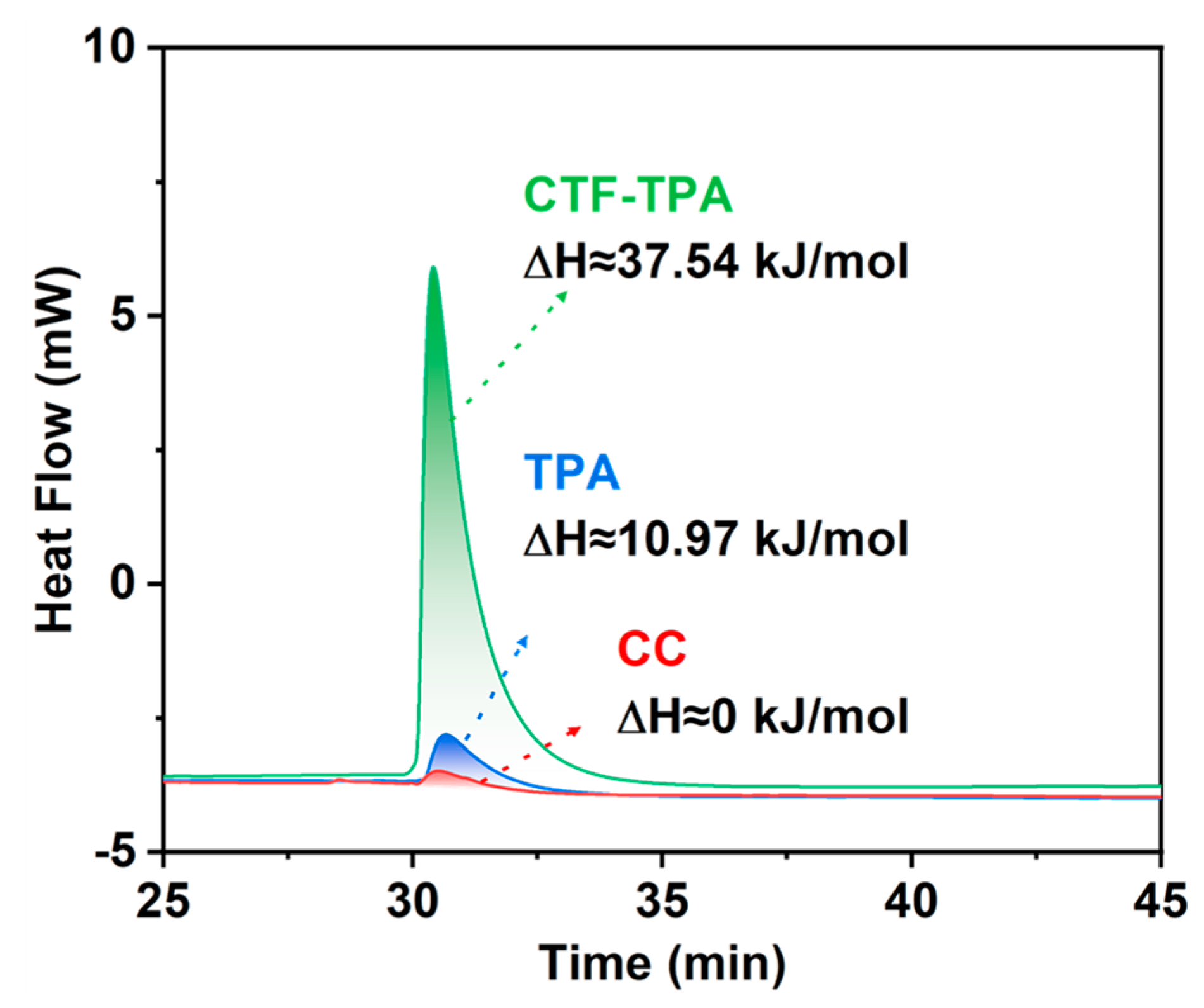
3.4. Catalyst–Substrate Adsorption
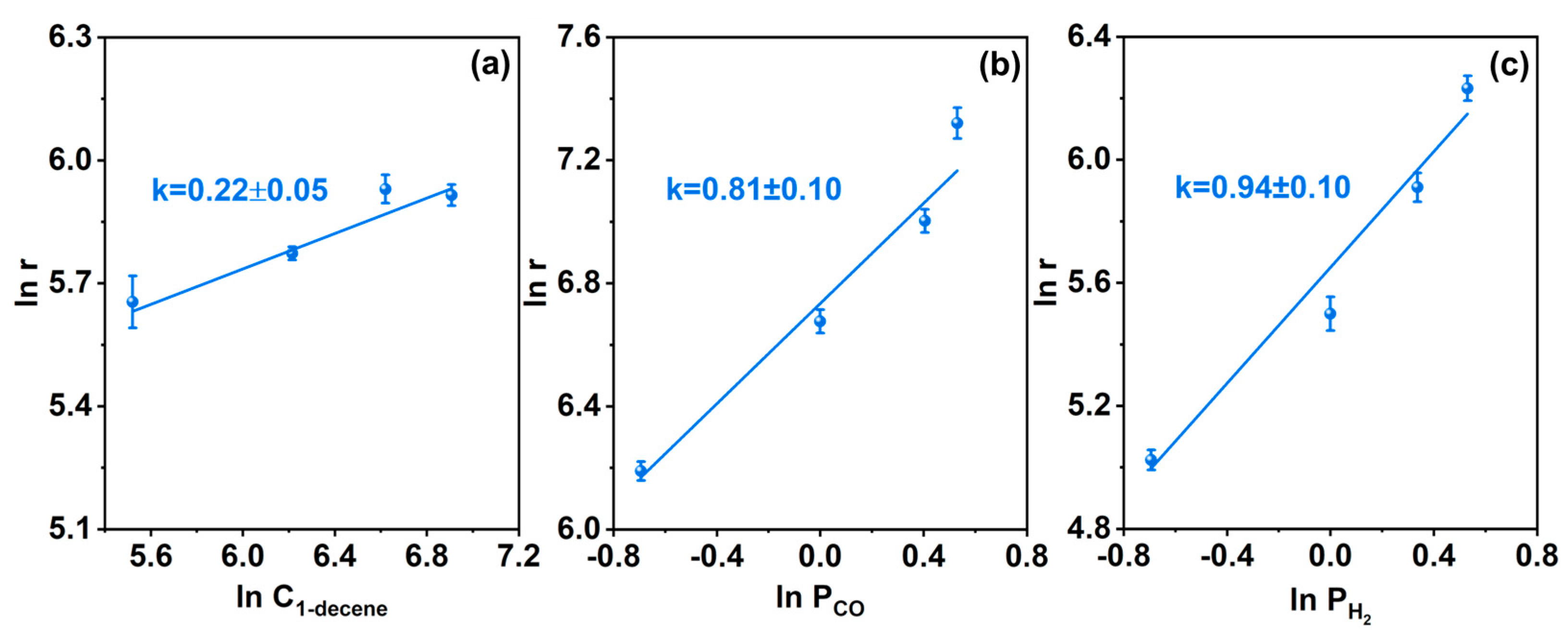
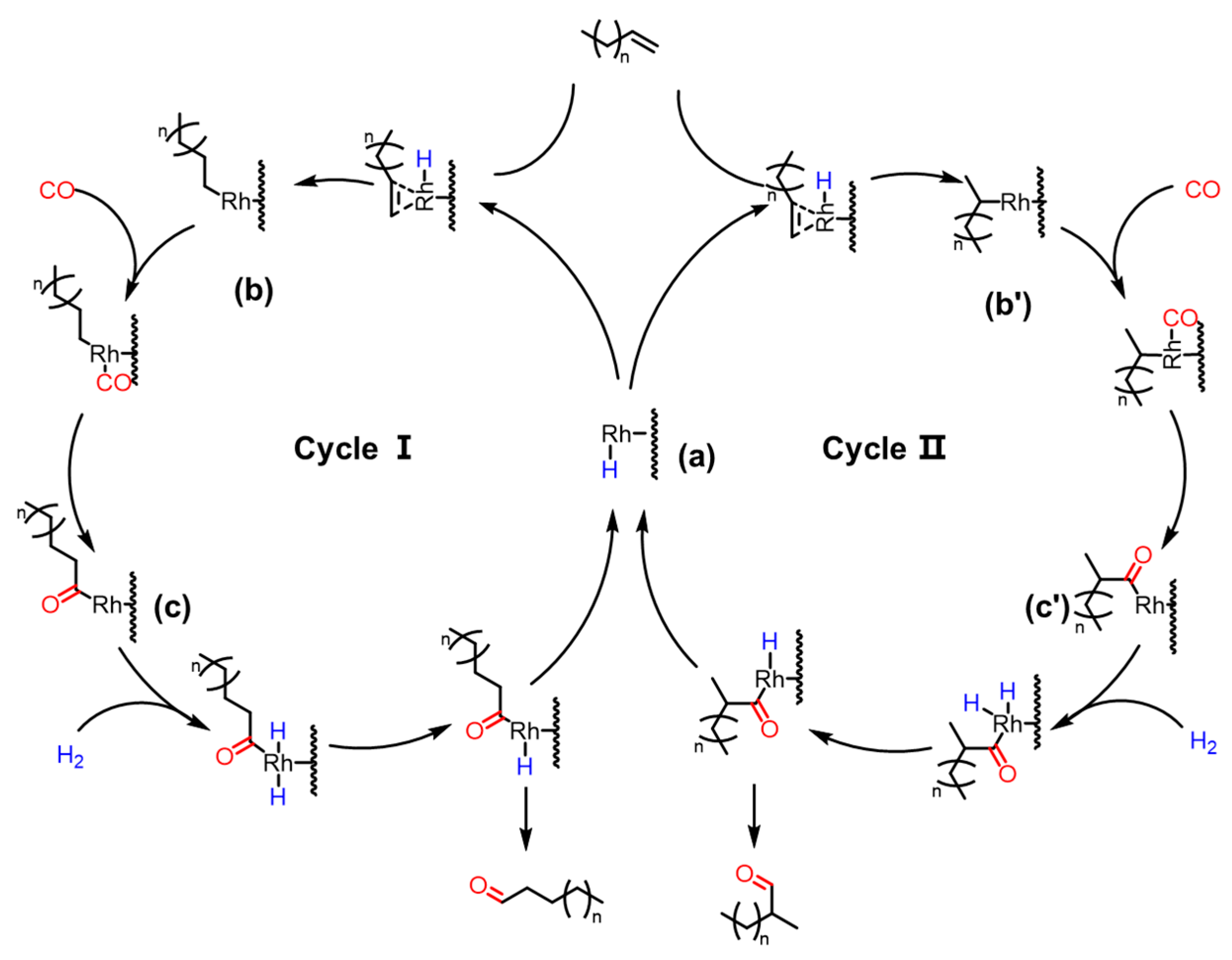
3.5. Mechanism of Olefin Hydroformylation over Rh/CTF-TPA
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhao, K.; Wang, X.; He, D.; Wang, H.; Qian, B.; Shi, F. Recent Development Towards Alkene Hydroformylation Catalysts Integrating Traditional Homo- and Heterogeneous Catalysis. Catal. Sci. Technol. 2022, 12, 4962–4982. [Google Scholar] [CrossRef]
- Nurttila, S.S.; Linnebank, P.R.; Krachko, T.; Reek, J.N.H. Supramolecular Approaches to Control Activity and Selectivity in Hydroformylation Catalysis. ACS Catal. 2018, 8, 3469–3488. [Google Scholar] [CrossRef]
- Fleischer, I.; Dyballa, K.M.; Jennerjahn, R.; Jackstell, R.; Franke, R.; Spannenberg, A.; Beller, M. From Olefins to Alcohols: Efficient and Regioselective Ruthenium-Catalyzed Domino Hydroformylation/Reduction Sequence. Angew. Chem. Int. Ed. 2013, 52, 2949–2953. [Google Scholar] [CrossRef]
- Dydio, P.; Detz, R.J.; de Bruin, B.; Reek, J.N.H. Beyond Classical Reactivity Patterns: Hydroformylation of Vinyl and Allyl Arenes to Valuable β- and γ-Aldehyde Intermediates Using Supramolecular Catalysis. J. Am. Chem. Soc. 2014, 136, 8418–8429. [Google Scholar] [CrossRef] [PubMed]
- Franke, R.; Selent, D.; Boerner, A. Applied Hydroformylation. Chem. Rev. 2012, 112, 5675–5732. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Simon, C.; Gramage-Doria, R.; Raoufmoghaddam, S.; Parella, T.; Costas, M.; Ribas, X.; Reek, J.N.H. Enantioselective Hydroformylation by a Rh-Catalyst Entrapped in a Supramolecular Metallocage. J. Am. Chem. Soc. 2015, 137, 2680–2687. [Google Scholar] [CrossRef]
- Dydio, P.; Detz, R.J.; Reek, J.N.H. Precise Supramolecular Control of Selectivity in the Rh-Catalyzed Hydroformylation of Terminal and Internal Alkenes. J. Am. Chem. Soc. 2013, 135, 10817–10828. [Google Scholar] [CrossRef] [PubMed]
