Abstract
The first-principles methods, based on the density function theory, are performed to calculate the properties of pure and doped Al3Sc. The structural stability, and mechanical and electronic properties of L12-Al3Sc1−xMx (M = Zr, Ti, Y, and Li) have been investigated. A negative formation enthalpy for L12-Al3Sc1–xMx indicated that all doped structures were stable, and Al24Sc6Zr2 was found to be the most stable. The elastic constants, elastic moduli and Debye temperatures of Al3Sc, with different doping elements and different doping concentrations, were calculated to explore the influences of doping on the mechanical properties and Debye temperatures of Al3Sc. Furthermore, the calculated results suggested that both Al24Sc6Zr2 and Al24Sc6Ti2 could optimize the mechanical properties. Finally, the electronic properties based on the analyses of densities of states and electron density distributions, have been performed, to explain the underlying mechanisms for the structural and mechanical properties of the L12-Al3Sc1–xMx structures.
1. Introduction
In the past decades, Al-Sc alloys have attracted great attention, due to higher strength and stiffness, compared with pure Al [1,2]. The addition of Sc can greatly improve the mechanical properties of Al alloy, since a large number of nanoparticles (Al3Sc) are formed in the aging process [3,4,5], and Al3Sc with a cubic L12 structure has a small lattice mismatch with α-Al. However, the industrial application of Al3Sc has been limited because of the high cost of Sc, and the low solubility of Sc in Al. As a result, it is necessary to find an element to replace Sc, which can improve the mechanical properties of the alloy and reduce its cost.
In recent years, researcher has shown that doping has a certain impact on the structural characteristics and mechanical properties of alloys, through experimental investigations [6,7,8,9] and theoretical calculations [10,11,12]. The behaviors and properties of the alloying elements in Al3Sc, have been studied by experiments. For example, Fuller et al. studied the replacement of Sc by Zr in Al-Sc alloys, and found that the coarsening resistance was increased at higher temperatures [13]. Dalen et al. studied the effects of Ti additions on the structural and creep properties of Al-Sc alloys [14]. Seidman et al. indicated that the addition of Li has resulted in an increased driving force for precipitate nucleation [15]. Harada et al. compared the thermal expansion and creep properties of Al3Sc and Al3(Sc, Y) [16,17]. Up to now, the microstructure, compression, fracture behavior, elastic and optical properties of Al3Sc have been widely researched [18,19] by the density functional theory(DFT) method. Furthermore, the structural, electronic, mechanical, and thermodynamic properties of Al3Sc, under different pressures and temperatures, have been calculated [20,21,22]. Moreover, the effect of transition metals on the structural stability of Al3Sc1-xMx was studied by the special quasi random structures method [23]. In addition, the properties of Al3Sc1−xMx (M=Zr, Ti) with different concentrates, were performed [24,25]. Nevertheless, the influences of the doping elements on the mechanical and electronic properties of the Al-Sc-M system have required further investigations.
In this work, we calculated the structural stability and mechanical properties of the L12-Al3Sc1–xMx (M = Zr, Ti, Y, and Li) structures with concentrations of 3.125 at.% and 6.25 at.%. Second, the mechanical properties and electronic properties of the L12-Al3Sc1–xTMx structures have been intensively discussed in relation to their electronic properties. This investigation could provide theoretical guidance to the application of Al-Sc-based alloys.
2. Materials and Methods
All calculations were performed on the basis of the density functional theory (DFT) with the Vienna Ab Initio Simulation Package (VASP) [26]. The pseudopotential in the reciprocal space was described by the projector-augmented wave (PAW) method [27]. The generalized gradient approximation (GGA), with the Perdew-Burke-Ernzerhof (PBE) [28] function was applied to describe the exchange-correlation potential. Both the k-space integral and plane-wave basis were chosen to ensure that the total energy was converged. The convergence criterion for the self-consistent field energy was set to be 5 × 10−6 eV/atom. For the plane wave expansion, a kinetic cutoff energy of 500 eV was considered to be sufficient. The geometry optimization was terminated when the Hellman–Feynman force on each atom was smaller than the 0.003 eV/nm. The integral in the Brillouin zone was sampled by the Monkhorst-Pack method [29], with the k-point mesh of 21 × 21 × 21 for Al3Sc and 11 × 11 × 11 for the 2 × 2 × 2 supercells. All calculations were carried out with the potentials for Al(3s23p1), Li(1s22s1), Sc(3s23p63d14s2), Ti(3s23p63d24s2), Zr(4s24p64d25s2), and Y(4s24p64d15s2), as the valence electrons. Overall, all calculations were operated in 0 K, with the equivalent hydrostatic pressure.
3. Results
3.1. Structural Stability
The Al3Sc phase had a cubic structure (Space Group: Pm3m (No. 221)), which contained 3 Al atoms and 1 Sc atom, in a unit cell. Based on the Al3Sc phase, a 2 × 2 × 2 supercell (Figure 1a) was constructed to investigate the effects of the doped element (M = Zr, Ti, Y, and Li). In addition, the effects of the doping concentrations (at.%) were also considered, which included 3.125% (Figure 1b) and 6.25% (Figure 1c). The structures of the surpercells used in this work are shown in Figure 1, and the optimized structural parameters are listed in Table 1.
