Abstract
An efficient approach to the synthesis of chiral selenoureas consisting of Cinchona alkaloid scaffolds was described. The new selenoureas were assessed as bifunctional organocatalysts in the asymmetric Michael addition reactions under mild conditions. The best results were obtained for selenoureas bearing the 4-fluorophenyl group. These catalysts promoted the reactions with enantioselectivities of up to 96% ee. Additionally, the catalytic performance of the thiourea and selenourea counterpart was compared.
1. Introduction
In recent years, growing attention has been focused on the synthesis and applications of selenourea derivatives. The structures of selenoureas are closely related to those of analogs of oxygen and sulfur compounds [1,2,3], but the presence of larger, more polarizable selenium results in a change of significant properties. They possess various biological properties, such as antioxidant [4,5], antibacterial and antifungal [6], antileishmanial [7], pesticidal [8], and anti-urease activity [9]. Selenoureas also demonstrate enzyme inhibition, free-radical scavenging, and anticancer activity [10,11]. These compounds are very useful starting materials for the synthesis of selenium-containing heterocycles [12,13] and are used for anion binding and recognition [14,15]. To the best of our knowledge, only a few examples of chiral selenoureas have been reported in the literature [16,17]. Currently, chiral bis-selenourea is used as a strong hydrogen-bonding donor for highly efficient chiral recognition of a diverse range of tertiary alcohols [18]. Still, little is known about the application of selenoureas as hydrogen-bond donors in organocatalysis. Bolm and co-workers were the first to apply a selenourea derivative as a chiral catalyst in the asymmetric Michael addition of α-nitrocyclohexane to aryl nitroalkenes [19].
Chiral bifunctional organocatalysts, which incorporate a thiourea group as H-bond donors with Lewis base, are commonly used in asymmetric catalysis. Both parts of the catalysts simultaneously activate the electrophile and nucleophile leading to the high stereocontrol of the reaction [20,21]. Nowadays, thioureas are widely recognized as highly useful and powerful organocatalysts that promote a diverse range of reactions with excellent enantioselectivity [22,23,24]. Cinchona alkaloids with an exceptionally good chiral backbone have been used for the synthesis of many successful catalysts [25]. Inspired by the improved catalytic activity of the thioureas in comparison to ureas, we have taken interest in the modification of the “privileged” Cinchona alkaloid scaffolds by substituting the thiourea with a selenourea moiety.
Here, we report the approach to the efficient synthesis of a series of novel Cinchona alkaloid-derived selenoureas, and we also demonstrate their organocatalytic potential in asymmetric Michael additions.
2. Results and Discussion
2.1. Preparation of Compounds
Cinchona alkaloid-based selenoureas were prepared by coupling reactions of 9-epi-aminoalkaloids with aryl isoselenocyanates. Although there are some methods in the literature for the preparation of selenoureas [4,10,26], the best procedure still turns out to be the reaction of isoselenocyanates with amines. The crucial step is the preparation of isoselenocyanates, which, unlike their sulfur analogs, are not commercially available. Several strategies towards their synthesis have been reported [27,28,29]. The oxidation of corresponding isocyanides with elemental black selenium in a suitable solvent seems to be the most reliable method [13,16,30,31].
The first coupling partner for the synthesis of desired selenoureas was Cinchona alkaloid-based amines 2a–e, which were prepared in good yields (65–75%) from naturally occurring Cinchona alkaloids, namely quinine (QN), cinchonidine (CD), quinidine (QD), and the respective dihydro derivatives (dihydroquinine (DHQN)/dihydroquinidine (DHQD)) in a three-step sequence via mesylation [32], azidation with NaN3 in warm DMF followed by SN2 displacement [33], and then treatment with triphenylphosphine (Staudinger reduction) to amine (Scheme 1) [34].
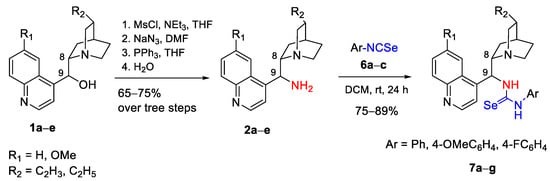
Scheme 1.
General synthetic route for compounds 7a–g.
Non-commercial aryl isoselenocyanates 6a–c, the second coupling partner, were synthesized in good yields (46–68%) in a three-step protocol (Scheme 2) that involved N-formylation of the commercially available aryl amines 3a–c with formic acid in toluene, conversion of the formamides 4a–c into isocyanides 5a–c upon dehydration with POCl3 in the presence of NEt3 in dichloromethane (DCM) [35]. The last step included the reaction of the isocyanides with black selenium in DCM at 45 °C in darkness.
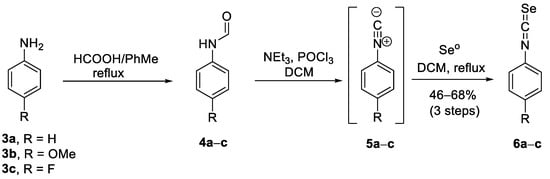
Scheme 2.
Synthesis of aryl isoselenocyanates 6a–c.
The coupling of the aryl isoselenocyanates 6a–c with 9-epi-aminoalkaloids 2a–e in DCM at 45 °C in the darkness resulted in the formation of the desired selenoureas 7a–g in high yields (75–89%) (Scheme 1), (Figure 1). All new compounds were fully characterized by spectroscopic methods (IR, 1H, and 13C NMR, HRMS). Copies of 1H, 13C NMR and IR spectra are presented in Supplementary Materials.
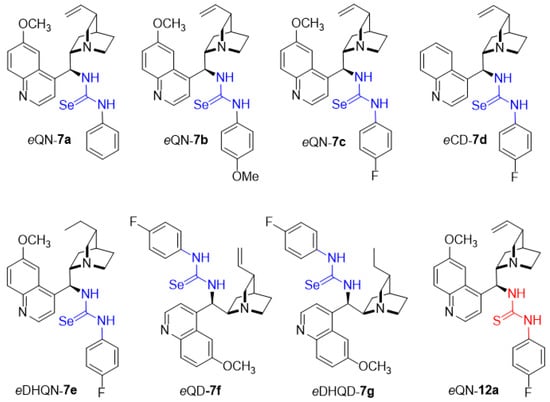
Figure 1.
Structures of the Cinchona-based selenoureas.
In an alternative approach, we intended to introduce the isoselenocyanate moiety in the Cinchona alkaloid skeleton. 9S-Amino-deoxyquinine 2a was chosen for the transformation into isoselenocyanate. Amine 2a was converted into 9S-formylamino-deoxyquinine 8a by treatment with the excess of methyl formate in quantitative yield (Scheme 3). The spectroscopic data of compound 8a can be found in Supplementary Materials, Figures S15 and S16.
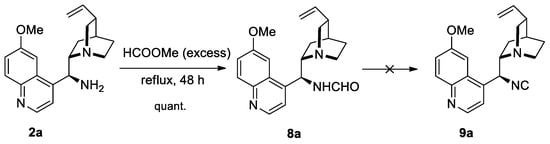
Scheme 3.
Unsuccessful attempts to prepare isocyanide 9a.
Surprisingly, dehydration of formamide 8a using the previously applied POCl3/NEt3 system did not provide the desired isocyanide, and the amine 2a was fully recovered. Due to this unsatisfying result, other known methods to convert formamide 8a to isocyanide 9a were tested. Hence, the procedures using the triphosgene/NEt3/DCM and PPh3/CCl4/NEt3 system as well as Vilsmeier and Burgess reagents turned out to be ineffective [17,30,36].
