Summary
Management of pulmonary hypertension—news from Dana Point. Recently, the 4th WHO conference on pulmonary hypertension (PH) was hold in Dana Point. Thereby, new treatment algorhythms were developed. Specific PH therapy with prostanoids, endothelin-receptor antagonists and phosphodiesterase-5-inhibitors is now also recommended for patients with pulmonary arterial hypertension (PAH) in NYHA/WHO class II. Combination therapy is recommended for PAH patients not sufficiently responding to monotherapy, however, the best combination has still to be investigated in clinical trials. If PAH patients are stable on therapy, closely supervised training can be recommended. For chronic thromboembolic PH (CTEPH), pulmonary endarterectomy remains the treatment of choice. CTEPH patients not eligible for surgery may profit from drug therapy in analogy to PAH patients. Patients with PH due to chronic heart or lung diseases should be treated as efficiently as possible for their underlying condition. Many new compounds for the treatment of PH are currently under investigation, including sGC stimulators, thyrosin kinase inhibitors and serotonin antagonists.
Einleitung
Im Februar 2008 fand in Dana Point, Kalifornien, USA, das vierte «WHO Meeting on Pulmonary Hypertension» statt. Die Therapieempfehlungen hieraus haben einen besonderen Stellenwert, da sie den internationalen Standard repräsentieren und zugleich als Vorlage für nationale und internationale Leitlinien gelten. Somit werden sich die gemeinsamen Leitlinien der European Society of Cardiology (ESC) und European Respiratory Society (ERS) zur Therapie der pulmonalen Hypertonie, die im Herbst 2009 erscheinen werden, eng an die Empfehlungen aus Dana Point anlehnen. Dieser Artikel gibt einen zusammenfassenden Überblick bezüglich den zu erwartenden Neuerungen in Bezug auf die Therapie der pulmonalen Hypertonie.
Empfehlungen zur Behandlung der Pulmonalarteriellen Hypertonie (PAH)
Im Rahmen des dritten «WHO Meeting on Pulmonary Hypertension» in Venedig 2003 wurden die idiopathische, die familiäre und die pulmonale Hypertonie, assoziiert mit Krankheiten wie Kollagenosen, kongenitalen Herzvitien, portaler Hypertonie und HIV, um die wichtigsten zu nennen, unter dem Begriff pulmonal-arterielle Hypertonie (PAH) zusammengefasst []. Gegenüber den Therapieempfehlungen der PAH aus dem WHO-Meeting von Venedig 2003 [] werden die Empfehlungen aus Dana Point einige entscheidende Weiterentwicklungen präsentieren.
Nicht-medikamentöse Therapie
Kürzlich konnte gezeigt werden, dass gezieltes und individuell angepasstes Training bei PAH-Patienten nicht nur komplikationsarm möglich ist, sondern auch eine deutliche Verbesserung der körperlichen Belastbarkeit und anderer Parameter der Lebensqualität bewirkt []. Somit können supervidierte Trainingsprogramme für PAH-Patienten empfohlen werden. Es sei aber in diesem Rahmen klar darauf hingewiesen, dass hier keineswegs irgendein Trainingsprogramm, sondern ein kontrolliertes, spezifisches Training, überwacht durch ein erfahrenes PAH-Team, gemeint ist.
Medikamentöse Therapie
Für die medikamentöse Therapie der PAH gilt weiterhin, dass sie ausschliesslich an erfahrenen Expertenzentren nach Verifizierung der korrekten Diagnose mittels Rechtsherzkatheter eingeleitet werden soll. Hierbei soll zumindest bei Patienten mit idiopathischer PAH eine Vasoreagibilitätstestung mittels kurzwirksamen, pulmonal-selektiven Vasodilatatoren (inhalatives Stickstoffmonoxid oder Iloprost) erfolgen. Falls sich hiermit der mittlere pulmonal-arterielle Druck um mindestens 10 mm Hg auf ≤ 40 mm Hg bei normalem Herzzeitvolumen senken lässt, kann mit KalziumKanal-Blockern behandelt werden []. Bei solchen «Responders» kommt es gelegentlich unter KalziumKanal-Blockern zu einer dramatischen klinischen und hämodynamischen Verbesserung, wie sie mit anderen PAH-Medikamenten selten gesehen wird. Es ist jedoch anzumerken, dass auch solche Akut-Responder engmaschig überwacht werden müssen, da ein Teil nicht längerfristig auf die Kalzium-Kanal-Blocker-Therapie anspricht. Diese Patienten, wie auch die grosse Mehrzahl der PAH-Patienten, die in der Akuttestung keine «Responder» sind, werden mit Prostanoiden, Endothelin-Rezeptor-Antagonisten oder Phosphodiesterase-5Inhibitoren behandelt [,,].