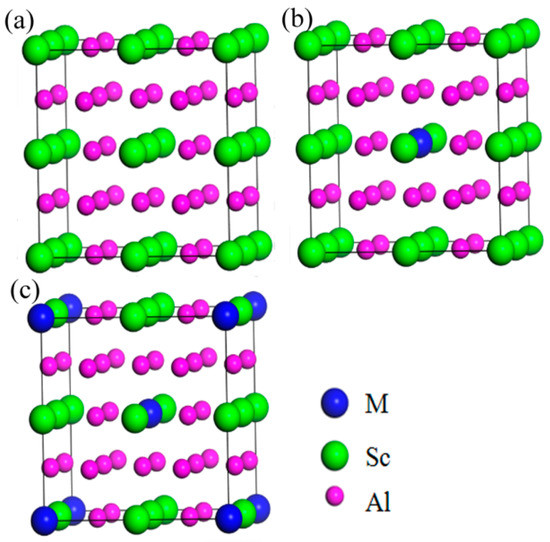
Figure 1.
Structures of Al3Sc (a) 2 × 2 × 2 surpercell; doped with the alloying element (M = Sc, Zr, Ti, Y, Li) at different doping concentrations, (b) 3.125%, and (c) 6.25%. Blue, green, and pink balls represent (M = Zr/Ti/Y/Li), Sc, and Al atoms, accordingly.

Table 1.
The crystal parameters (Å), density (ρ/g·cm−3), and heat of formation (ΔHf/eV·atom−1) of L12-Al3Sc1–xMx at the ground state.
It is well-known that a lower formation energy implies a more stable structure. In order to study the influence of the doped elements on the structural stability, the enthalpies of formation (ΔHf) of the Al3Sc structure, before and after doping, were calculated with the following formula:
where Etotal is the total energy of the doped structure; n stands for the total number of atoms in the Al-Sc-M system; Ei and Ni are the energies per atom of species i, and the corresponding number of atoms in the doped structure. The energies per atom of Al, Sc, and M (M = Zr/Ti/Y/Li) were calculated from bulk Al with an FCC structure, a bulk Sc and M (M = Zr/Ti/Y) with an HCP structure, and a bulk M (M = Li) with a BCC structure.
The calculation results of ΔHf are listed in Table 1. The negative ΔHf indicates that the doped structure could stably form at 0 K; all structures were stable. However, the ΔHf values of the doped structures were greater than that of pure Al3Sc, except for the Al-Sc-Zr system. This meant that Zr doping could improve stability, while the Ti/Y/Li dopings might reduce the structural stability. This was consistent with results from previous studies [23,30]. In addition, it was found that the stability of Al24Sc6Zr2 was higher in comparison to Al24Sc7Zr. Except for the Zr addition, the higher concentration dopings should have caused a lower stability of the structure.
3.2. Elastic Properties
The elastic properties could provide necessary information on the resistance of the material to extrinsically applied stress. For the cubic crystals, there were three independent elastic constants (i.e., C11, C12, and C44). The calculated results of the elastic constants are listed in Table 2.
Table 2.
Calculated elastic constants Cij for L12-Al3Sc1–xMx at the ground state.
Notably, the results of Al3Sc were close to the published DFT calculations [15] and experimental measurements [31], implying the reliability of the calculation results. The criterions for the mechanical stability of the cubic crystal [32] were estimated using Equation (2):
The calculated results showed that the elastic constants of all structures could satisfy the above stability conditions, indicating that all structures had mechanical stability.
The calculated elastic constants as a function of doping concentration for Al3Sc are exhibited in Figure 2. The C11 values of Al3Sc, before and after doping, were larger than the other elastic constants (Figure 2a), suggesting that the axes compression resistances were stronger. The C11 values in the Al24Sc7M system were obviously decreased, which proved that the axes pressure resistances were reduced. However, Al24Sc6M2 have found to be quite complex. For example, Al24Sc6Y2 and Al24Sc6Li2 have smaller C11 values, Al24Sc6Zr2 had a similar C11 value, and Al24Sc6Ti2 possessed a larger C11 value. On the other hand, both Al24Sc7M and Al24Sc6M2 exhibited higher C12 values (Figure 2b), suggesting that the Poisson effect was enhanced [25]. Additionally, the Al-Sc-M systems had shown a slightly reduced tendency for C44 (Figure 2c), especially for the Al24Sc6M2, indicating that the resistance to shear deformation was gently diminished.
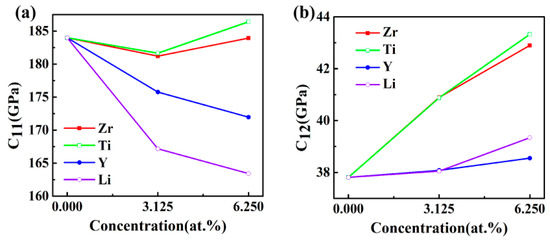
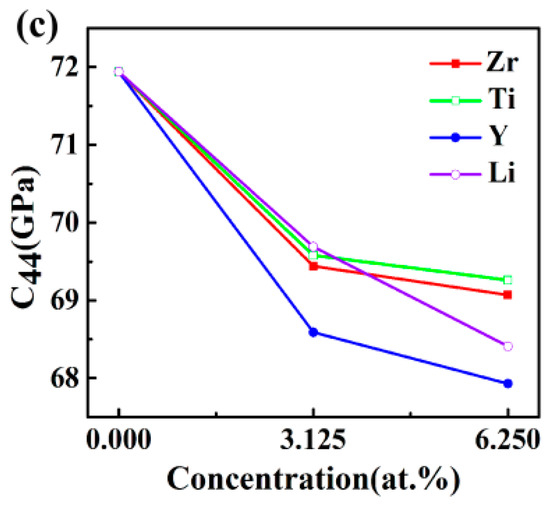
Figure 2.