- Neves, A.C.B.; Calvete, M.J.F.; Pinho e Melo, T.M.V.D.; Pereira, M.M. Immobilized Catalysts for Hydroformylation Reactions: A Versatile Tool for Aldehyde Synthesis. Eur. J. Org. Chem. 2012, 2012, 6309–6320. [Google Scholar] [CrossRef]
- Zhang, X.; Yan, T.; Hou, H.; Yin, J.; Wan, H.; Sun, X.; Zhang, Q.; Sun, F.; Wei, Y.; Dong, M.; et al. Regioselective Hydroformylation of Propene Catalysed by Rhodium-Zeolite. Nature 2024, 629, 597. [Google Scholar] [CrossRef]
- Samanta, P.; Sole-Daura, A.; Rajapaksha, R.; Wisser, F.M.; Meunier, F.; Schuurman, Y.; Sassoye, C.; Mellot-Draznieks, C.; Canivet, J. Heterogenized Molecular Rhodium Phosphine Catalysts within Metal-Organic Frameworks for Alkene Hydroformylation. ACS Catal. 2023, 13, 4193–4204. [Google Scholar] [CrossRef]
- Yang, D.-H.; Tao, Y.; Ding, X.; Han, B.-H. Porous Organic Polymers for Electrocatalysis. Chem. Soc. Rev. 2022, 51, 761–791. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Liu, G. Porous Organic Materials Offer Vast Future Opportunities. Nat. Commun. 2020, 11, 4984. [Google Scholar] [CrossRef] [PubMed]
- Ussia, M.; Pumera, M. Towards Micromachine Intelligence: Potential of Polymers. Chem. Soc. Rev. 2022, 51, 1558–1572. [Google Scholar] [CrossRef]
- Das, S.; Heasman, P.; Ben, T.; Qiu, S. Porous Organic Materials: Strategic Design and Structure-Function Correlation. Chem. Rev. 2017, 117, 1515–1563. [Google Scholar] [CrossRef]
- Puthiaraj, P.; Lee, Y.-R.; Zhang, S.; Ahn, W.-S. Triazine-Based Covalent Organic Polymers: Design, Synthesis and Applications in Heterogeneous Catalysis. J. Mater. Chem. A 2016, 4, 16288–16311. [Google Scholar] [CrossRef]
- Kuhn, P.; Forget, A.; Su, D.; Thomas, A.; Antonietti, M. From Microporous Regular Frameworks to Mesoporous Materials with Ultrahigh Surface Area: Dynamic Reorganization of Porous Polymer Networks. J. Am. Chem. Soc. 2008, 130, 13333–13337. [Google Scholar] [CrossRef] [PubMed]
- Tahir, N.; Krishnaraj, C.; Leus, K.; Van der Voort, P. Development of Covalent Triazine Frameworks as Heterogeneous Catalytic Supports. Polymers 2019, 11, 1326. [Google Scholar] [CrossRef]