Als wichtigste Neuerung aus Dana Point wird nun erstmals auch für PAH-Patienten im funktionellen Stadium NYHA II eine gezielte Therapie empfohlen. Hintergrund dieser Empfehlung sind Erkenntnisse aus drei randomisierten, plazebokontrollierten Studien: In der EARLY-Studie wurden PAH-Patienten mit NYHAII während sechs Monaten mit dem Endothelin-RezeptorAntagonisten Bosentan behandelt []. In der VerumGruppe nahm der pulmonal-vaskuläre Widerstand gegenüber Plazebo signifikant ab, und die Zeit bis zu einer klinischen Verschlechterung verlängerte sich signifikant, so dass gegenüber Plazebo nach sechs Monaten Therapie eine relative Risikoreduktion von 77% erreicht werden konnte. Somit konnte die EARLYStudie erstmalig belegen, dass mit einer gezielten Therapie in früheren Stadien die Progression der Erkrankung verlangsamt werden kann. In der ARIES-Studie wurde Ambrisentan, ein Endothelin-Rezeptor-Antagonist, der selektiv am Endothelin-A-Rezeptor angreift, untersucht []. Hierbei konnte gezeigt werden, dass sich die 6-Minuten-Gehstrecke, definiert als primärer Studienendpunkt, bei Patienten im NYHA-IIund -III-Stadium nach 12-wöchiger Therapie signifikant verbesserte. Gleiches wurde auch für den Phosphodiesterase-5-Inhibitor Sildenafil in der SUPER1-Studie gezeigt []. In Europa sind die EndothelinRezeptor-Antagonisten Bosentan undAmbrisentan die einzigen Substanzen, die für die Behandlung der PAH im Stadium II zugelassen sind, in der Schweiz ist dies bisher nur Bosentan, die Zulassung für Ambrisentan wird jedoch demnächst folgen. Für Patienten im NYHA-III-Stadium werden Endothelin-RezeptorAntagonisten, Phosphodiesterase-5-Inhibitoren und inhaliertes Iloprost mit dem höchsten Evidenzgrad empfohlen. Im funktionellen Stadium IV gilt international kontinuierlich intravenöses verabreichtes Epoprostenol nach wie vor als Therapie der Wahl.Auch andere intravenös oder subkutan verabreichte Prostanoide sowie Kombinationen mit Endothelin-RezeptorAntagonisten und Phosphodiesterase-5-Inhibitoren können im funktionellen Stadium IV eingesetzt werden.
Eine wichtige Neuerung aus Dana Point ist, dass der Kombinationstherapie aus obengenannten Substanzen ein wesentlich höherer Stellenwert bei unzureichendem Ansprechen auf eine Monotherapie zukommt. Hierbei wird ein unzureichende Ansprechen definiert als Nichterreichen folgender klinischer Ziele: ein stabiles klinisches Bild, vorzugsweise in den funktionellen Stadien NYHA I und II, ohne Hinweise auf eine Rechtsherzinsuffizienz, keine Synkopen, akzeptable Leistungsfähigkeit mit einer 6-Minuten-Gehstrecke >400 m (bei Patienten unter 40 Jahren gar >500 m). Regelmässige Rechtsherzkatheteruntersuchungen werden zur Verlaufskontrolle empfohlen, wobei hier eine kompensierte Rechtsherzfunktion mit normalen rechtsventrikulären Füllungsdrücken und einem normalen Herzzeitvolumen entscheidender sind als die Höhe der pulmonal-arteriellen Druckwerte, da letztere bei zunehmender Rechtsherzinsuffizienz wieder abnehmen können. Spezifische Intervalle für Rechtsherzkatheteruntersuchungen werden nicht angegeben, im klinischenAlltag richten sich diese vorwiegend nach dem Krankheitsverlauf und dem Therapieansprechen. Bezüglich der Auswahl der Kombinationspartner werden keine spezifischen Empfehlungen gegeben, da es bislang noch keine kontrollierten Langzeitstudien zur Kombinationstherapie gibt.
Empfehlungen zur Behandlung anderer Formen der PH
Von der PAH streng abzugrenzen sind andere Formen der PH, so die PH bei chronischer Linksherzbelastung (Gruppe II), die PH bei chronischen Lungenkrankheiten (Gruppe III) oder die chronisch-thromboembolische PH (Gruppe 4).