The elastic constants Cij (GPa) of L12-Al3Sc doped with the element M (M = Zr, Ti, Y, and Li) on the dependence of doping concentration: (a) C11, (b) C12, and (c) C44.
The polycrystalline elastic moduli are the important performance parameters of engineering materials, such as bulk modulus (B), shear modulus (G), Young’s modulus (E), and Poisson’s ratio (ν). The bulk modulus and shear modulus were determined using the Voigt-Reuss-Hill method [33]. For the cubic structure, the bulk modulus (B) and shear modulus (G) were calculated from Equations (3) and (4), respectively:
Young’s modulus (E) and Poisson’s ratio (ν) were determined by B and G, and their expressions are shown in Equations (5) and (6) [34]:
The results of the elastic moduli are listed in Table 3. It is well-known that B is a measure of the degree to which a material deforms under hydrostatic pressure [35]. G indicates the material’s resistance to shear strain [35]. E is a representation of the stiffness of the material [36].
Table 3.
Elastic moduli B, G, E, ν, B/G, H, A and ΘD for L12-Al3Sc1–xMx, at the ground state.
The influences of doped elements at different concentrations on the elastic modulus of Al3Sc are displayed in Figure 3a–c. The values of B doped with Zr and Ti increased with a growing doping concentration, while the values of B doped with Y and Li decreased with an increasing doping concentration. When the doping concentration was constant, Ti doped Al-Sc-M compounds had the maximum B values, implying that the addition of Ti into Al3Sc resulted in minimal deformation, under certain external pressure.
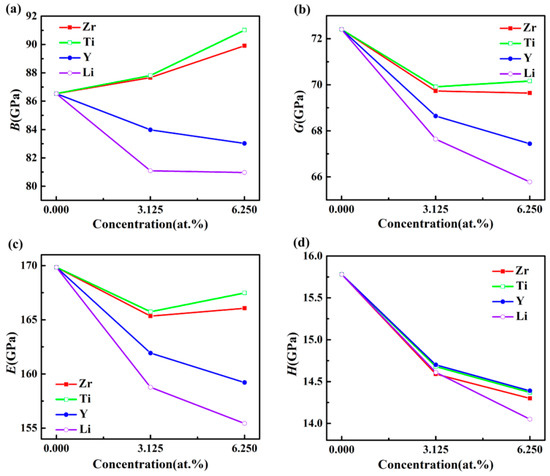
Figure 3.
The elastic moduli B (a), G (b), E (c), and hardness H (GPa), (d) of L12–Al3Sc doped with element M (M = Zr, Ti, Y, and Li), as a function of the doping concentration.
Furthermore, the doped Al-Sc-M compounds displayed lower G and E, meaning that the shear strain resistance and stiffness were reduced. In addition, the E values of Al3Sc doped with Zr and Ti were increased, when the doping concentrations increased from 3.125% to 6.25%, indicating that the higher concentration doping showed a better performance for the Al-Sc-M (M = Zr/Ti).
The investigation of the stiffness could be completed by providing the microhardness parameter (H), given by the following relation [37]:
Figure 3d exhibits the H values of Al3Sc with different doping elements and concentrations. It is clearly observed that the H values of the doped Al3Sc decreased with an increasing concentration. For example, Al24Sc6Ti2 presented the maximum H value. Although the H values of the doped Al3Sc were decreased, the reduced magnitude was small, compared with that of Al3Sc. Considering the cost of Sc, the doped Al3Sc was more valuable for industrial application.
The B/G ratio was used to distinguish the ductility and brittleness of compounds [38]. The greater value of B/G corresponded to a better ductility in the material. The results shown in Figure 4a indicate that the L12-Al3Sc structure displayed a higher ductility, after doping. This conclusion could be proved by the Cauchy pressure (C12−C44) [39]. Furthermore, the B/G values for the doped Al3Sc increased with the elevated doping concentration, which demonstrated that a higher doping concentration enhances the ductility of Al3Sc. In addition, Poisson’s ratio (v) reflected the transverse deformation for the material [40]. It could be clearly observed that the changing trends of ν with doping elements and concentrations were similar to that of B/G (Figure 4). It is common knowledge that a material exhibits better ductility, when the Poisson’s ratio is large. The calculated results demonstrated that a better ductility of Al3Sc doped with Zr/Ti was exhibited, compared to Y/Li.
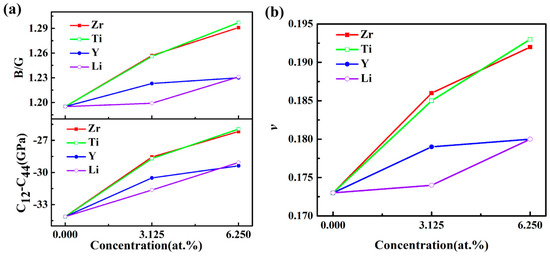
Figure 4.
(a) B/G and (C12 – C44) and (b) ν of L12-Al3Sc doped with element M (M = Zr, Ti, Y, and Li) as a function of the doping concentration.