- Din, I.U.; Nasir, Q.; Garba, M.D.; Alharthi, A.I.; Alotaibi, M.A.; Usman, M. A Review of Preparation Methods for Heterogeneous Catalysts. Mini-Rev. Org. Chem. 2022, 19, 92–110. [Google Scholar] [CrossRef]
- Sun, G.; Chen, W.; Li, Y.; Fan, S.; Lv, J.; Zhao, T. In Situ Synthesis of Rh@Nax Catalyst for 1-Hexene Hydroformylation. Fuel 2024, 373, 132327. [Google Scholar] [CrossRef]
- Flores, C.; Batalha, N.; Marcilio, N.R.; Ordomsky, V.V.; Khodakov, A.Y. Influence of Impregnation and Ion Exchange Sequence on Metal Localization, Acidity and Catalytic Performance of Cobalt BEA Zeolite Catalysts in Fischer-Tropsch Synthesis. Chemcatchem 2019, 11, 568–574. [Google Scholar] [CrossRef]
- Zhang, H.Z.; Qiu, J.S.; Liang, C.H.; Li, Z.L.; Wang, X.N.; Wang, Y.P.; Feng, Z.C.; Li, C. A Novel Approach to Co/Cnts Catalyst Via Chemical Vapor Deposition of Organometallic Compounds. Catal. Lett. 2005, 101, 211–214. [Google Scholar] [CrossRef]
- Kuhn, P.; Antonietti, M.; Thomas, A. Porous, Covalent Triazine-Based Frameworks Prepared by Ionothermal Synthesis. Angew. Chem. Int. Ed. 2008, 47, 3450–3453. [Google Scholar] [CrossRef]
- Yu, S.-Y.; Mahmood, J.; Noh, H.-J.; Seo, J.-M.; Jung, S.-M.; Shin, S.-H.; Im, Y.-K.; Jeon, I.-Y.; Baek, J.-B. Direct Synthesis of a Covalent Triazine-Based Framework from Aromatic Amides. Angew. Chem. Int. Ed. 2018, 57, 8438–8442. [Google Scholar] [CrossRef]
- Ren, S.; Bojdys, M.J.; Dawson, R.; Laybourn, A.; Khimyak, Y.Z.; Adams, D.J.; Cooper, A.I. Porous, Fluorescent, Covalent Triazine-Based Frameworks Via Room-Temperature and Microwave-Assisted Synthesis. Adv. Mater. 2012, 24, 2357–2361. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Yang, L.-M.; Wang, X.; Guo, L.; Cheng, G.; Zhang, C.; Jin, S.; Tan, B.; Cooper, A. Covalent Triazine Frameworks Via a Low-Temperature Polycondensation Approach. Angew. Chem. Int. Ed. 2017, 56, 14149–14153. [Google Scholar] [CrossRef]