Further transformations of the quinine-derived amine 2a applying the Hofmann isonitrile synthesis protocol [37] modified by Mąkosza [38] followed for the preparation of novel isonitrile 9a. The reaction of 2a with chloroform in a biphasic system, 25% aqueous sodium hydroxide solution, and DCM and TEBAC (triethylbenzylammonium chloride) as a phase-transfer catalyst, afforded compound 9a with a conversion of approximately 50% (Scheme 4). Attempts to isolate detected products were unsuccessful because isocyanide 9a hydrolyzed during column chromatography with the formation of formamide 8a.
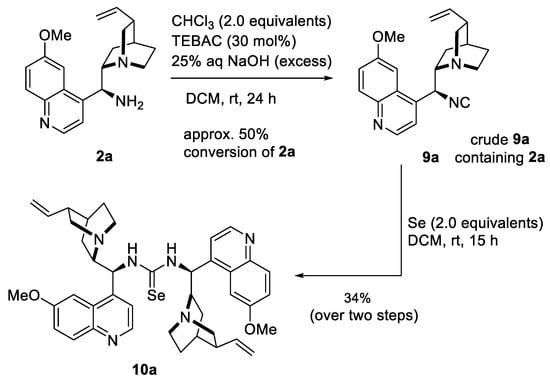
Scheme 4.
The synthetic route for dimeric alkaloid selenourea 10a.
Accordingly, we turned our attention to converting the resulting crude product into dimeric alkaloid selenourea 10a. The addition of selenium to the crude mixture of isocyanide 9a and unreacted amine 2a in DCM led to a moderate yield of product 10a (Scheme 4) (spectral data available in Supplementary Materials, Figures S17 and S18).
In contrast, the one-pot transformation of the amine 2a into isoselenocyanate 11a under phase-transfer catalysis did not lead to the formation of the desired product 11a (Scheme 5). In this case, a mixture of compounds was formed.
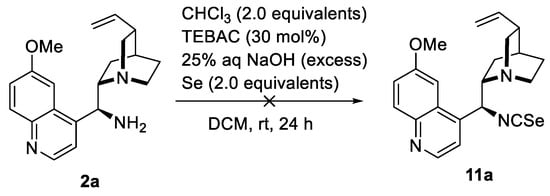
Scheme 5.
Unsuccessful attempts to prepare isoselenocyanate 11a.
2.2. Catalytic Activity of Tested Compounds
With the aforementioned set of chiral selenoureas in hand, we examined their catalytic abilities as bifunctional organocatalysts in the asymmetric Michael addition. We chose the Michael reaction of nitromethane to trans-chalcone developed by Soós et al. [39] as the model reaction.
The screening experiments are summarized in Table 1. The results showed that the selenourea catalysts 7a–g afforded Michael adducts with good to excellent enantioselectivity ranging from 70% to 95% (Table 1, entries 1–7). The influence of substituents in the aromatic ring was also noted for the quinine-based selenourea. As expected, compounds bearing the electron-withdrawing substituent in the phenyl ring delivered a superior effect on the catalyzed reactions in comparison to their counterparts possessing electron-neutral and electron-donating groups. It is known that pKa values of catalysts correlate well with their hydrogen-bond donating abilities [40]. Electron-withdrawing substituents increase the acidity of the double hydrogen bond donor and subsequently, improve the electrophile activation ability [41,42]. There, the application of unsubstituted (entry 1), 4-methoxy- (entry 2), and 4-fluoro-(entry 3) derivatives gave 81, 70, and 95% ee, respectively. Better stereoselectivity was achieved for (8S,9S)-selenourea derivatives 7c–e (quinine series) as compared to (8R,9R) quinidine series (Table 1, entries 3–5 vs. entries 6 and 7). These results demonstrate that the configurations of the alkaloid determine the stereochemical result of the addition. The absence of the methoxyl group at the C6′ position in cinchonidine-derived selenourea 7d did not considerably affect the outcome of the catalyzed reaction compared to the selenourea catalyst 7c, which possesses this group (91% ee, entry 4 vs. 95% ee, entry 3). The yields of the reaction were poor to moderate (up to 46%), slightly better for selenourea derivatives of the quinine series. Higher yields were observed for dihydro alkaloid-based selenoureas 7e and 7g (36–46% ee, entries 5 and 7 vs. 22–26% ee, entries 3,4, and 6).

Table 1.
Asymmetric Michael addition of nitromethane to trans-chalcone a.
To directly compare the catalytic utility of sulfur and selenium catalyst counterparts, the sulfur analog 12a (Figure 1) of the most potent selenourea catalyst 7c was synthesized (copies of NMR an IR spectra are gathered in Supplementary Materials). When thiourea 12a was tested a similar result was observed, with slightly lower enantioselectivity (Table 1, entry 8).
All used selenoureas 7a–g decomposed under the reaction conditions to corresponding carbodiimides (noticeable precipitation of black selenium).
Based on the experimental results and the previous mechanism studies reported by Pedrosa and coworkers [44], a plausible transition state model for the Michael reactions of chalcone 13 and nitromethane catalyzed by 7c was proposed (Figure 2). In the transition state, the selenourea moiety of the catalyst activates nitromethane by hydrogen bonding while the bridgehead nitrogen atom of the quinine unit activates trans-chalcone. While in the transition state, a Re-face attack is favored, giving the R-configured product.
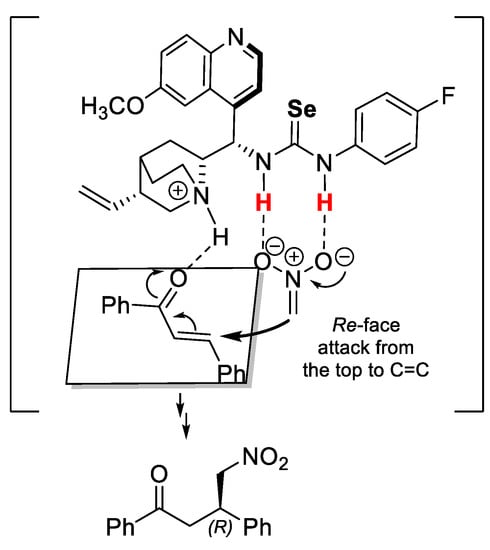
Figure 2.
A proposed transition state.
Next, Cinchona-derived selenoureas were applied as organocatalysts for promoting the sulfa-Michael reactions of less reactive thioacetic acid to trans-chalcones [45]. All selenourea catalysts afforded the product 15 with high yields ranging from 90% to 99% with up to 21% ee under mild reaction conditions (Table 2). Compounds 7c–g, possessing the 4-fluorophenyl group, were found to be the most potent among probed catalysts. Selenourea 7c delivered better stereocontrol in the reaction compared to the sulfur counterpart 12a (Table 2, 21% ee vs. 17% ee, entries 3 and 8). Selenourea catalysts of (8S,9S) the quinine series 7c–e offered product 15 under better stereocontrol (Table 2, 17–21% ee, entries 3–5 vs. 12–14% ee, entries 6,7). No noticeable catalyst decomposition was observed under the reaction conditions.
Table 2.
Sulfa-Michael addition of thioacetic acid to trans-chalcone a.
The moderate results of conversion (Michael addition) and enantioselectivity (sulfa-Michael addition) indicate that the reaction conditions could be possibly further optimized to improve the catalytic activity of synthesized organocatalysts and to avoid their decomposition (e.g., by altering reaction time and temperature or the used solvent).