PH bei chronischen Lungenkrankheiten
Das Auftreten einer PH bei chronisch-obstruktiver Lungenkrankheit (COPD) und Lungenfibrose ist sehr häufig, je nach Definition ist dies bei 30–80% der Patienten der Fall [,]. Die PH bei Lungenkrankheiten ist üblicherweise im Vergleich zur PAH relativ mild, die mittleren pulmonalen Druckwerte liegen häufig zwischen 25 und 30 mm Hg in Ruhe. Unter körperlicher Belastung steigen die pulmonal-arteriellen Druckwerte jedoch meist stark an, zusammen mit einer deutlichen Verschlechterung des Gasaustausches. Dies führt zu einer deutlich eingeschränkten Leistungsfähigkeit und einer hiermit verminderten Lebensqualität. Zudem ist selbst eine leichte PH bei Lungenkrankheiten mit einer schlechten Prognose assoziiert [,]. Das Einsetzen der obenerwähnten «PAH-Medikamente» wird bei PH aufgrund von Lungenkrankheiten nicht empfohlen, da keine Studien existieren, die die Sicherheit und Wirksamkeit bei diesen Patienten hinreichend dokumentieren. Es ist zu Bedenken, dass diese vasodilatativ-wirkenden Substanzen bei Lungenkrankheiten die Gefahr einer Zunahme des intrapulmonalen Rechts-links-ShuntVolumens in sich bergen und hierdurch zu einer Verschlechterung des Gasaustausches führen können. Lediglich bei einem kleinen Teil der Patienten mit COPD, bei denen die PH unverhältnismässig stark ausgeprägt ist («out-of-proportion» PH), könnte ein Therapieversuch mit obigen Substanzen unter strenger Kontrolle des Gasaustausches gewagt werden. Es handelt sich dabei um COPD-Patienten mit vergleichsweise leicht eingeschränkter funktionellen Erstsekundenkapazität (FEV1 ≥50%) und schwerer pulmonaler Hypertonie (pulmonaler Mitteldruck ≥40 mm Hg). Es wäre sehr wünschenswert, wenn die Therapie der PH bei COPD, insbesondere auch die «out-of proportion» PH, im Rahmen von kontrollierten Studien untersucht würde.
PH bei chronischer Linksherzinsuffizienz
Bei der Linksherzinsuffizienz kann es aufgrund von erhöhten enddiastolischen Füllungsdrücken der linken Herzkammer und/oder des linken Vorhofes zur passiven Erhöhung des pulmonal-venösen und -kapillären Druckes und somit zu einer PH (post-kapilläre PH) kommen. Bei diesen Patienten sind der transpulmonale Druckgradient und der Widerstand im Lungenkreislauf normal. Vorrangiges Therapieziel ist hier die optimale Therapie der Linksherzinsuffizienz. Bei einem Teil der Patienten mit Linksherzinsuffizienz finden sich aber disproportional hohe pulmonal-arterielle Druckwerte mit erhöhten transpulmonalen Gradienten und erhöhtem pulmonal-arteriellem Widerstand. Die Pathogenese dieser disproportionalen PH bei Linksherzinsuffizienz ist noch weitgehend unklar. Es ist denkbar, dass bei chronischer Linksherzinsuffizienz infolge fortgeschrittener Strukturpathologie des Herzens trotz Therapie keine normalen Füllungsdrücke erzielt werden, insbesondere nicht unter Belastung. Bei längerem Bestehen der dadurch induzierten pulmonal-venösen PH könnte es somit auf der pulmonal-arteriellen Seite zu einem strukturellen Gefässumbau kommen, so dass die PH sich zunehmend «fixiert» und diese sich histopathologisch kaum mehr von einer PAH unterscheidet. Bisher gibt es keine Studien, die gezielt die spezifische Langzeittherapie dieser Formen der PH unter Berücksichtigung der transpulmonalen Gradienten untersucht haben. Daher kann auch eine Therapie mit obenbeschriebenen «PAH-Medikamenten» momentan nicht generell empfohlen werden, eine Therapie im Rahmen von entsprechenden Studien wäre sehr wünschenswert.