- Bojdys, M.J.; Jeromenok, J.; Thomas, A.; Antonietti, M. Rational Extension of the Family of Layered, Covalent, Triazine-Based Frameworks with Regular Porosity. Adv. Mater. 2010, 22, 2202–2205. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Tian, C.; Mahurin, S.M.; Chai, S.-H.; Wang, C.; Brown, S.; Veith, G.M.; Luo, H.; Liu, H.; Dai, S. A Superacid-Catalyzed Synthesis of Porous Membranes Based on Triazine Frameworks for CO2 Separation. J. Am. Chem. Soc. 2012, 134, 10478–10484. [Google Scholar] [CrossRef]
- Yu, M.; Wang, X.; Yang, X.; Zhao, Y.; Jiang, J.-X. Conjugated Microporous Copolymer Networks with Enhanced Gas Adsorption. Polym. Chem. 2015, 6, 3217–3223. [Google Scholar] [CrossRef]
- Zhou, Y.-B.; Wang, Y.-Q.; Ning, L.-C.; Ding, Z.-C.; Wang, W.-L.; Ding, C.-K.; Li, R.-H.; Chen, J.-J.; Lu, X.; Ding, Y.-J.; et al. Conjugated Microporous Polymer as Heterogeneous Ligand for Highly Selective Oxidative Heck Reaction. J. Am. Chem. Soc. 2017, 139, 3966–3969. [Google Scholar] [CrossRef]
- Xiong, S.; Fu, X.; Xiang, L.; Yu, G.; Guan, J.; Wang, Z.; Du, Y.; Xiong, X.; Pan, C. Liquid Acid-Catalysed Fabrication of Nanoporous 1,3,5-Triazine Frameworks with Efficient and Selective CO2 Uptake. Polym. Chem. 2014, 5, 3424–3431. [Google Scholar] [CrossRef]
- Wang, D.; Zeng, G.; Fang, J.; Li, H.; Chen, H.; Ma, J.; Dong, Z. Catalytic Hydroformylation of Alkenes to Branched Aldehydes Promoted by Water on Rh Nanoclusters-Anchored Porous Triphenylphosphine Frameworks. Chem. Eng. J. 2024, 482, 148860. [Google Scholar] [CrossRef]
- Jurado, L.; Esvan, J.; Luque-Alvarez, L.A.A.; Bobadilla, L.F.F.; Odriozola, J.A.; Posada-Perez, S.; Poater, A.; Comas-Vives, A.; Axet, M.R. Highly Dispersed Rh Single Atoms over Graphitic Carbon Nitride as a Robust Catalyst for the Hydroformylation Reaction. Catal. Sci. Technol. 2023, 13, 1425–1436. [Google Scholar] [CrossRef]
- Uner, O.; Bayrak, Y. The Effect of Carbonization Temperature, Carbonization Time and Impregnation Ratio on the Properties of Activated Carbon Produced from Arundo Donax. Microporous Mesoporous Mater. 2018, 268, 225–234. [Google Scholar] [CrossRef]