3. Conclusions
We have developed a synthetic route to the novel achievement of Cinchona-based selenoureas and the strategy for producing dimeric alkaloid selenoureas. To our knowledge, this study represents the first example of a promising application of chiral Cinchona alkaloid-derived selenoureas as bifunctional organocatalysts in asymmetric Michael addition reactions. Our preliminary attempts resulted in low to excellent enantioselectivities and yields under mild reaction conditions. Further investigations of the catalytic performance in other asymmetric reactions with this type of bifunctional selenoureas are currently underway in our laboratory.
4. Materials and Methods
4.1. General Information
Solvents were distilled, and other reagents were used as received. Reactions were monitored by thin-layer chromatography (TLC) on silica gel 60 F-254 precoated plates (Merck, Darmstadt, Germany), and spots were visualized with a UV lamp. Products were purified by standard column chromatography on silica gel 60 (230–400 mesh) (Merck). Optical rotations at 578 nm were measured using an Optical Activity Ltd. (Huntington, UK) Model AA-5 automatic polarimeter. Melting points were determined using a Boëtius hot-stage apparatus (PHMK VEB Analytic, Dresden, Germany). 1H and 13C NMR (600 MHz and 151 MHz, respectively) spectra were recorded in CDCl3 on Bruker Avance DRX 300 and NMR Bruker Avance II 600 MHz (Bruker, Billerica, MA, USA). IR spectra were measured using the Vertex 70V vacuum FT-IR spectrometer (Bruker Optics, Ettlingen, Germany). High-resolution mass spectra (HRMS) were recordered using electrospray ionization mode on the Waters LCT Premier XE TOF spectrometer (Waters Corporation, Milford, MA, USA). The enantiomeric ratios of the samples were determined by chiral high-performance liquid chromatography (HPLC) measurements (Thermo Fisher Scientific, Waltham, MA, USA) using Chiracel AD-H or Chiralpak AS-H chiral columns. The configuration of the products was assigned by comparison to literature data.
4.2. Preparation of Starting Compounds
The starting Cinchona alkaloids (QN, CD, QD, DHQN, DHQD) were commercially available (Buchler GmbH). Cinchona alkaloid mesylates were prepared by a standard procedure [26]. 9-Amino-(9-deoxy)-epialkaloids 2a–e were synthesized according to a general procedure described in the literature [33,34]. Aryl isoselenocyanates 6a–c were prepared according to the known procedure [35,46,47].
4.3. General Procedure for Selenourea Synthesis
9-Amino-(9-deoxy)-epialkaloid 2 (1.00 mmol, 1.00 equiv.) was dissolved in CH2Cl2 (5.0 mL), and a solution of appropriate aryl isoselenocyanate 6 (1.00 mmol, 1.00 equiv.) in CH2Cl2 (1.0 mL) was added. The mixture was stirred for 15 h at rt under argon, in darkness. The solvent was evaporated in vacuo and the residue was purified by chromatography on silica gel (CH2Cl2/MeOH, 10:1) to afford the desired product 7. The structure of selenoureas 7 were confirmed by spectroscopic methods (IR, 1H, and 13C NMR) and high- resolution mass spectrometry (data available in Supplementary Materials).
4.3.1. N-[(8S,9S)-6′-Methoxycinchonan-9-yl]-N′-Phenylselenourea eQN-7a
Yield 85%, pale yellow solid, mp 106–108 °C, Rf = 0.66 (CH2Cl2/MeOH 10:1).= −164.5 (c 0.22, CH2Cl2). 1H NMR (600 MHz, CDCl3): δ 9.64 (br s, 1H), 8.44 (s, 1H), 7.97 (d, J = 9.1 Hz, 1H), 7.88 (br s, 1H), 7.40–7.34 (m, 3H), 7.32–7.27 (m, 1H), 7.25 (d, J = 7.8 Hz, 2H), 7.19 (br s, 1H), 6.15 (br s, 1H), 5.63 (dt, J = 17.2, 8.6 Hz, 1H), 4.98−4.90 (m, 2H), 3.96 (s, 3H), 3.52–3.38 (m, 1H), 3.29 (br s, 1H), 3.11 (t, J = 12.1 Hz, 1H), 2.75 (d, J = 13.9 Hz, 1H), 2.71–2.62 (m, 1H), 2.36–2.20 (m, 1H), 1.75–1.65 (m, 2H), 1.64–1.59 (m, 1H), 1.32 (t, J = 11.7 Hz, 1H), 0.97 (dd, J = 14.3, 6.4 Hz, 1H) ppm. 13C NMR (150 MHz, CDCl3): δ 179.2, 157.8, 147.5 (2C overlapped), 144.8, 140.5, 137.5, 131.6, 129.6 (2C overlapped), 128.1, 127.2 (2C overlapped), 125.6, 122.0, 119.9, 115.0, 102.7, 61.0, 58.5, 55.8, 55.1, 41.7, 39.1, 27.4, 27.2, 25.5 ppm. IR: 3157, 2930, 2862, 1620, 1589, 1507, 1473, 1431, 1314, 1297, 1259, 1225, 1028, 915, 850, 835, 823, 758, 693 cm−1. HRMS (ESI): m/z calculated for C27H31N4O80Se [M+H]+: 507.1663, found: 507.1657.
4.3.2. N-[(8S,9S)-6′-Methoxycinchonan-9-yl]-N′-(4-Methoxyphenyl)Selenourea eQN-7b
Yield 82%, pale yellow solid, mp 86–88 °C, Rf = 0.60 (CH2Cl2/MeOH 10:1).= −219.2 (c 0.30, CH2Cl2). 1H NMR (600 MHz, CDCl3): δ 8.72 (br s, 1H), 8.59 (s, 1H), 8.00 (d, J = 9.0 Hz, 1H), 7.80 (br s, 1H), 7.37 (d, J = 9.0 Hz, 1H), 7.25 (br s, 1H), 7.17 (d, J = 8.3 Hz, 2H), 6.91 (d, J = 8.2 Hz, 2H), 6.02 (br s, 1H), 5.63 (dt, J = 17.5, 8.8 Hz, 1H), 5.00–4.91 (m, 2H), 3.96 (s, 3H), 3.83 (s, 3H), 3.42 (br s, 2H), 3.18 (t, J = 12.2 Hz, 1H), 2.74 (br s, 2H), 2.31 (s, 1H), 1.78–1.60 (m, 3H), 1.36 (s, 1H), 1.03 (s, 1H) ppm. 13C NMR (150 MHz, CDCl3): δ 179.1, 158.7, 157.8, 147.5, 145.3, 144.7, 140.9, 131.5, 130.3, 128.2, 127.8 (2C overlapped), 122.1, 119.7, 114.9, 114.8 (2C overlapped), 102.8, 61.1, 58.4, 55.9, 55.6, 55.2, 41.7, 39.3, 27.7, 27.3, 25.6 ppm. IR: 3237, 2931, 2865, 1621, 1507, 1240, 1227, 1166, 1029, 917, 826, 716, 643 cm−1. HRMS (ESI): m/z calculated for C28H33N4O280Se [M+H]+: 537.1771, found: 537.1769.