The elastic anisotropy has an important implication in engineering science, since it is highly correlated with the possibility to induce micro cracks in materials [41]. The expression of elastic anisotropy (A) is shown below:
It is noted that the material is isotropic when A is equal to 1. The degree to which the value of A deviates from 1 represents the strength of the anisotropy of the material. The A values of Al-Sc-M (M = Zr/Ti/Y/Li) were calculated, and the results are listed in Table 3. The results showed that Al3Sc had exhibited anisotropic behavior, before and after doping. Moreover, the A values for the doped Al3Sc were more deviated from 1, when the doping concentrations varied from 3.125% to 6.25%. It is worth noting that the deviation degree of Al-Sc-Li system was the largest. For example, Al24Sc6Li2 possessed the strongest anisotropy.
3.3. Debye Temperature
Debye temperature (ΘD) is a fundamental parameter for the material’s thermodynamic properties. It is correlated with many physical properties (i.e., specific heat, elastic constant, and melting temperature). The ΘD value of a solid can usually be calculated from the sound velocity. ΘD is related to the modulus of elasticity in Anderson’s model [42]. Then, ΘD is defined as [43]:
where h, k, NA, n, ρ, M, , , and denote Planck’s, Boltzmann’s and Avogadro’s constants, total number of atoms, density, molecular weight, average sound velocity, transverse sound velocity, and longitudinal sound velocity, respectively.
In the Debye theory, ΘD is the temperature of a crystal’s highest normal mode of vibration. That is, the highest temperature can be achieved due to a single normal vibration. It is well-known that a higher ΘD corresponds to a better thermal conductivity of a material. Figure 5 describes ΘD of Al3Sc, at different doping elements and concentrations. The ΘD values of the Al-Sc-Li structure increased with an increasing concentration. On the contrary, the ΘD values of Al-Sc-M (M = Zr/Y) decrease with the increasing concentration. For the Al-Sc-Ti structure, the ΘD values were a little higher than Al3Sc. However, the ΘD had decreased slightly, when the concentration increased from 3.125% to 6.25%. The higher ΘD values of the Al-Sc-Li structure indicated that their thermal conductivities were better, compared to other structures.
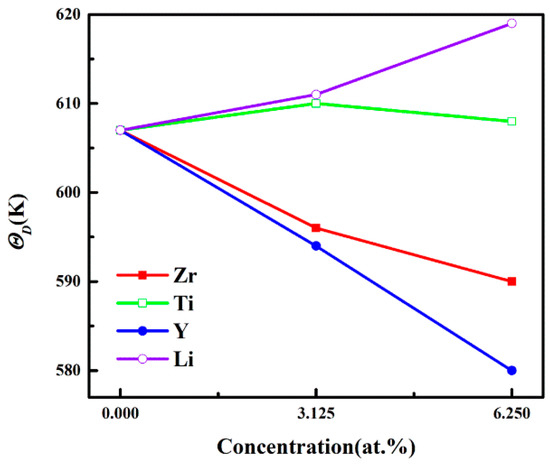
Figure 5.
The ΘD values of L12-Al3Sc doped with element M (M = Zr, Ti, Y, and Li) as a function of the doping concentration.
Based on the above calculation results, it was noticeable that the species and concentrations of the doped elements had intensive impacts on the mechanical properties of Al3Sc. In order to obtain high performance compounds, it was important to select appropriate doped element and concentration. The mechanical properties of Al3Sc before and after doping were intensively compared. It was demonstrated that doping Ti/Zr could better optimize the performances of the Al3Sc structure. This conclusion was consistent with previous reports [14,17,23]. Furthermore, the mechanical properties of Al-Sc-Ti were slightly better than that of Al-Sc-Zr. This discrepancy with the result, measured through experiments, might have arisen from a different temperature. It is worth noting that the experimental measurement was generally operated at 573 K, whereas, this work was completed at 0 K. To be more important, the structure with a higher doping concentration (6.25%) had a higher performance over the lower doping concentration (3.125%).
3.4. Electronic Properties
To gain a better understanding of the doping effects at the electronic level, the total densities of states (TDOS) and partial densities of states (PDOS) were calculated in this work. The calculated TDOS are shown in Figure 6. The Fermi energy level (Ef) represented by a dotted line was set to zero. It could be clearly seen that the TDOS of the structure was not zero at the Fermi level, indicating that the structures had good metallic properties [2]. Meanwhile, the TDOS of Al24Sc6M2 (M = Zr/Ti/Y/Li) were expensed in the energy scales.
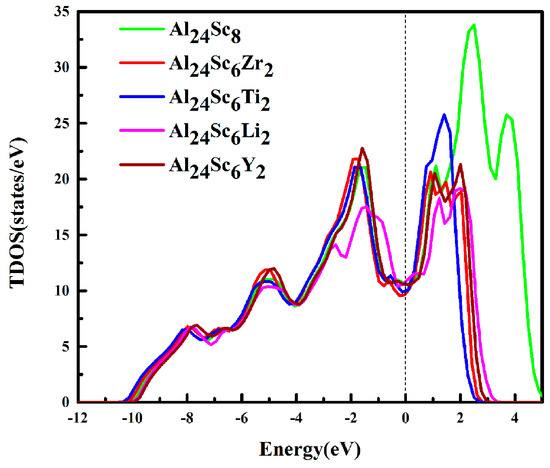
Figure 6.
Total density of states (TDOSs) of the Al3Sc and Al24Sc6M2 (M = Sc, Zr, Ti, Li and Y) alloys.