- Puthiaraj, P.; Ahn, W.-S. Synthesis of Copper Nanoparticles Supported on a Microporous Covalent Triazine Polymer: An Efficient and Reusable Catalyst for O-Arylation Reaction. Catal. Sci. Technol. 2016, 6, 1701–1709. [Google Scholar] [CrossRef]
- Puthiaraj, P.; Ahn, W.-S. Facile Synthesis of Microporous Carbonaceous Materials Derived from a Covalent Triazine Polymer for CO2 Capture. J. Energy Chem. 2017, 26, 965–971. [Google Scholar] [CrossRef]
- Raza, A.A.; Ravi, S.; Tajudeen, S.S.; Sheriff, A.K.I. Sulfonated Covalent Triazine Polymer Loaded with Pd Nanoparticles as a Bifunctional Catalyst for One Pot Hydrogenation Esterification Reaction. J. Solid State Chem. 2021, 302, 122417. [Google Scholar] [CrossRef]
- Lim, H.; Cha, M.C.; Chang, J.Y. Preparation of Microporous Polymers Based on 1,3,5-Triazine Units Showing High CO2 Adsorption Capacity. Macromol. Chem. Phys. 2012, 213, 1385–1390. [Google Scholar] [CrossRef]
- Li, B.; Fang, J.; Xu, D.; Zhao, H.; Zhu, H.; Zhang, F.; Dong, Z. Atomically Dispersed Co Clusters Anchored on N-Doped Carbon Nanotubes for Efficient Dehydrogenation of Alcohols and Subsequent Conversion to Carboxylic Acids. Chemsuschem 2021, 14, 4536–4545. [Google Scholar] [CrossRef]
- Mokhtari, N.; Khataei, M.M.; Dinari, M.; Monjezi, B.H.; Yamini, Y. Imine-Based Covalent Triazine Framework: Synthesis, Characterization, and Evaluation Its Adsorption. Mater. Lett. 2020, 263, 127221. [Google Scholar] [CrossRef]
- Peng, X.; Wei, L.; Jing, X.; Cui, L.; Wu, J.; Meng, G.; Liu, Z.; Guo, X. Stimuli-Responsive Nano-Polymer Composite Materials Based on the Triazine Skeleton Structure Used in Drug Delivery. JOM 2019, 71, 308–314. [Google Scholar] [CrossRef]