4.3.3. N-(4-Fluorophenyl)-N′-[(8S,9S)-6′-Methoxycinchonan-9-yl]Selenourea eQN-7c
Yield 86%, pale yellow solid, mp 104–105 °C, Rf = 0.55 (CH2Cl2/MeOH 10:1).= −142.5 (c 0.24, CH2Cl2). 1H NMR (600 MHz, CDCl3): δ 9.10 (br s, 1H), 8.57 (s, 1H), 8.00 (d, J = 9.2 Hz, 1H), 7.80 (br s, 1H), 7.38 (dd, J = 9.2, 2.6 Hz, 1H), 7.28–7.17 (m, 3H), 7.07 (t, J = 8.3 Hz, 2H), 6.07 (br s, 1H), 5.64 (dq, J = 17.5, 9.5, 8.4 Hz, 1H), 5.02–4.93 (m, 2H), 3.97 (s, 3H), 3.43 (br s, 1H), 3.31 (br s, 1H), 3.16 (t, J = 12.1 Hz, 1H), 2.73 (br s, 2H), 2.32 (br s, 1H), 1.72 (br s, 2H), 1.66 (br s, 1H), 1.37 (t, J = 12.3 Hz, 1H), 1.00 (br s, 1H) ppm. 13C NMR (150 MHz, CDCl3) δ 179.7, 161.4 (d, JC–F = 247.7 Hz), 158.0, 147.5 (2C overlapped), 144.9, 140.6, 133.7, 131.7, 128.2, 128.1 (2C overlapped), 122.12, 119.7, 116.6 (d, JC–F = 22.5 Hz, 2C overlapped), 115.2, 102.8, 61.1, 55.9, 55.2, 50.8, 41.8, 39.2, 27.6, 27.3, 25.6 ppm. IR: 3169, 2931, 2864, 1620, 1504, 1473, 1320, 1217, 1153, 1029, 916, 830, 643 cm−1. HRMS (ESI): m/z calculated for C27H30N4O80SeF [M+H]+: 525.1570, found: 525.1578.
4.3.4. N-[(8S,9S)-Cinchonan-9-yl]-N′-(4-Fluorophenyl)Selenourea eCD-7d
Yield 88%, pale yellow solid, mp. 66–68 °C, Rf = 0.62 (CH2Cl2/MeOH 10:1). = −173.8 (c 0.32, CH2Cl2). 1H NMR (600 MHz, CDCl3): δ 8.93 (s, 1H), 8.78 (s, 1H), 8.47 (s, 1H), 8.13 (d, J = 8.4 Hz, 1H), 7.73 (ddd, J = 8.4, 6.8, 1.2 Hz, 1H), 7.64 (dd, J = 9.7, 5.5 Hz, 1H), 7.35 (s, 1H), 7.30–7.21 (m, 2H), 7.09 (t, J = 8.3 Hz, 2H), 6.06 (s, 1H), 5.61 (dq, J = 17.8, 8.9, 7.3 Hz, 1H), 5.07–4.82 (m, 2H), 3.42 (m, 2H), 3.26–3.11 (m, 1H), 2.77 (s, 2H), 2.34 (s, 1H), 1.74 (m, 3H), 1.35 (t, J = 11.9 Hz, 1H), 1.07 (s, 1H) ppm. 13C NMR (150 MHz, CDCl3): δ 180.0, 161.3 (d, JC–F = 247.5 Hz), 150.0, 148.5, 146.2, 140.2, 133.7, 130.4, 129.4, 127.9 (2C overlapped), 127.0, 126.8, 124.2, 119.7, 116.4 (d, JC–F = 22.9 Hz, 2C overlapped), 115.2, 61.2, 58.3, 54.9, 41.5, 38.9, 27.2, 27.1, 25.1 ppm. IR: 3168, 2938, 2864, 1589, 1504, 1320, 1289, 1214, 914, 831, 797, 752, 639 cm−1. HRMS (ESI): m/z calculated for C26H28N480SeF [M+H]+: 495.1463, found: 495.1463.
4.3.5. N-[4-Fluorophenyl]-N′-[(8S,9S)-10,11-Dihydro-6′-Methoxycinchonan-9-yl]Selenourea eDHQN-7e
Yield 85%, pale yellow solid, mp 62−64 °C, Rf = 0.54 (CH2Cl2/MeOH 10:1). = −165.7 (c 0.14, CH2Cl2). 1H NMR (600 MHz, CDCl3): δ 8.88 (br s, 1H), 8.71 (s, 1H), 8.03 (d, J = 9.2 Hz, 1H), 7.89 (br s, 1H), 7.40 (dd, J = 9.1, 2.7 Hz, 2H), 7.30 (d, J = 7.0 Hz, 2H), 7.05 (t, J = 8.3 Hz, 2H), 6.32 (s, 1H), 3.99 (s, 3H), 3.64 (s, 1H), 3.32 (s, 1H), 2.89 (s, 1H), 2.60 (s, 1H), 2.20 (d, J = 10.4 Hz, 1H), 1.77 (br m, 4H), 1.45 (s, 1H), 1.24 (q, J = 7.9 Hz, 2H), 1.13 (s, 1H), 0.81 (t, J = 7.3 Hz, 3H) ppm. (150 MHz, CDCl3): δ 180.0, 161.2 (d, JC–F = 247.6 Hz), 158.0, 147.5, 144.8, 144.5, 133.9, 131.6, 127.9 (3C overlapped), 122.1, 119.9, 116.3 (d, JC–F = 23.2 Hz, 2C overlapped), 102.7, 60.9, 56.6, 55.9, 50.7, 41.9, 36.6, 27.6, 27.0, 25.1, 24.9, 11.8 ppm. IR: 3167, 2929, 2864, 1621, 1505, 1473, 1315, 1218, 1029, 917, 829, 715, 644 cm−1. HRMS (ESI): m/z calculated for C27H32N4O80SeF [M+H]+: 527.1727, found: 527.1732.
4.3.6. N-(4-Fluorophenyl)-N′-[(8R,9R)-6′-Methoxycinchonan-9-yl]Selenourea eQD-7f
Yield 81%, pale yellow solid, mp 71–73 °C, Rf = 0.43 (CH2Cl2/MeOH 10:1). = +331.1 (c 0.18, CH2Cl2). 1H NMR (600 MHz, CDCl3): δ 9.04 (br s, 1H), 8.63 (s, 1H), 8.00 (d, J = 9.5 Hz, 1H), 7.77 (br s, 1H), 7.45–7.19 (m, 4H), 7.07 (t, J = 8.3 Hz, 2H), 6.17 (s, 1H), 5.90 (dq, J = 16.3, 5.9 Hz, 1H), 5.23 (m, 2H), 3.97 (s, 3H), 3.31 (s, 2H), 3.18–2.75 (m, 4H), 2.49–2.24 (m, 1H), 1.58 (m, 2H), 1.36 (s, 1H), 1.03 (s, 1H) ppm. 13C NMR (150 MHz, CDCl3): δ 180.1, 161.3 (d, JC–F = 247.0 Hz), 158.1, 147.5 (2C), 144.9, 139.5, 133.8, 131.7, 128.1, 127.8 (2C overlapped), 122.6, 119.6, 116.4 (d, JC–F = 23.3 Hz, 2C overlapped), 115.7, 102.4, 61.3, 55.9, 50.9, 48.8, 47.4, 38.7, 27.2, 26.0, 25.1 ppm. IR: 3169, 2934, 2868, 1620, 1589, 1505, 1473, 1323, 1310, 1319, 1028, 917, 849, 827, 795, 715 cm−1. HRMS (ESI): m/z calculated for C27H30N4O80SeF [M+H]+: 525.1570, found: 525.1575.