In der Praxis ist es oft nicht ganz einfach, eine PH aufgrund einer diastolischen Dysfunktion des linken Ventrikels von einer PAH zu unterscheiden. Eine eingehendeAbklärung mit einem Herzkatheter in einem erfahrenen Zentrum ist hier für die Diagnose unabdingbar. In Dana Point wurde ein diagnostischer Algorithmus entwickelt, der sich gezielt mit diesem Problem befasst.
Chronische thromboembolische PH (CTEPH)
Für die CTEPH gilt die pulmonale Endarterektomie immer noch als Therapie der ersten Wahl, da hiermit meist eine exzellente Lebensqualität, eine Belastbarkeit und häufig eine Normalisierung der Ruhehämodynamik erreicht werden kann. Diese sehr schwierige und komplikationsträchtige Operation sollte jedoch ausgewählten Chirurgen mit viel Erfahrung auf diesem Gebiet vorbehalten sein, da die Operationsmortalität auch in erfahrenen Händen mit 4–10% relativ hoch ist [,]. Für Patienten, bei denen Kontraindikationen für diese Operation bestehen (distale Form der CTEPH oder Komorbiditäten), werden medikamentöse Therapieoptionen analog der PAH evaluiert. Die Effektivität von Prostanoiden, Endothelin-Rezeptor-Antagonisten und Phosphodiesterase-5-Inhibitoren bei inoperabler CTEPH konnte bereits in mehreren, zum Teil jedoch unkontrollierten Studien gezeigt werden [,]. Es ist zu erwarten, dass für die inoperable CTEPH bald medikamentöse Therapieempfehlungen in Analogie zur PAH formuliert werden können.
Neue Substanzen zur Therapie der PAH
Die Therapie der PAH hat sich in den letzten Jahren durch Einführung der neuen PAH-Medikamente (Prostanoide, Endothelin-Rezeptor-Antagonisten, Phosphodiesterase-5-Inhibitoren) dramatisch verbessert. Kontrollierte Studien zeigen, dass durch die genannten Wirkstoffe die klinische Symptomatik vermindert, die körperliche Belastbarkeit gesteigert und die Krankheitsprogression verzögert werden können. Zudem kann im Vergleich zu historischen Kontrollkollektiven auch die Prognose verbessert werden []. Die Krankheit PAH ist jedoch mit den derzeit zur Verfügung stehenden Mitteln nicht heilbar. Zudem weisen viele Patienten trotz intensiver Behandlung mit den derzeit zur Verfügung stehenden Mitteln immer noch erhebliche Beschwerden auf, die mit einer deutlichen Einschränkung der Lebensqualität und mit einer begrenzten Lebenserwartung einhergehen. Daher sind noch viele Anstrengungen nötig, um die Versorgung von Patienten mit PAH weiter zu verbessern. Einige vielversprechende neue Wirkstoffe aus verschiedenen Substanzklassen befinden sich bereits in klinischer Erprobung. Diese werden nachstehend kurz geschildert.
sGC-Stimulatoren
Der NO-Signalweg spielt eine wichtige Rolle für die Regulation des Gefässtonus in der Lungenstrombahn. NO vermittelt durch die Aktivierung der löslichen Guanylatzyklase (soluble guanylate cyclase = sGC) die Bildung von zyklischem Guanosinmonophosphat (cGMP) in glatten Muskelzellen und führt somit zur Vasorelaxation (Figure 1). PAH-Patienten weisen in der Lungenstrombahn eine verminderte Bioverfügbarkeit von NO auf, was mit einer reduzierten Fähigkeit zur Gefässerweiterung einhergeht. Phosphodiesterase5-Inhibitoren setzten hier an, indem sie den Phosphodiesterase-5-vermittelten Abbau von cGMP hemmen und so zur pulmonalen Gefässerweiterung führen. Jedoch ist deren therapeutische Wirksamkeit durch die verminderte Bioverfügbarkeit von NO möglicherweise begrenzt. Ein attraktiver Ansatz ist daher, die direkte Stimulation der sGC mit sogenannten sGC-Stimulatoren, die unabhängig von NO wirken. Im Lungengewebe von idiopathischen PAH-Patienten findet sich eine deutlich erhöhte sGC-Expression gegenüber von Gesunden, und in tierexperimentellen Studien konnte die Wirksamkeit des oral verfügbaren sGC-Stimulators BAY 63–2521 gezeigt werden [,,]. So führte BAY 63–2521 zu einer partiellen Rückbildung der PH, der resultierenden Rechtsherzhypertrophie und des pulmonal-vaskulären Remodelings. Die Wirksamkeit dieser Substanz wird nun in klinischen Studien bei PAHPatienten geprüft, erste Resultate werden voraussichtlich bald vorliegen.