- Qiao, X.; Li, Q.; Schaugaard, R.N.; Noffke, B.W.; Liu, Y.; Li, D.; Liu, L.; Raghavachari, K.; Li, L.-S. Well-Defined Nanographene-Rhenium Complex as an Efficient Electrocatalyst and Photocatalyst for Selective CO2 Reduction. J. Am. Chem. Soc. 2017, 139, 3934–3937. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Hu, W.; Zhang, X.; He, P.; Pattengale, B.; Liu, C.; Cendejas, M.; Hermans, I.; Zhang, X.; Zhang, J.; et al. 2D Covalent Organic Frameworks as Intrinsic Photocatalysts for Visible Light-Driven CO2 Reduction. J. Am. Chem. Soc. 2018, 140, 14614–14618. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Zhang, S.; Zhang, L.; Liu, X.; Jia, Z.; Li, L.; Ta, N.; Wang, A.; Liu, W.; Wang, A.; et al. Suppressing Metal Leaching and Sintering in Hydroformylation Reaction by Modulating the Coordination of Rh Single Atoms with Reactants. J. Am. Chem. Soc. 2024, 146, 11955–11967. [Google Scholar] [CrossRef] [PubMed]
- Lang, R.; Li, T.; Matsumura, D.; Miao, S.; Ren, Y.; Cui, Y.-T.; Tan, Y.; Qiao, B.; Li, L.; Wang, A.; et al. Hydroformylation of Olefins by a Rhodium Single-Atom Catalyst with Activity Comparable to RhCl(PPh3)3. Angew. Chem. Int. Ed. 2016, 55, 16054–16058. [Google Scholar] [CrossRef]
- Sun, T.; Liang, Y.; Luo, W.; Zhang, L.; Cao, X.; Xu, Y. A General Strategy for Kilogram-Scale Preparation of Highly Crystal-line Covalent Triazine Frameworks. Angew. Chem. Int. Ed. 2022, 61, e202203327. [Google Scholar] [CrossRef]
- Zhao, K.; Wang, H.; Wang, X.; Li, T.; Dai, X.; Zhang, L.; Cui, X.; Shi, F. Confinement of Atomically Dispersed Rh Catalysts Within Porous Monophosphine Polymers for Regioselective Hydroformylation of Alkenes. J. Catal. 2021, 401, 321–330. [Google Scholar] [CrossRef]
- Bauer, G.; Ongari, D.; Tiana, D.; Gaumann, P.; Rohrbach, T.; Pareras, G.; Tarik, M.; Smit, B.; Ranocchiari, M. Metal-Organic Frameworks as Kinetic Modulators for Branched Selectivity in Hydroformylation. Nat. Commun. 2020, 11, 1059. [Google Scholar] [CrossRef]
- Shi, Y.; Hu, X.; Zhu, B.; Zhang, S.; Huang, W. Hydroformylation of 1-Octene over Nanotubular TiO2-Supported Amorphous Co-B Catalysts. Chem. Res. Chin. Univ. 2015, 31, 851–857. [Google Scholar] [CrossRef]