4.3.7. N-[4-Fluorophenyl]-N′-[(8R,9R)-10,11-Dihydro-6′-Methoxycinchonan-9-yl]Selenourea eDHQD-7g
Yield 78%, pale yellow solid, mp 88–90 °C, Rf = 0.40 (CH2Cl2/MeOH 10:1). = +142.5 (c 0.24, CH2Cl2). 1H NMR (600 MHz, CDCl3): δ 9.03 (br s, 1H), 8.70 (s, 1H), 8.02 (d, J = 9.2 Hz, 1H), 7.89 (br s, 1H), 7.40 (d, J = 11.1 Hz, 2H), 7.38–7.30 (m, 2H), 7.04 (t, J = 8.5.Hz, 2H), 6.40 (s, 1H), 3.97 (s, 3H), 3.71–3.45 (m, 1H), 3.13 (br m, 3H), 3.03–2.86 (m, 1H), 1.88–1.34 (m, 7H), 1.09 (s, 1H), 0.94 (s, 3H) ppm. 13C NMR (150 MHz, CDCl3): δ 180.2, 161.1 (d, JC–F = 245.5 Hz), 158.0, 147.5, 144.8, 144.2, 133.8, 131.7, 128.0, 127.6 (2C overlapped), 122.4, 119.9, 116.2 (d, JC–F = 21.7 Hz, 2C overlapped), 102.3, 61.2, 55.7, 49.5, 48.7, 36.8, 35.0, 26.2, 25.9, 25.3, 24.6, 11.9 ppm. IR: 3181, 2931, 2867, 1621, 1505, 1473, 1322, 1219, 1153, 1029, 848, 829, 715 cm−1. HRMS (ESI): m/z calculated for C27H32N4O80SeF [M+H]+: 527.1727, found: 527.1735.
4.4. Preparation of N-[(8S,9S)-6′-Methoxycinchonan-9-yl]Formamide 8a
Amine 2a (0.323 g, 1.00 mmol) was placed in a Teflon-capped ampoule and dissolved in an excess of methyl formate (4.00 mL). The mixture was heated to 45 °C for 48 h. The solvent was removed in vacuo, yielding compound 8a as a solidifying oil quantitatively (0.352 g), pale yellow solid, Rf = 0.25 (CH2Cl2/MeOH 10:1). = +32.5° (c 0.20, CH2Cl2). 1H NMR (600 MHz, CDCl3): δ 8.73 (dd, J = 4.6, 1.8 Hz, 1H), 8.20 (s, 1H), 8.02 (dd, J = 9.2, 1.9 Hz, 1H), 7.63 (s, 1H), 7.39 (dt, J = 9.2, 2.3 Hz, 1H), 7.33 (d, J = 4.6 Hz, 1H), 5.75 (dt, J = 17.6, 9.0 Hz, 1H), 5.50 (s, 1H), 5.06–4.97 (m, 2H), 3.98 (s, 4H), 3.36–3.18 (m, 3H), 2.85−2.71 (m, 2H), 2.35 (d, J = 7.9 Hz, 2H), 1.72 (q, J = 3.0 Hz, 1H), 1.65 (t, J = 8.1 Hz, 2H), 1.55 (t, J = 12.0 Hz, 1H), 0.94 (dd, J = 14.5, 6.2 Hz, 1H) ppm. 13C NMR (150 MHz, CDCl3): δ 161.0, 158.1, 147.5, 144.9, 144.1, 141.1, 131.8, 128.3, 122.0, 119.1, 114.9, 101.9, 59.5, 56.1, 55.8, 49.6, 41.1, 39.4, 27.7, 27.4, 26.5 ppm. IR: 3226, 3073, 2937, 2864, 1938, 1672, 1621, 1591, 1475, 1434, 1383, 1365, 1229, 1028, 916, 852, 828, 714, 678, 636, 616 cm−1. HRMS (ESI): m/z calculated for C21H26N3O2 [M+H]+: 352.2021, found: 352.2025.
4.5. Preparation of N,N′-bis[(8S,9S)-6′-Metoxycinchonan-9-yl]Selenourea 10a
9S-Amino-9-deoxyquinine 2a (0.324 g, 1.00 mmol, 1.00 equiv.) was dissolved in CH2Cl2 (6 mL) and 25% aqueous solution of NaOH (3 mL), triethylbenzylammonium chloride (TEBAC) (68.5 mg, 0.30 mmol, 30.0 mol%) and CHCl3 (0.239 g, 160 μL, 2.00 mmol, 2.00 equivalents) were added. The mixture was stirred for 24 h at rt and water (10 mL) was added and the mixture was extracted with CH2Cl2 (3 × 15 mL). The combined organic phases were dried over Na2SO4 and evaporated. The residue was dissolved in CH2Cl2 (10 mL) and selenium (0.158 g, 2.00 mmol, 2.00 equivalents) was added and the solution was stirred overnight under argon. The solvent was removed in vacuo and the residue was purified by chromatography in the dark (silica gel, CH2Cl2/MeOH 10:1 → MeOH) to afford 0,136 g (37%) of dimeric selenourea 10a as a pale yellow oil, Rf = 0.05 (CH2Cl2:MeOH 10:1), = −130.5° (c 0.11, CH2Cl2). 1H NMR (600 MHz, CDCl3): δ 8.78 (br s, 1H), 8.67 (br s, 1H), 8.12 (br s, 1H), 7.97 (d, J = 9.1 Hz, 1H), 7.49 (br s, 1H), 7.40–7.32 (m, 1H), 6.48 (br s, 1H), 5.58 (br s, 1H), 4.90 (br s, 2H), 3.88 (s, 3H), 3.18 (br s, 1H), 2.90 (br s, 2H), 2.41 (s, 1H), 2.31 (s, 1H), 2.17 (s, 1H), 1.76 (s, 1H), 1.51 (br m, 2H), 1.38 (s, 1H), 0.73 (s, 1H) ppm. IR (KBr): 3250, 2931, 1867, 1621, 1590, 1508, 1473, 1449, 1316, 1261, 1227, 1029, 917, 850, 758, 693, 494 cm−1. HRMS (ESI): m/z calculated for C41H49N6O280Se [M+H]+: 737.3087, found: 737.3089. 13C NMR spectrum could not be obtained due to fast decomposition of the compound in solutions.
4.6. Preparation of N-(4-Fluorophenyl)-N′-[(8S,9S)-6′-Methoxycinchonan-9-yl]Thiourea eQN-12a
To the stirred solution of 9S-amino-9-deoxyquinine 2a (0.323 g, 1.00 mmol) in CH2Cl2 (5 mL) was added dropwise a solution of 4-fluorophenyl isoselenocyanate 6c (0.153 g, 1.00 mmol) in CH2Cl2 (1.00 mL) under nitrogen atmosphere. The resulting mixture was stirred for 15 h. The solvent was removed in vacuo and the residue was purified by column chromatography on silica gel (CH2Cl2/MeOH 10:1) to afford thiourea 0.286 g (60%) of compound 12 as pale yellow solid, mp 108–110 °C, Rf = 0.45 (CH2Cl2/MeOH 10:1).= −160.8 (c 0.24, CH2Cl2). 1H NMR (600 MHz, CDCl3): δ 8.88 (br s, 1H), 8.63 (d, J = 4.5 Hz, 1H), 8.18 (br s, 1H), 8.00 (d, J = 9.2 Hz, 1H), 7.81 (br s, 1H), 7.38 (dd, J = 9.2, 2.6 Hz, 1H), 7.34 (d, J = 4.5 Hz, 1H), 7.28–7.23 (m, 2H), 7.03 (t, J = 8.5 Hz, 2H), 6.10 (br s, 1H), 5.61 (ddd, J = 17.3, 10.2, 7.1 Hz, 1H), 5.04–4.94 (m, 2H), 3.96 (s, 3H), 3.56 (br s, 1H), 3.48 (br s, 1H), 3.24 (dd, J = 13.8, 10.2 Hz, 1H), 2.82 (td, J = 13.2, 11.5, 5.3 Hz, 2H), 2.38 (d, J = 8.6 Hz, 1H), 1.88–1.66 (m, 3H), 1.44 (ddt, J = 13.5, 10.2, 3.3 Hz, 1H), 1.02 (dd, J = 14.4, 6.2 Hz, 1H) ppm. 13C NMR (150 MHz, CDCl3): δ 181.2, 160.9 (d, J = 246.6 Hz), 158.1, 147.6, 144.8, 144.2, 139.6, 134.0, 131.6, 128.2, 127.3, 122.2, 119.8, 116.1 (d, J = 22.6 Hz), 115.7, 102.5, 60.8, 55.9, 55.0, 50.6, 41.9, 38.6, 27.1, 26.8, 25.3 ppm. IR: 3265, 2932, 2879, 1621, 1505, 1473, 1331, 1218, 1029, 990, 917, 831, 716, 551, 507, 452 cm−1. HRMS (ESI): m/z calculated for C27H30N4OSF [M+H]+: 477.2124, found: 477.2120.