The structures of Al3Sc, before and after doping, had a very wide pseudogap around Ef, implying that Al-Sc-M intermetallic compounds had strong covalent bonds, indicating that it had a good stability [44]. Moreover, the widths of the pseudogap for Al3Sc and Al-Sc-M (M = Zr, Ti, Y, and Li) structures were 2.65 eV, 2.74 eV, 2.53 eV, 2.59 eV, and 2.23 eV, respectively. It was indeed seen that Al24Sc6Zr2 was slightly wider than the pseudogap of Al3Sc, while that of Al24Sc6M2 (M = Ti/Y/Li) was slightly narrower. This proved that Al24Sc6Zr2 had a stronger covalent bond, which was the most stable [13,14,23]. However, the stability of M (M = Ti/Y/Li) doped Al3Sc was weakened. This was consistent with the calculation of the formation energy in Table 1.
The reason for this consequence, the Zr-d orbital provided more valence electrons to hybridize with the Al-p orbital than the Sc-d orbital, while the Ti-d orbital provided fewer valence electrons. At the same time, there was a stronger d-d bond interaction between the Sc and Zr atoms, which effectively enhanced the ductility of the material [45]. For the Al24Sc6Li2 structure, the Li atom replaced the Sc atom, and reduced the p-d hybridization. The number of bonding electrons per atom in Al3Sc and Al-Sc-M (M = Zr, Ti, Y, and Li) structures in the low energy region are shown in Figure 7, which were 2.94, 3.022, 2.994, 2.943, and 2.811, accordingly (energy range between−12 eV and Fermi levels). It is well-known that a higher number of bonding electrons implies an increased structural stability [34,46]. Thus, a stronger electron interaction should occur in Al24Sc6Zr2, and Al24Sc6Zr2 should have a larger structural stability. It was considered that as Zr had more valence electrons, it resulted in stronger electron interactions between the Zr-d orbital and the Al-p orbital, as well as between (Sc, Zr)-d [45,47].
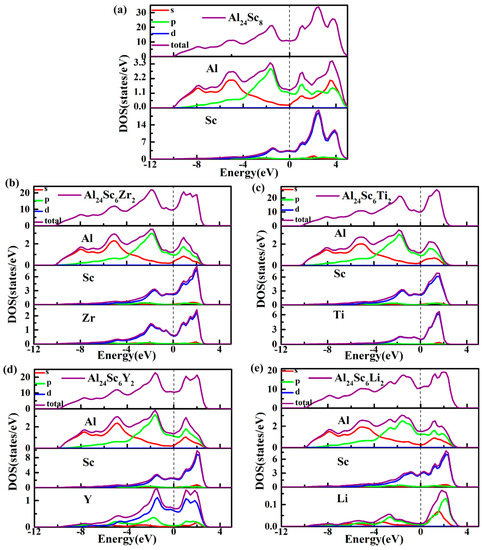
Figure 7.
Total density of states (TDOSs) and Partial density of states (PDOSs) of (a) Al3Sc and (b–e) Al24Sc6M2 (M = Zr, Ti, Y, and Li) alloys. TDOSs for different structures and different elements are represented by purple lines. PDOSs for different orbitals are denoted by s in red, p in green and d in blue.
To further illustrate the contribution of each atomic orbital to TDOS, the PDOS of each atom were calculated, as shown in Figure 7. The main bonding peaks of Al24Sc6M2 were predominantly derived from the Al-s and Al-p orbitals, in the energy range between −12 eV and −4 eV (Figure 7b–e), making the TDOS of Al24Sc6M2 almost coincident with the Al3Sc.
It could be clearly observed that, from −4 eV to 5 eV, the TDOS was mainly contributed by the strong hybridization of the Al(Li)-p and Sc(Zr/Ti/Y)-d orbitals, and a small contribution of Al-s was also observed. Additionally, there was a large overlap in the entire energy range, leading to a strong pd hybridization. The pseudogap was generated by the hybridization of Al-p and M-d (M = Sc, Zr, Ti, Y). In other words, there was a strong covalent bond in the Al-Sc-M (M = Zr, Ti, Y, and Li) structure. In addition, the PDOS of Zr/Ti/Y-d orbitals were different from the Sc-d orbital, suggesting that the doping elements should have taken effect on the TDOS. Below the Fermi level, the peaks of Zr-d orbital moved toward to the lower energy level (Figure 7b), contributing to the enhancement of the bonding states of Al24Sc6Zr2. Regardless, the Sc-d orbital of Al24Sc6Li2 moved toward the higher level (Figure 7e). Moreover, the magnitudes of Li-s/p orbitals were tiny, compared to the other atomic orbitals. This implied that there was a weaker covalent bond in Al24Sc6Li2 for the subdued hybridization between Li and Sc atoms.
For a deeper insight into the atomic bonding of the doped structures, the valence electron density distribution were also investigated. For example, the charge densities on the (100) and (110) planes, for each cell, are shown in Figure 8, in which the contour lines are plotted from 0.015 to 0.04 e/Å3 with 0.0025 e/Å3 interval.
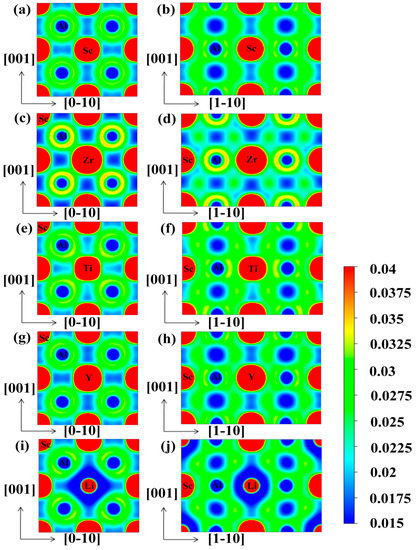
Figure 8.