- Wang, Y.; Jiang, M.; Tian, T.; Sun, Z.; Wu, G.; Wang, Y.; Guo, X.; He, W.; Ding, J.; Yan, L.; et al. A Highly Active Phosphine Oxides-Containing Porous Organic Polymer Supported Co Catalyst for Hydroformylation of 2-Octene. Mol. Catal. 2024, 567, 114459. [Google Scholar] [CrossRef]
- Chen, M.; Gupta, G.; Ordonez, C.W.; Lamkins, A.R.; Ward, C.J.; Abolafia, C.A.; Zhang, B.; Roling, L.T.; Huang, W. Intermetallic Nanocatalyst for Highly Active Heterogeneous Hydroformylation. J. Am. Chem. Soc. 2021, 143, 20907–20915. [Google Scholar] [CrossRef]
- Lan, Y.; Lu, Y.; Yun, D.; Xia, C.; Qian, B.; Liu, J. Nitrogen-containing Porous Organic Polymer Supported Rhodium Catalyst for Hydroformylation of Olefins. Chemistryselect 2023, 8, e202301779. [Google Scholar] [CrossRef]
- Tan, M.; Ishikuro, Y.; Hosoi, Y.; Yamane, N.; Ai, P.; Zhang, P.; Yang, G.; Wu, M.; Yang, R.; Tsubaki, N. Pph3 Functionalized Rh/Rgo Catalyst for Heterogeneous Hydroformylation: Bifunctional Reduction of Graphene Oxide by Organic Ligand. Chem. Eng. J. 2017, 330, 863–869. [Google Scholar] [CrossRef]
- Tao, L.; Zhong, M.; Chen, J.; Jayakumar, S.; Liu, L.; Li, H.; Yang, Q. Heterogeneous Hydroformylation of Long-Chain Alkenes in IL-in-Oil Pickering Emulsion. Green Chem. 2018, 20, 188–196. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, W.; Wang, S.; Gao, Z.; Luo, Z.; Wang, X.; Zeng, R.; Li, A.; Li, H.; Wang, M.; et al. Atomic-Level Insights in Optimizing Reaction Paths for Hydroformylation Reaction over Rh/Coo Single-Atom Catalyst. Nat. Commun. 2016, 7, 14036. [Google Scholar] [CrossRef]
- Liu, B.; Huang, N.; Wang, Y.; Lan, X.; Wang, T. Promotion of Inorganic Phosphorus on Rh Catalysts in Styrene Hydroformylation: Geometric and Electronic Effects. ACS Catal. 2021, 11, 1787–1796. [Google Scholar] [CrossRef]
- Amsler, J.; Sarma, B.B.; Agostini, G.; Prieto, G.; Plessow, P.N.; Studt, F. Prospects of Heterogeneous Hydroformylation with Supported Single Atom Catalysts. J. Am. Chem. Soc. 2020, 142, 5087–5096. [Google Scholar] [CrossRef]