4.7. General Procedure for the Michael Addition of Nitromethane to Trans-Chalcones
Trans-chalcone 13 (0.104 g, 0.50 mmol, 1.00 equivalent and the respective catalyst (10.0 mol%) were loaded to an ampoule. Next, toluene (0.30 mL) was added and the mixture was stirred for 10 min under argon. Then, nitromethane (91.6 mg, 80.5 μL, 1.50 mmol, 3.00 equivalents) was added by syringe and the reaction was further stirred for 96 h at room temperature. The reaction mixture was directly purified by chromatography (silica gel, hexane/EtOAc 3:1) to afford respective Michael adduct 14. The enantiomeric excess was determined by HPLC (Chiralcel AD-H column, hexane/i-PrOH 9:1, flow rate 1.0 mL/min, λ = 254 nm); t(S) = 14.7 min, t(R) = 20.5 min. The spectral and HPLC data (Supplementary Materials, Figures S21–S29) were in agreement with the reported values [43].
4.8. General Procedure for the Sulfa-Michael Addition of Thioacetic Acid to Trans-Chalcone
A reaction tube was loaded with the respective catalyst (10.0 mol%), trans-chalcone 13 (52.1 mg, 0.25 mmol, 1.00 equivalent), and diethyl ether (1.25 mL) under argon. After 10 min stirring at rt, thioacetic acid (38.1 mg, 35.2 μL 0.50 mmol, 2.00 equivalents) was added by syringe, and the stirring was continued for 4 h at rt. The reaction mixture was directly purified by chromatography (silica gel, hexane/EtOAc 3:1) to afford respective Michael adduct 15. Enantiomeric excess values were determined by chiral HPLC (Chiralcel AS-H column, hexane/i-PrOH 9:1, flow rate 1.0 mL/min, λ = 254 nm); t(R) = 9.7 min, t(S) = 15.0 min. The spectral and HPLC data (Supplementary Materials, Figures S30–S38) were in agreement with the reported values [44].
Supplementary Materials
The following are available online at https://www.mdpi.com/1996-1944/14/3/600/s1. Figure S1. 1H and 13C NMR spectra of N-[(8S,9S)-6′-methoxycinchonan-9-yl]-N′-phenylselenourea eQN-7a, Figure S2. IR spectra of N-[(8S,9S)-6′-methoxycinchonan-9-yl]-N′-phenylselenourea eQN-7a, Figure S3. 1H and 13C NMR spectra of N-[(8S,9S)-6′-methoxycinchonan-9-yl]-N′-[4-methoxyphenyl]selenourea eQN-7b, Figure S4. IR spectra of N-[(8S,9S)-6′-methoxycinchonan-9-yl]-N′-[4-methoxyphenyl]selenourea eQN-7b, Figure S5. 1H and 13C NMR spectra of N-[4-fluorophenyl]-N′-[(8S,9S)-6′-methoxycinchonan-9-yl]selenourea eQN-7c, Figure S6. IR spectra of N-[4-fluorophenyl]-N′-[(8S,9S)-6′-methoxycinchonan-9-yl]selenourea eQN-7c, Figure S7. 1H and 13C NMR spectra of N-[4-fluorophenyl]-N′-[(8S,9S)-cinchonan-9-yl]selenourea eCD-7d, Figure S8. IR spectra of N-[4-fluorophenyl]-N′-[(8S,9S)-cinchonan-9-yl]selenourea eCD-7d, Figure S9. 1H and 13C NMR spectra of N-[4-fluorophenyl]-N′-[(8S,9S)-10,11-dihydro-6′-methoxycinchonan -9-yl]selenourea eDHQN-7e, Figure S10. IR spectra of N-[4-fluorophenyl]-N′-[(8S,9S)-10,11-dihydro-6′-methoxycinchonan-9-yl]selenourea eDHQN-7e, Figure S11. 1H and 13C NMR spectra of N-[4-fluorophenyl]-N′-[(8R,9R)-6′-methoxycinchonan -9-yl]selenourea eQD-7f, Figure S12. IR spectra of N-[4-fluorophenyl]-N′-[(8R,9R)-6′- methoxycinchonan-9-yl]selenourea eQD-7f, Figure S13. 1H and 13C NMR spectra of N-[4-fluorophenyl]-N′-[(8R,9R)-10,11-dihydro-6′-methoxycinchonan-9-yl]selenourea eDHQD-7g, Figure S14. IR spectra of N-[4-fluorophenyl]-N′-[(8R,9R)-10,11-dihydro-6′-methoxycinchonan-9-yl]selenourea eDHQD-7g, Figure S15. 1H and 13C NMR spectra of N-[(8S,9S)-6′-methoxycinchonan-9-yl]formamide 8a, Figure S16. IR spectra of N-[(8S,9S)-6′-methoxycinchonan-9-yl]formamide 8a, Figure S17. 1H NMR spectra of N,N′-bis[(8S,9S)-6′-metoxycinchonan-9-yl]selenourea 10a, Figure S18. IR spectra of N,N′-bis[(8S,9S)-6′- metoxycinchonan-9-yl]selenourea 10a, Figure S19. 1H and 13C NMR spectra of N-[4-fluorophenyl]-N′-[(8S,9S)-6′-methoxycinchonan-9-yl]thiourea eQN-12a, Figure S20. IR spectra of N-[4-fluorophenyl]-N′-[(8S,9S)-6′-methoxycinchonan-9-yl]thiourea eQN-12a, Figure S21. HPLC chromatogram for 14: sample obtained with catalyst eQN-7a, Figure S22. HPLC chromatogram for 14: sample obtained with catalyst eQN-7b, Figure S23. HPLC chromatogram for 14: sample obtained with catalyst eQN-7c, Figure S24. HPLC chromatogram for 14: sample obtained with catalyst eCD-7d, Figure S25. HPLC chromatogram for 14: sample obtained with catalyst eDHQN-7e, Figure S26. HPLC chromatogram for 14: sample obtained with catalyst eQD-7f, Figure S27. HPLC chromatogram for 14: sample obtained with catalyst eDHQD-7g, Figure S28. HPLC chromatogram for 14: sample obtained with catalyst eQN-12a, Figure S29. HPLC chromatogram for racemic 14 without catalyst 7, Figure S30. HPLC chromatogram for 15: sample obtained with catalyst eQN-7a, Figure S31. HPLC chromatogram for 15: sample obtained with catalyst eQN-7b, Figure S32. HPLC chromatogram for 15: sample obtained with catalyst eQN-7c, Figure S33. HPLC chromatogram for 15: sample obtained with catalyst eCD-7d, Figure S34. HPLC chromatogram for 15: sample obtained with catalyst eDHQN-7e, Figure S35. HPLC chromatogram for 15: sample obtained with catalyst eQD-7f, Figure S36. HPLC chromatogram for 15: sample obtained with catalyst eDHQD-7g, Figure S37. HPLC chromatogram for 15: sample obtained with catalyst eQN-12a, Figure S38. HPLC chromatogram for racemic 15 without catalyst 7.