The electron density contour plots (Unit: e/Å3) on the (100) and (110) planes for L12-Al3Sc (a,b), L12-Al24Sc6Zr2 (c,d), L12-Al24Sc6Ti2 (e,f), L12-Al24Sc6Y2 (g,h), and L12-Al24Sc6Li2 (i,j).
Figure 8a,b display the charge distribution of pure Al3Sc on the (100) and (110) planes, respectively. It was clearly observed that the charge densities of the neighboring Al-Al, Al-Sc, and Sc-Sc had overlaps, especially between the Al-Sc, indicating that there were strong covalent bonds in Al3Sc. Moreover, these covalent bonds were mainly generated by the hybridization between Al-p and Sc-d orbitals. Compared with (110) plane, the charge distribution between the neighboring Al-Sc on the (100) surface was weak, which proved that the covalent bond on the (110) plane was stronger, leading to a brittle fracture. This was caused by the difference in the local symmetries between the (100) and (110) plane. This feature was consistent with previous reports [36]. The charge densities of Al24Sc6Zr2 (100) and the (110) planes are shown in Figure 8c,d, respectively. The overlap of the charge density between the neighboring Al-Zr was increased, which meant that the covalent bond was enhanced. Moreover, Zr was slightly less electronegative than Sc. On this account, Al-Zr exhibited weaker ionic bond properties. This property could be analyzed by the Bader charge [48,49]. The calculation results showed that 0.38 electrons were transferred from Sc to Al in Al3Sc. Nevertheless, the charge transferred from (Sc, Zr) to Al was reduced to 0.34, and the charge distribution on the Sc atom remained unchanged. However, the bonding difference between (100) and (110) planes was decreased, which was profitable for the improvement of the ductility of Al24Sc6Zr2 [24]. The charge distributions of the Al3Sc doped with Ti and Y are shown in Figure 8e–h, accordingly. In contrast to the Al24Sc6Zr2, the overlaps of charge density between Al-Ti/Y, Sc-Ti, and Sc-Sc were reduced, implying that the covalent bonds were weakened. The charge density distributions of Al3Sc doped with Li are shown in Figure 8i,j. It was observed that the charge density overlap between Sc-Li decreased, indicating that Sc-Li exhibited a weaker covalent bond, which was mainly contributed by the hybridization of the Sc-d and Li-p states.
4. Conclusions
In order to explore the effects of the doped elements (M) on the mechanical properties of Al3Sc, both, the structural stability and mechanical properties of Al3Sc with different doping elements and concentrations, in combination with the influence of the higher doping concentration on the electronic properties of Al3Sc were systematically investigated using the first-principles methods. Based on the results of this study, the following conclusions could be deduced. First, it was observed that the Al3Sc structure could be stable after doping. For instance, Al-Sc-Zr had the highest stability, and Al24Sc6Zr2 performed better on stability, compared to Al24Sc7Zr. On the other hand, Al-Sc-M (M = Ti, Y, Li) reduced the stability of Al3Sc, and Al24Sc6M2 performed worse on stability, compared with Al24Sc7M. The calculated elastic constants of Al3Sc, before and after doping, showed its mechanical stability. Moreover, the calculated B/G results revealed that the doped Al3Sc with a higher concentration exhibited a better ductility, especially, when doped with Zr and Ti. It was noted that, the calculated results of the elastic modulus B, G, E, and ν suggested that both Al24Sc6Zr2 and Al24Sc6Ti2 displayed better mechanical properties. Additionally, the TDOS and PDOS analyses indicated that the doped Al3Sc had a pseudogap and a strong covalent bonding, which was due to the strong pd state hybridization. Among them, the maximum pseudogap existed in Al24Sc6Zr2, indicating its best stability. This conclusion was consistent with the calculated formation enthalpy. Ultimately, the obtained results could provide an important theoretical basis for a wide application of the Al-Sc alloy.
Author Contributions
Conceptualization, D.C., X.L. and M.W.; Methodology, X.L. and M.W.; Software, X.L. and M.W.; Formal Analysis, D.C., C.X., Y.W., X.L. and M.W.; Investigation, D.C. and X.L.; Data Curation, D.C., X.L. and M.W.; Writing—Original Draft Preparation, D.C.; Writing—Review & Editing, C.X., Y.W., X.L. and M.W.; Funding Acquisition, Y.W.