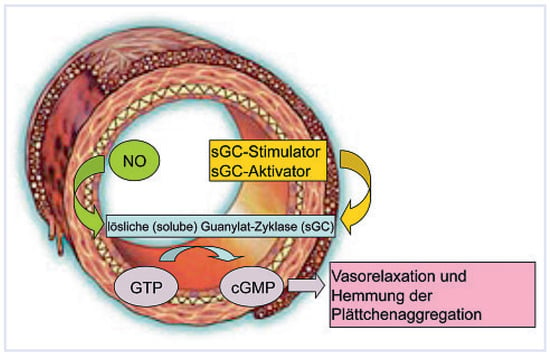
Figure 1.
Stickstoffmonoxid (Nitric Oxide, NO) ist der wichtigste körpereigene Vasodilatator und wirkt über die Aktivierung der löslichen Guanylatzyklase (sGC). Aktivatoren bzw. Stimulatoren der sGC führen über die Erhöhung der intrazellulären cGMP-Spiegel NO-unabhängig zur Gefässerweiterung.
Thyrosin-Kinase-Inhibitoren
In den fortgeschrittenen Stadien der PH findet sich histologisch ein ausgeprägtes pulmonal-vaskuläres Remodeling. Dieses beruht auf der durch Wachstumsfaktoren induzierten Stimulation von glatten Muskelzellen und deren Schutz vor programmiertem Zelltod (Apoptose). Diese Wachstumsfaktoren vermitteln ihre Effekte durch die Aktivierung spezifischer RezeptorThyrosinkinasen, die eine intrinsische TyrosinkinaseAktivität besitzen (Figure 2). Durch Hemmung dieser Tyrosinkinase-Aktivität mittels spezifischer Tyrosinkinase-Inhibitoren werden die ligandeninduzierte Aktivierung von Rezeptor-Thyrosinkinasen und somit wachstumsfaktorvermittelte zelluläre Effekte (Proliferation, Migration, Schutz vorApoptose) inhibiert. Ein solcher Wachstumsfaktor und seine Rezeptoren, der bei PH vermehrt exprimiert wird, ist der «platelet derived growth factor» [,]. In verschiedenen Tiermodellen der PAH konnte gezeigt werden, dass durch den Tyrosinkinase-Inhibitor Imatinib sowohl die strukturellen Gefässwandveränderungen in der Lungenstrombahn als auch die Ausbildung einer PH vermindert werden []. Darüber hinaus liess sich auch eine bereits bestehende PH zurückbilden. Die Wirksamkeit von Imatinib bei fortgeschrittener, therapierefraktärer PAH wurde bereits in Einzelfallberichten publiziert []. Ein mögliches Problem könnte hierbei allerdings die für Imatinib beschriebene kardiale Toxizität darstellen. Eine Phase-II-Studie, bei der Imatinib zusätzlich zur etablierten PAH-Therapie über 24 Wochen mit Plazebo verglichen wurde, wurde kürzlich abgeschlossen, die Resultate hiervon werden bald erwartet. Zwischenzeitlich werden weitere Kinase-Inhibitoren, wie z.B. der kombinierte Thyrosin- und Serin-/ThreoninKinase-Inhibitor Sorafenib bezüglich ihrer Wirksamkeit bei PH untersucht. Erste tierexperimentelle Studien zeigten bereits eine günstige Wirkung [].
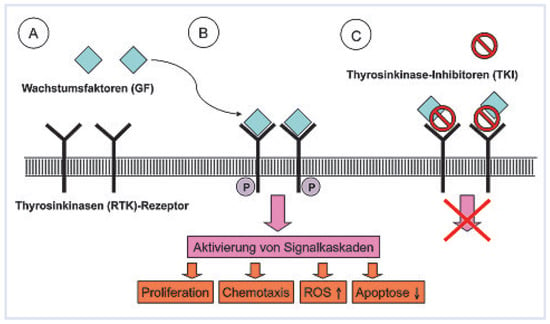
Figure 2.
(A) Keine Wachstumsfaktoren an transmembranöse Rezeptor-Thyrosinkinasen gebunden, somit niedrige Kinaseaktivität. (B) Wachstumsfaktoren binden an extrazelluläre Bindungsstellen der Rezeptor-Thyrosinkinasen und führen so zur Aktivierung von Signalkaskaden. (C) Tyhrosinkinase-Inhibitoren hemmen die wachstumsfaktorvermittelte Aktivierung von Signalkaskaden.