Author Contributions
Conceptualization, M.Z.-B.; methodology, M.Z.-B. and J.N.; investigation, J.N. and M.Z.-B.; data curation, M.Z.-B. and J.N.; writing—original draft preparation, M.Z.-B.; writing—review and editing, M.Z.-B. and J.N.; supervision, M.Z.-B.; project administration, M.Z.-B.; funding acquisition, M.Z.-B. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data are available on request from the corresponding author.
Acknowledgments
Authors are grateful for the financial support given by the Polish Ministry of Science and Higher Education by subvention activity for the Faculty of Chemistry at Wrocław University of Science and Technology.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bondi, A. Van der Waals volumes and radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Bharatam, P.V.; Moudgil, R.; Kaur, D. Electron delocalization in isocyanates, formamides, and ureas: Importance of orbital interactions. J. Phys. Chem. A 2003, 107, 1627–1634. [Google Scholar] [CrossRef]
- Bibelayi, D.; Lundemba, A.S.; Allen, F.H.; Galek, P.T.A.; Pradon, J.; Reilly, A.M.; Groomb, C.R.; Yav, Z.G. Hydrogen bonding at C=Se acceptors in selenoureas,selenoamides and selones. Acta Cryst. 2016, B72, 317–325. [Google Scholar] [CrossRef]
- Merino-Montiel, P.; Maza, S.; Martos, S.; López, Ó.; Maya, I.; Fernández-Bolaños, J.G. Synthesis and antioxidant activity of O-alkyl selenocarbamates, selenoureas and selenohydantoins. Eur. J. Pharm. Sci. 2013, 48, 582–592. [Google Scholar] [CrossRef]
- Ruberte, A.C.; Ramos-Inza, S.; Aydillo, C.; Talavera, I.; Encío, I.; Plano, D.; Sanmartín, C. Novel N,N′-disubstituted acylselenoureas as potential antioxidant and cytotoxic agents. Antioxidants 2020, 9, 55. [Google Scholar] [CrossRef]
- Hussain, R.A.; Badshah, A.; Tahir, M.N.; Hassan, T.U.; Bano, A. Synthesis, chemical characterization, DNA binding, antioxidant, antibacterial, and antifungal activities of ferrocence incorporated selenoureas. J. Biochem. Mol. Toxicol. 2014, 28, 60–68. [Google Scholar] [CrossRef]
- Díaz, M.; de Lucio, H.; Moreno, E.; Espuelas, S.; Aydillo, C.; Jiménez-Ruiz, A.; Toro, M.A.; Gutiérrez, K.J.; Martínez-Merino, V.; Cornejo, A.; et al. Novel urea, thiourea and selenourea derivatives of diselenides: Synthesis and leishmanicidal activity. Antimicrob. Agents Chemoth. 2019, 63, e02200-18. [Google Scholar] [CrossRef]
- Zakrzewski, J.; Krawczyk, M. Synthesis and pesticidal properties of thio and seleno analogs. Phosphorus Sulfur Silicon Relat. Elem. 2009, 184, 1880–1903. [Google Scholar] [CrossRef]
- Sivapriya, K.; Suguna, P.; Banerjee, A.; Saravanan, V.; Rao, D.N.; Chandrasekaran, S. Facile one-pot synthesis of thio and selenourea derivatives: A new class of potent urease inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 6387–6391. [Google Scholar] [CrossRef]
- Hussain, R.A.; Badshah, A.; Shah, A. Synthesis and biological applications of selenoureas. Appl. Organomet. Chem. 2014, 28, 61–73. [Google Scholar] [CrossRef]
- Alcolea, V.; Plano, D.; Karelia, D.N.; Palop, J.A.; Amin, S.; Sanmartín, C.; Sharma, A.K. Novel seleno- and thio-urea derivatives with potent in vitro activities against several cancer cell lines. Eur. J. Med. Chem. 2016, 113, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Koketsu, M.; Ishihara, H. Synthesis of 1,3-selenazine and 1,3-selenazole and their biological activities. Curr. Org. Chem. 2003, 7, 175–185. [Google Scholar] [CrossRef]
- Ninomiya, M.; Garud, D.R.; Koketsu, M. Selenium-containing heterocycles using selenoamides, selenoureas, selenazadines, and isoselenocyanates. Heterocycles 2010, 81, 2027–2055. [Google Scholar] [CrossRef]
- Casula, A.; Llopis-Lorente, A.; Garau, A.; Isaia, F.; Kubicki, M.; Lippolis, V.; Sancenon, F.; Martinez-Manez, R.; Owczarzak, A.; Santi, C.; et al. A new class of silica-supported chromo-fluorogenic chemosensors for anion recognition based on a selenourea scaffold. Chem. Commun. 2017, 53, 3729–3732. [Google Scholar] [CrossRef] [PubMed]
- Picci, G.; Mocci, R.; Ciancaleoni, G.; Lippolis, V.; Zielińska-Błajet, M.; Caltagirone, C. Bis-selenoureas for anion binding: A 1H NMR and theoretical study. ChemPlusChem. 2020, 85, 1389–1395. [Google Scholar] [CrossRef]
- López, Ó.; Maza, S.; Ulgar, V.; Maya, I.; Fernández-Bolaños, J.G. Synthesis of sugar-derived isoselenocyanates, selenoureas, and selenazoles. Tetrahedron 2009, 65, 2556–2566. [Google Scholar] [CrossRef]
- Chennakrishnareddy, G.; Nagendra, G.; Hemantha, H.P.; Das, U.; Guru Row, T.N.; Sureshbabu, V.; Chennakrishnareddy, G.; Nagendra, G.; Hemantha, H.P.; Das, U.; et al. Isoselenocyanates derived from Boc/Z-amino acids: Synthesis, isolation, characterization, and application to the efficient synthesis of unsymmetrical selenoureas and selenoureidopeptidomimetics. Tetrahedron 2010, 66, 6718–6724. [Google Scholar] [CrossRef]
- Bian, G.; Yang, S.; Huang, H.; Zong, H.; Song, L.; Fan, H.; Sun, X. Chirality sensing of tertiary alcohols by a novel strong hydrogen-bonding donor—selenourea. Chem. Sci. 2016, 7, 932–938. [Google Scholar] [CrossRef]
- Jörres, M.; Schiffers, I.; Atodiresei, I.; Bolm, C. Asymmetric Michael additions of α–nitrocyclohexanone to aryl nitroalkenes catalyzed by natural amino acid-derived bifunctional thioureas. Org. Lett. 2012, 14, 4518–4521. [Google Scholar] [CrossRef]
- Connon, S.J. Asymmetric catalysis with bifunctional cinchona alkaloid-based urea and thiourea organocatalysts. Chem. Commun. 2008, 22, 2499–2510. [Google Scholar] [CrossRef]
- Lu, L.Q.; An, L.Q.; Chen, J.R.; Xiao, W.J. Dual activation in organocatalysis: Design of tunable and bifunctional organocatalysts and their applications in enantioselective reactions. Synlett 2012, 4, 490–508. [Google Scholar] [CrossRef]