Funding
This work was sponsored by the National Key Research and Development Program of China (Grant No.2018YFB1106302), and the project (Grant No. 2017WAMC002), sponsored by the Anhui Province Engineering Research Center of Aluminum Matrix Composites (China).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Asta, M.; Ozoliņš, V. Structural, vibrational, and thermodynamic properties of Al-Sc alloys and intermetallic compounds. Phys. B 2001, 64, 094104. [Google Scholar] [CrossRef]
- Asta, M.; Ozolins, V.; Woodward, C. A first-principles approach to modeling alloy phase equilibria. JOM 2001, 53, 16–19. [Google Scholar] [CrossRef]
- Marquis, E.A.; Seidman, D.N. Nanoscale structural evolution of Al3Sc precipitates in Al(Sc) alloys. Acta Mater. 2001, 49, 1909–1919. [Google Scholar] [CrossRef]
- Ebrahimi, Z.; Ebrahimi, H. Effects of elastic contributions on the evolution of nano-structure Al3Sc phase: A phase-field study. Sci. Iran. 2016, 23, 1539–1548. [Google Scholar] [CrossRef][Green Version]
- Raghukiran, N.; Sujith, R.; Agrawal, H.; Shabadi, R.; Kumar, R. In situ age hardening and grain refinement in as-sprayed Al-Sc binary alloy deposits. J. Alloys Compd. 2018, 735, 1596–1602. [Google Scholar] [CrossRef]
- Kundu, S.; Thirunavukarasu, G.; Chatterjee, S.; Mishra, B. Effect of Bonding Temperature on Phase Transformation of Diffusion-Bonded Joints of Duplex Stainless Steel and Ti-6Al-4V Using Nickel and Copper as Composite Intermediate Metals. Met. Mater. Trans. A 2015, 46, 5756–5771. [Google Scholar] [CrossRef]
- Jiang, C.; Sordelet, D.; Gleeson, B. Effects of Pt on the elastic properties of B2 NiAl: A combined first-principles and experimental study. Acta Mater. 2006, 54, 2361–2369. [Google Scholar] [CrossRef]
- Jia, M.; Zheng, Z.; Gong, Z. Microstructure evolution of the 1469 Al-Cu-Li-Sc alloy during homogenization. J. Alloys Compd. 2014, 614, 131–139. [Google Scholar] [CrossRef]
- Lai, J.; Zhang, Z.; Chen, X.-G. Precipitation strengthening of Al-B4C metal matrix composites alloyed with Sc and Zr. J. Alloy. Compd. 2013, 552, 227–235. [Google Scholar] [CrossRef]
- Mo, Y.; Pang, M.; Yang, W.; Zhan, Y. Effects of alloying elements on structural, electronic and mechanical properties of AlSc2 by first-principles calculations. Comput. Mater. Sci. 2013, 69, 160–167. [Google Scholar] [CrossRef]
- Park, N.; Lee, S.-C.; Cha, P.-R. Effects of alloying elements on the stability and mechanical properties of Fe3Al from first-principles calculations. Comput. Mater. Sci. 2018, 146, 303–309. [Google Scholar] [CrossRef]
- Tian, J.; Zhao, Y.; Hou, H.; Wang, B. The effect of alloying elements on the structural stability, mechanical properties, and Debye temperature of Al3Li: A first-principles study. Materials 2018, 11, 1471. [Google Scholar] [CrossRef] [PubMed]
- Fuller, C.B.; Seidman, D.N.; Dunand, D.C. Mechanical properties of Al(Sc,Zr) alloys at ambient and elevated temperatures. Acta Mater. 2003, 51, 4803–4814. [Google Scholar] [CrossRef]
- Van Dalen, M.E.; Dunand, D.C.; Seidman, D.N. Effects of Ti additions on the nanostructure and creep properties of precipitation-strengthened Al-Sc alloys. Acta Mater. 2005, 53, 4225–4235. [Google Scholar] [CrossRef]
- Mao, Z.; Chen, W.; Seidman, D.; Wolverton, C. First-principles study of the nucleation and stability of ordered precipitates in ternary Al-Sc-Li alloys. Acta Mater. 2011, 59, 3012–3023. [Google Scholar] [CrossRef]
- Harada, Y.; Dunand, D.; Dunand, D. Thermal expansion of Al3Sc and Al3(Sc0.75X0.25). Scr. Mater. 2003, 48, 219–222. [Google Scholar] [CrossRef]
- Harada, Y.; Dunand, D.; Dunand, D. Creep properties of Al3Sc and Al3(Sc, X) intermetallics. Acta Mater. 2000, 48, 3477–3487. [Google Scholar] [CrossRef]
- Li, D.L.; Chen, P.; Yi, J.X.; Tang, B.Y.; Peng, L.M.; Ding, W.J. Ab initio study on the thermal properties of the fcc Al3Mg and Al3Sc alloys. J. Phys. D Appl. Phys. 2009, 42, 225407. [Google Scholar] [CrossRef]
- Hu, W.-C.; Liu, Y.; Li, D.-J.; Zeng, X.-Q.; Xu, C.-S. Mechanical and thermodynamic properties of Al3Sc and Al3Li precipitates in Al-Li-Sc alloys from first-principles calculations. Phys. B Condens. Matter. 2013, 427, 85–90. [Google Scholar] [CrossRef]
- Chen, D.; Chen, Z.; Wu, Y.; Wang, M.; Ma, N.; Wang, H. First-principles investigation of mechanical, electronic and optical properties of Al3Sc intermetallic compound under pressure. Comput. Mater. Sci. 2014, 91, 165–172. [Google Scholar] [CrossRef]
- Pan, R.-K.; Wang, H.-C.; Shao, L.; Zheng, J.; Pan, X.-Z.; Tang, B.-Y. Temperature dependence of elastic properties of L12-Al3Sc: A first-principles study. Comput. Mater. Sci. 2016, 111, 424–429. [Google Scholar] [CrossRef]