Serotonin-Rezeptor-Antagonisten
Das Serotonin-System spielt eine wichtige Rolle in der Pathogenese der PH []. Serotonin führt über 5-HT1Bund 5-HT2A-Rezeptoren zur pulmonalen Vasokonstriktion und hemmt via 5-HT2B-Rezeptoren die Zellproliferation und Bindung von Elastase und TGF-b (Figure 3). Via den Serotonin-Transporter (5-HTT) vermittelt Serotonin zudem die Hyperplasie der glatten Muskelzellen. Bei PH wird insbesondere der 5-HT2B-Rezeptor vermehrt exprimiert. Terguride (transdihydrolisuride) ist als partieller Dopamin-Antagonist in Japan zur Therapie der Hyperprolaktinämie zugelassen und ist für diese Indikation gut verträglich. Terguride hemmt zudem via 5-HT2B- und 5-HT2A-Rezeptoren das Serotonin-System und hat auch antiadrenerge Eigenschaften. Insgesamt wirkt Terguride antiproliferativ, antithrombotisch und antifibrotisch und führt zur Relaxation von glatten Muskelzellen. Terguride senkte dosisabhängig den pulmonal-arteriellen Druck bei Ratten, bei denen zuvor mittels Monocrotalin eine PH induziert wurde. Terguride-behandelte Tiere wiesen entsprechend histologisch eine geringere Muskularisierung der Pulmonalarterien und weniger Rechtsherzhypertrophie auf. Zurzeit wird in Europa eine Phase-II-Studie (TERAPH) durchgeführt, bei der auch die Schweiz mit dem PH-Zentrum Zürich vertreten ist. Im Rahmen dieser Studie werden PAH-Patienten zusätzlich zur etablierten Therapie für 12 Wochen mit Terguride oder Plazebo behandelt. Primärer Endpunkt ist der pulmonale Gefässwiderstand, gemessen im Rechtsherzkatheter, sekundäre Endpunkte sind hämodynamische Parameter (pulmonal-arterieller Druck, Herzindex) und klinische Parameter (6-Minuten-Gehstrecke, NYHA/WHO-Klasse) und Biomarker (NTproBNP). Kürzlich wurde Terguride von der amerikanischen FDA als «Orphan»-Arzneimittel zur PAH eingestuft.
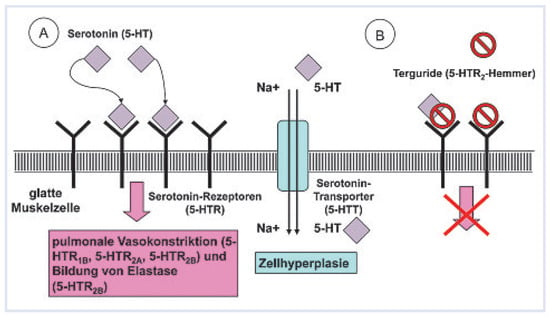
Figure 3.
(A) Serotoninvermittelte Effekte bei pulmonaler Hypertonie auf eine glatte Gefässmuskelzelle: Vasokonstriktion und Proliferation, vermittelt durch die Serotonin-Rezeptoren sowie Muskelzellhyperplasie, vermittelt durch den Serotonin-Transporter. (B) Der Serotonin-Rezeptor-Antagonist hemmt die Vasokonstriktion und Zellproliferation.
Im Sinne der translationellen Forschung sollen vielversprechende Ergebnisse aus Tiermodellen möglichst rasch beim Menschen geprüft werden. Es ist bekannt, dass Patienten, die an kontrollierten Studien teilnehmen, eine verbesserte Prognose haben, selbst wenn sie Plazebo erhalten, zum Teil durch die Studienbetreuung, zum Teil auch, weil sie nach Studienende das Medikament in Rahmen der «open-label extension»-Phase weiter erhalten können. Es ist deshalb wichtig, dass PAH-Patienten in enger Zusammenarbeit mit Zentren betreut werden, die von Pharmafirmen für die entsprechenden internationalen Studien berücksichtigt werden.
Zusammenfassung für die Praxis
Die während des vierten WHO-Meetings betreffend pulmonaler Hypertonie (PH) in Dana Point erarbeiteten Therapieempfehlungen beinhalten einige Neuerungen für Patienten mit pulmonal-arterieller Hypertonie (PAH), aber auch für Patienten mit anderen Formen der PH.