- Serdyuk, O.V.; Heckel, C.M.; Tsogoeva, S. Bifunctional primary amine-thioureas in asymmetric organocatalysis. Org. Biomol. Chem. 2013, 11, 7051–7071. [Google Scholar] [CrossRef] [PubMed]
- Parvin, T.; Yadava, R.; Choudhury, L.H. Recent applications of thiourea-based organocatalysts in asymmetric multicomponent reactions (AMCRs). Org. Biomol. Chem. 2020, 18, 5513–5532. [Google Scholar] [CrossRef] [PubMed]
- Steppeler, F.; Iwan, D.; Wojaczyńska, E.; Wojaczyński, J. Chiral thioureas–preparation and significance in asymmetric synthesis and medicinal chemistry. Molecules 2020, 25, 401. [Google Scholar] [CrossRef]
- Song, C.E. (Ed.) Cinchona Alkaloids in Synthesis and Catalysis: Ligands, Immobilization and Organocatalysis; Wiley: Weinheim, Germany, 2009. [Google Scholar]
- Ishihara, H.; Koketsu, M.; Fukuta, Y.; Nada, F. Reaction of lithium aluminum hydride with elemental selenium: its application as a selenating reagent into organic molecules. J. Am. Chem. Soc. 2001, 123, 8408–8409. [Google Scholar] [CrossRef]
- Zhou, Y.; Denk, M.K. Synthesis and reactivity of subvalent compounds. Part 13: Reaction of triethyl orthoformate with amines and selenium—a convenient one-step three-component synthesis for selenoureas. Tetrahedron Lett. 2003, 44, 1295–1299. [Google Scholar] [CrossRef]
- Koketsu, M.; Takakura, N.; Ishihara, H. Efficient synthesis of selenoureas from the corresponding carbodiimides. Synth Commun. 2002, 32, 3075–3079. [Google Scholar] [CrossRef]
- Takikawa, Y.; Watanabe, H.; Sasaki, R.; Shimada, K. Conversion of amides, N,N,N′,N′-tetramethylurea, and esters to the corresponding selenoxo compounds by treatment with bis(trimethylsilyl) selenide and BF3·OEt2. Bull. Chem. Soc. Jpn. 1994, 67, 876–878. [Google Scholar] [CrossRef]
- Fernández-Bolaños, J.G.; López, Ó.; Ulgar, V.; Maya, I.; Fuentes, J. Synthesis of O-unprotected glycosyl selenoureas. A new acces to bicyclic sugar isoureas. Tetrahedron Lett. 2004, 45, 4081–4084. [Google Scholar] [CrossRef]
- Zakrzewski, J.; Huras, B.; Kiełczewska, A. Synthesis of isoselenocyanates. Synthesis 2016, 48, 85–96. [Google Scholar] [CrossRef]
- Zielińska-Błajet, M.; Kucharska, M.; Skarżewski, J. Simple preparation of enantiomeric Michael adducts of thiophenol to chalcones: Easily available new chiral building blocks. Synthesis 2006, 7, 1176–1182. [Google Scholar] [CrossRef]
- Kacprzak, K.; Gierczyk, B. Clickable 9-azido-(9-deoxy)-Cinchona alkaloids: Synthesis and conformation. Tetrahedron Asymmetry 2010, 21, 2740–2745. [Google Scholar] [CrossRef]
- Brunner, H.J.; Bügler, H.J.; Nuber, B. Preparation of 9-amino(9-deoxy)cinchona alkaloids. Tetrahedron Asymmetry 1995, 6, 1699–1702. [Google Scholar] [CrossRef]
- Sharma, S.; Maurya, R.A.; Min, K.I.; Jeong, G.Y.; Kim, D.P. Odorless isocyanide chemistry: An integrated microfluidic system for a multistep reaction sequence. Angew. Chem. Int. Ed. Engl. 2013, 52, 7564–7568. [Google Scholar] [CrossRef] [PubMed]
- Walborsky, H.M.; Niznik, G.E. Synthesis of isonitriles. J. Org. Chem. 1972, 37, 187–190. [Google Scholar] [CrossRef]
- Hofmann, A.W. Ueber eine neue reihe von homologen der cyanwasserstoffsäure. Liebigs Ann. Chem. 1868, 146, 107–119. [Google Scholar] [CrossRef]
- Mąkosza, M.; Wawrzyniewicz, M. Reactions of organic anions. XXIV. Catalytic method for preparation of dichlorocyclopropane derivatives in aqueous medium. Tetrahedron Lett. 1969, 10, 4659–4662. [Google Scholar] [CrossRef]
- Vakulya, B.; Varga, S.; Csámpai, A.; Soós, T. Highly enantioselective conjugate addition of nitromethane to chalcones using bifunctional Cinchona organocatalysts. Org. Lett. 2005, 7, 1967–1969. [Google Scholar] [CrossRef]
- Li, X.; Deng, H.; Zhang, B.; Li, J.; Zhang, L.; Luo, S.; Cheng, J.P. Physical organic study of structure–activity– enantioselectivity relationships in asymmetric bifunctional thiourea catalysis: Hints for the design of new organocatalysts. Chem. Eur. J. 2010, 16, 450–455. [Google Scholar] [CrossRef]
- Jakab, G.; Tancon, C.; Zhang, Z.; Lippert, K.M.; Schreiner, P.R. (Thio)urea organocatalyst equilibrium acidities in DMSO. Org. Lett. 2012, 14, 1724–1727. [Google Scholar] [CrossRef]
- Zhang, Z.; Bao, Z.; Xing, H. N,N′-Bis[3,5-bis(trifluoromethyl)phenyl]thiourea: A privileged motif for catalyst development. Org. Biomol. Chem. 2014, 12, 3151–3162. [Google Scholar] [CrossRef] [PubMed]
- Corey, E.J.; Zhang, F.Y. Enantioselective Michael addition of nitromethane to alpha, beta-enones catalyzed by chiral quaternary ammonium salts. A simple synthesis of (R)-baclofen. Org. Lett. 2000, 2, 4257–4259. [Google Scholar] [CrossRef]
- Manzano, R.; Andrés, J.M.; Álvarez, R.; Muruzábal, M.D.; de Lera, Á.R.; Pedrosa, R. Enantioselective conjugate addition of nitro compounds to α,β-unsaturated ketones: An experimental and computational study. Chem. Eur. J. 2011, 17, 5931–5938. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zu, L.; Wang, J.; Wang, W. Organocatalytic enantioselective Michael addition of thioacetic acid to enones. Tetrahedron Lett. 2006, 47, 3145–3148. [Google Scholar] [CrossRef]
- Barton, D.H.R.; Parekh, S.I.; Tajbakhsh, M.; Theodorakis, E.A.; Chi-Lam, T. A convenient and high yielding procedure for the preparation of isoselenocyanates. Synthesis and reactivity of O-alkylselenocarbamates. Tetrahedron 1994, 50, 639–654. [Google Scholar] [CrossRef]
- Perez-Labrada, K.; Brouard, I.; . Mendez, I.; Rivera, D.G. Multicomponent synthesis of Ugi-type ceramide analogues and neoglycolipids from lipidic isocyanides. J. Org. Chem. 2012, 77, 4660–4670. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).