- Duan, Y.H.; Sun, Y.; Peng, M.J.; Zhou, S.G. Ab-initio investigations on elastic properties in L12 structure Al3Sc and Al3Y under high pressure. J. Alloys Compd. 2014, 585, 587–593. [Google Scholar] [CrossRef]
- Wang, R.N.; Ma, L.; Pan, R.K.; Luo, T.P.; Zhou, S.C.; Tang, B.Y. First-principles study of L12-Al3(Sc1−xTMx) alloys using special quasirandom structures. Comput. Mater. Sci. 2013, 79, 136–142. [Google Scholar] [CrossRef]
- Huang, Y.C.; Guo, X.F.; Ma, Y.L.; Shao, H.B.; Xiao, Z.B. Stabilities, electronic and elastic properties of L12-Al3(Sc1−x,Zrx) with different Zr content: A first-principles study. Phys. B Condens. Matter 2018, 548, 27–33. [Google Scholar] [CrossRef]
- Khenioui, Y.; Boulechfar, R.; Maazi, N.; Ghemid, S. FP-LAPW investigation of Al3(Sc1−xTix) alloys properties in L12 and D022 structures. Int. J. Mod. Phys. B 2018, 32, 1850167. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Qian, Y.; Xue, J.L.; Wang, Z.J.; Yang, Z.H.; Qian, P. Mechanical properties evaluation of Zr addition in L12-Al3(Sc1−xZrx) using first-principles calculation. JOM 2016, 68, 1293–1300. [Google Scholar] [CrossRef]
- Tian, T.; Wang, X.F.; Li, W. Ab initio calculations on elastic properties in L12 structure Al3X and X3Al-type (X= transition or main group metal) intermetallic compounds. Solid State Commun. 2013, 156, 69–75. [Google Scholar] [CrossRef]
- Shi, D.; Wen, B.; Melnik, R.; Yao, S.; Li, T. First-principles studies of Al-Ni intermetallic compounds. J. Solid State Chem. 2009, 182, 2664–2669. [Google Scholar] [CrossRef]
- Hill, R. The Elastic Behaviour of a Crystalline Aggregate. Proc. Phys. Soc. Sect. A 1952, 65, 349–354. [Google Scholar] [CrossRef]
- Zhou, L.J.; Su, K.H.; Wang, Y.L.; Zeng, Q.F.; Li, Y.L. First-principles study of the properties of Li, Al and Cd doped Mg alloys. J. Alloys Compd. 2014, 596, 63–68. [Google Scholar] [CrossRef]
- Young, A.F.; Sanloup, C.; Gregoryanz, E.; Scandolo, S.; Hemley, R.J.; Mao, H.-K. Synthesis of Novel Transition Metal NitridesIrN2andOsN2. Phys. Lett. 2006, 96, 155501. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.N.; Tang, B.Y.; Peng, L.M.; Ding, W.J. Ab initio study of the effect of Zr content on elastic and electronic properties of L12-Al3(Sc1−xZrx) alloys. Comput. Mater. Sci. 2012, 59, 87–93. [Google Scholar] [CrossRef]
- Teter, D.M. Computational Alchemy: The Search for New Superhard Materials. MRS Bull. 1998, 23, 22–27. [Google Scholar] [CrossRef]
- Pugh, S. XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Lond. Edinb. Dublin Philos. Mag. J. Sci. 1954, 45, 823–843. [Google Scholar] [CrossRef]
- Pettifor, D.G. Theoretical predictions of structure and related properties of intermetallics. Mater. Sci. Technol. 1992, 8, 345–349. [Google Scholar] [CrossRef]
- Mattesini, M.; Ahuja, R.; Johansson, B. Cubic Hf3N4 and Zr3N4: A class of hard materials. Phys. B 2003, 68, 184108. [Google Scholar]
- Tvergaard, V.; Hutchinson, J.W. Microcracking in Ceramics Induced by Thermal Expansion or Elastic Anisotropy. J. Am. Ceram. Soc. 1988, 71, 157–166. [Google Scholar] [CrossRef]
- Anderson, O.L. A simplified method for calculating the debye temperature from elastic constants. J. Phys. Chem. Solids 1963, 24, 909–917. [Google Scholar] [CrossRef]
- Ravindran, P.; Fast, L.; Korzhavyi, P.A.; Johansson, B.; Wills, J.; Eriksson, O. Density functional theory for calculation of elastic properties of orthorhombic crystals: Application to TiSi2. J. Appl. Phys. 1998, 84, 4891–4904. [Google Scholar] [CrossRef]
- Sun, Z.M.; Ahuja, R.; Schneider, J.M. Theoretical investigation of the solubility in (MxM’2−x)AlC (M and M’= Ti,V, Cr). Phys. Rev. B 2003, 68, 224112. [Google Scholar] [CrossRef]
- Tian, J.; Han, G.; Wei, H.; Zheng, Q.; Jin, T.; Sun, X.; Hu, Z. Effects of alloying elements on the electronic structure and ductility of NiAl compounds investigated by X-ray absorption fine structure. Philos. Mag. 2013, 93, 2161–2171. [Google Scholar] [CrossRef]
- Nylén, J.; Garcìa, F.G.; Mosel, B.; Pöttgen, R.; Häussermann, U. Structural relationships, phase stability and bonding of compounds PdSnn (n = 2, 3, 4). Solid State Sci. 2004, 6, 147–155. [Google Scholar] [CrossRef]
- Electronic structure and hybridization effects in Hume-Rothery alloys containing transition elements. Phys. Rev. B 1995, 52, 7920–7933. [CrossRef]
- Bader, R. Atoms in Moledules: A Quantum Theory; Oxford University Press: New York, NY, USA, 1990. [Google Scholar]
- Delaire, O.; Fultz, B. Charge redistribution and rhonon entropy of Vanadium alloys. Phys. Rev. Lett. 2006, 97, 245701. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).