Für PAH-Patienten in den funktionellen Stadien NYHA/WHO II wird nun eine Therapie mit Endothelin-Rezeptor-Antagonisten oder Phosphodiesterase5-Inhibitoren empfohlen. Bei unzureichendemAnsprechen auf eine Monotherapie mit einem der PAH-Medikamente wird nun der Kombinationstherapie ein grösserer Stellenwert eingeräumt. Letztere wird in der Schweiz schon seit längerem erfolgreich angewandt [] und soll in einem anerkannten PH-Zentrum erfolgen. Für stabile PAH-Patienten unter Therapie werden engmaschig supervidierte Trainingsprogramme empfohlen. Für Patienten mit CTEPH gilt die pulmonale Endarterektomie weiterhin als Therapie der ersten Wahl. Für Patienten, die sich hierfür nicht qualifizieren, gibt es immer mehr Evidenz, dass medikamentöse Therapien analog der PAH wirksam sind. Für Patienten mit PH infolge Herz- und Lungenkrankheiten gilt weiterhin, dass die Grundkrankheit nach allen Mitteln der Kunst möglichst gut behandelt wird. Bei disproportionaler PH kann ein Therapieversuch mit einem spezifischen PAH-Medikament erwogen werden. Dies sollte jedoch möglichst im Rahmen einer Studie geschehen. Nach wie vor ist die PH nicht heilbar, so dass weitere Verbesserungen der medikamentösen Therapie notwendig sind. Eine Reihe vielversprechender neuer Substanzen befindet sich derzeit in klinischer Erprobung. Erwähnenswert hierbei sind sGC-Stimulatoren, Tyrosinkinase-Inhibitoren und Serotonin-Antagonisten.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Simonneau, G.; Galie, N.; Rubin, L.J.; Langleben, D.; Seeger, W.; Domenighetti, G.; et al. Clinical classification of pulmonary hypertension. J. Am. Coll. Cardiol. 2004, 43, S5–S12. [Google Scholar] [CrossRef]
- Galie, N.; Seeger, W.; Naeije, R.; Simonneau, G.; Rubin, L.J. Comparative analysis of clinical trials and evidence-based treatment algorithm in pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2004, 43, S81–8. [Google Scholar] [CrossRef]
- Mereles, D.; Ehlken, N.; Kreuscher, S.; Ghofrani, S.; Hoeper, M.M.; Halank, M.; et al. Exercise and respiratory training improve exercise capacity and quality of life in patients with severe chronic pulmonary hypertension. Circulation 2006, 114, 1482–1489. [Google Scholar] [CrossRef]
- Gomberg-Maitland, M.; Olschewski, H. Prostacyclin therapies for the treatment of pulmonary arterial hypertension. Eur. Respir. J. 2008, 31, 891–901. [Google Scholar] [CrossRef]
- Wilkins, M.R.; Wharton, J.; Grimminger, F.; Ghofrani, H.A. Phosphodiesterase inhibitors for the treatment of pulmonary hypertension. Eur. Respir. J. 2008, 32, 198–209. [Google Scholar] [CrossRef]
- Dupuis, J.; Hoeper, M.M. Endothelin receptor antagonists in pulmonary arterial hypertension. Eur. Respir. J. 2008, 31, 407–415. [Google Scholar] [CrossRef]
- Galie, N.; Rubin, L.; Hoeper, M.; Jansa, P.; Al-Hiti, H.; Meyer, G.; et al. Treatment of patients with mildly symptomatic pulmonary arterial hypertension with bosentan (EARLY study): A double-blind, randomised controlled trial. Lancet 2008, 371, 2093–2100. [Google Scholar] [CrossRef] [PubMed]
- Galie, N.; Olschewski, H.; Oudiz, R.J.; Torres, F.; Frost, A.; Ghofrani, H.A.; et al. Ambrisentan for the treatment of pulmonary arterial hypertension: Results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation 2008, 117, 3010–3019. [Google Scholar] [CrossRef] [PubMed]
- Galie, N.; Ghofrani, H.A.; Torbicki, A.; Barst, R.J.; Rubin, L.J.; Badesch, D.; et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N. Engl. J. Med. 2005, 353, 2148–2157. [Google Scholar] [CrossRef]
- Chaouat, A.; Bugnet, A.S.; Kadaoui, N.; Schott, R.; Enache, I.; Ducolone, A.; et al. Severe pulmonary hypertension and chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2005, 172, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Behr, J.; Ryu, J.H. Pulmonary hypertension in interstitial lung disease. Eur. Respir. J. 2008, 31, 1357–1367. [Google Scholar] [CrossRef]