- Liu, B.; Huang, N.; Wang, Y.; Lan, X.; Wang, T. Regioselectivity Regulation of Styrene Hydroformylation over Rh-Based Phosphides: Combination of DFT Calculations and Kinetic Studies. Chem. Eng. J. 2022, 441, 136101. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, Z.; Hui, Y.; Wang, L.; Zhang, J.; Yi, X.; Chen, W.; Wang, C.; Wang, H.; Qin, Y.; et al. Rhodium Nanoparticles Supported on Silanol-Rich Zeolites Beyond the Homogeneous Wilkinson’s Catalyst for Hydroformylation of Olefins. Nat. Commun. 2023, 14, 2531. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Conv. (%) | Sel. (%) | L/B | TOF (h−1) 2 |
|---|---|---|---|---|
| Rh(CO)2(acac) | 26.9 | 48.8 | 1.83 | 925 |
| Rh/CTF-TPA | 57.5 | 55.8 | 2.51 | 1929 |
| Catalyst | Conv. (%) | Sel. (%) | L/B | TOF (h−1) 2 |
|---|---|---|---|---|
| 1-hexene | 85.9 | 85.4 | 2.47 | 3187 |
| 1-octene | 88.9 | 72.0 | 2.47 | 2960 |
| 1-decene | 57.5 | 84.3 | 2.51 | 1929 |
| 1-dodecene | 58.3 | 80.2 | 2.50 | 1992 |
| styrene | 60.1 | 100 | 0.70 | 2033 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Zhang, X.; Qin, G.; He, P.; Hao, Y. Boosting Hydroformylation via Reactant Enrichment in Covalent Triazine Frameworks with Atomically Dispersed Rh. Materials 2025, 18, 2691. https://doi.org/10.3390/ma18122691
Li X, Zhang X, Qin G, He P, Hao Y. Boosting Hydroformylation via Reactant Enrichment in Covalent Triazine Frameworks with Atomically Dispersed Rh. Materials. 2025; 18(12):2691. https://doi.org/10.3390/ma18122691
Chicago/Turabian StyleLi, Xinguo, Xiangjie Zhang, Gaolei Qin, Peng He, and Yajuan Hao. 2025. "Boosting Hydroformylation via Reactant Enrichment in Covalent Triazine Frameworks with Atomically Dispersed Rh" Materials 18, no. 12: 2691. https://doi.org/10.3390/ma18122691
APA StyleLi, X., Zhang, X., Qin, G., He, P., & Hao, Y. (2025). Boosting Hydroformylation via Reactant Enrichment in Covalent Triazine Frameworks with Atomically Dispersed Rh. Materials, 18(12), 2691. https://doi.org/10.3390/ma18122691