- Fedullo, P.F.; Auger, W.R.; Kerr, K.M.; Rubin, L.J. Chronic thromboembolic pulmonary hypertension. N. Engl. J. Med. 2001, 345, 1465–1472. [Google Scholar] [CrossRef]
- Jamieson, S.W.; Kapelanski, D.P.; Sakakibara, N.; Manecke, G.R.; Thistlethwaite, P.A.; Kerr, K.M.; et al. Pulmonary endarterectomy: Experience and lessons learned in 1,500 cases. Ann. Thorac. Surg. 2003, 76, 1457–1462. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, S.; Speich, R.; Domenighetti, G.; Geiser, T.; Aubert, J.D.; Rochat, T.; et al. Bosentan therapy for chronic thromboembolic pulmonary hypertension. A national open label study assessing the effect of Bosentan on haemodynamics, exercise capacity, quality of life, safety and tolerability in patients with chronic thromboembolic pulmonary hypertension (BOCTEPH-Study). Swiss Med Wkly. 2007, 137, 573–580. [Google Scholar] [PubMed]
- Hoeper, M.M.; Mayer, E.; Simonneau, G.; Rubin, L.J. Chronic thromboembolic pulmonary hypertension. Circulation 2006, 113, 2011–2020. [Google Scholar] [CrossRef]
- Fischler, M.; Speich, R.; Dorschner, L.; Nicod, L.; Domenighetti, G.; Tamm, M.; et al. Pulmonary hypertension in Switzerland: Treatment and clinical course. Swiss Med. Wkly. 2008, 138, 371–378. [Google Scholar]
- Dumitrascu, R.; Weissmann, N.; Ghofrani, H.A.; Dony, E.; Beuerlein, K.; Schmidt, H.; et al. Activation of soluble guanylate cyclase reverses experimental pulmonary hypertension and vascular remodeling. Circulation 2006, 113, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Evgenov, O.V.; Pacher, P.; Schmidt, P.M.; Hasko, G.; Schmidt, H.H.; Stasch, J.P. NO-independent stimulators and activators of soluble guanylate cyclase: Discovery and therapeutic potential. Nat. Rev. Drug Discov. 2006, 5, 755–768. [Google Scholar] [CrossRef]
- Schermuly, R.T.; Stasch, J.P.; Pullamsetti, S.S.; Middendorff, R.; Muller, D.; Schluter, K.D.; et al. Expression and function of soluble guanylate cyclase in pulmonary arterial hypertension. Eur. Respir. J. 2008, 32, 881–891. [Google Scholar] [CrossRef]
- Schermuly, R.T.; Dony, E.; Ghofrani, H.A.; Pullamsetti, S.; Savai, R.; Roth, M.; et al. Reversal of experimental pulmonary hypertension by PDGF inhibition. J. Clin. Invest. 2005, 115, 2811–2821. [Google Scholar] [CrossRef]
- Perros, F.; Montani, D.; Dorfmuller, P.; Durand-Gasselin, I.; Tcherakian, C.; Le Pavec, J.; et al. Platelet-derived growth factor expression and function in idiopathic pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2008, 178, 81–88. [Google Scholar] [CrossRef]
- Ghofrani, H.A.; Seeger, W.; Grimminger, F. Imatinib for the treatment of pulmonary arterial hypertension. N. Engl. J. Med. 2005, 353, 1412–1413. [Google Scholar] [CrossRef] [PubMed]
- Klein, M.; Schermuly, R.T.; Ellinghaus, P.; Milting, H.; Riedl, B.; Nikolova, S.; et al. Combined tyrosine and serine/threonine kinase inhibition by sorafenib prevents progression of experimental pulmonary hypertension and myocardial remodeling. Circulation 2008, 118, 2081–2090. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Morrell, N.W.; Archer, S.L.; Stenmark, K.R.; MacLean, M.R.; Lang, I.M.; et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2004, 43, 13S–24S. [Google Scholar] [CrossRef] [PubMed]
- Beyer, S.; Speich, R.; Fischler, M.; Maggiorini, M.; Ulrich, S. Long-term experience with oral or inhaled vasodilator combination therapy in patients with pulmonary hypertension. Swiss Med. Wkly. 2006, 136, 114–118. [Google Scholar] [CrossRef]
© 2009 by the author. Attribution-Non-Commercial-NoDerivatives 4.0.