
Abundant Species Diversity and Essential Functions of Bacterial Communities Associated with Dinoflagellates as Revealed from Metabarcoding Sequencing for Laboratory-Raised Clonal Cultures



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Strain Information, Grouping Details, and Sample Collection
2.2. DNA Extraction and PCR Amplification
2.3. Illumina MiSeq Sequencing
2.4. Sequencing Data Processing and Bioinformatic Analyses
2.5. Functional Annotation of the Presented Common Bacterial Communities
3. Results
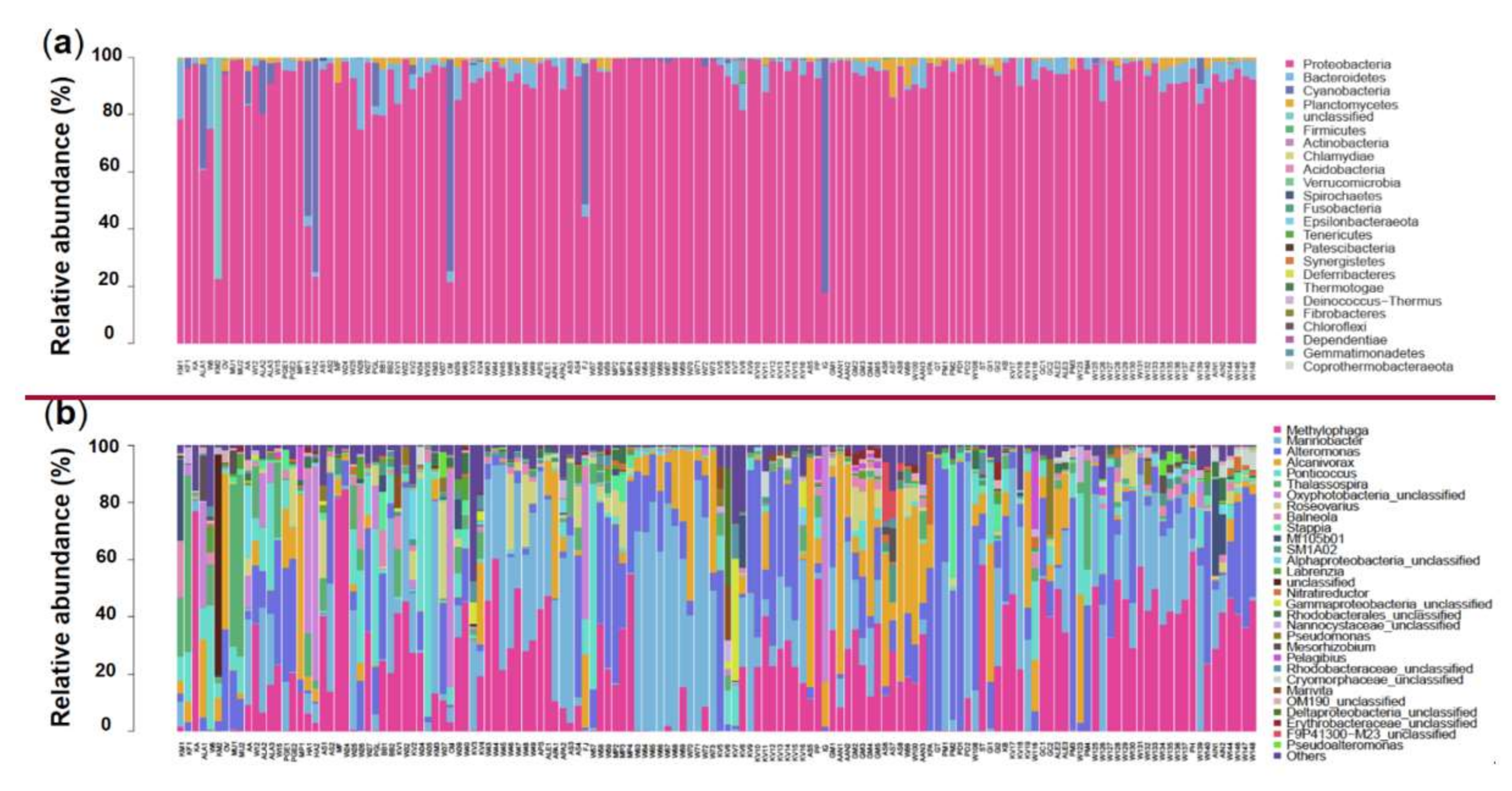
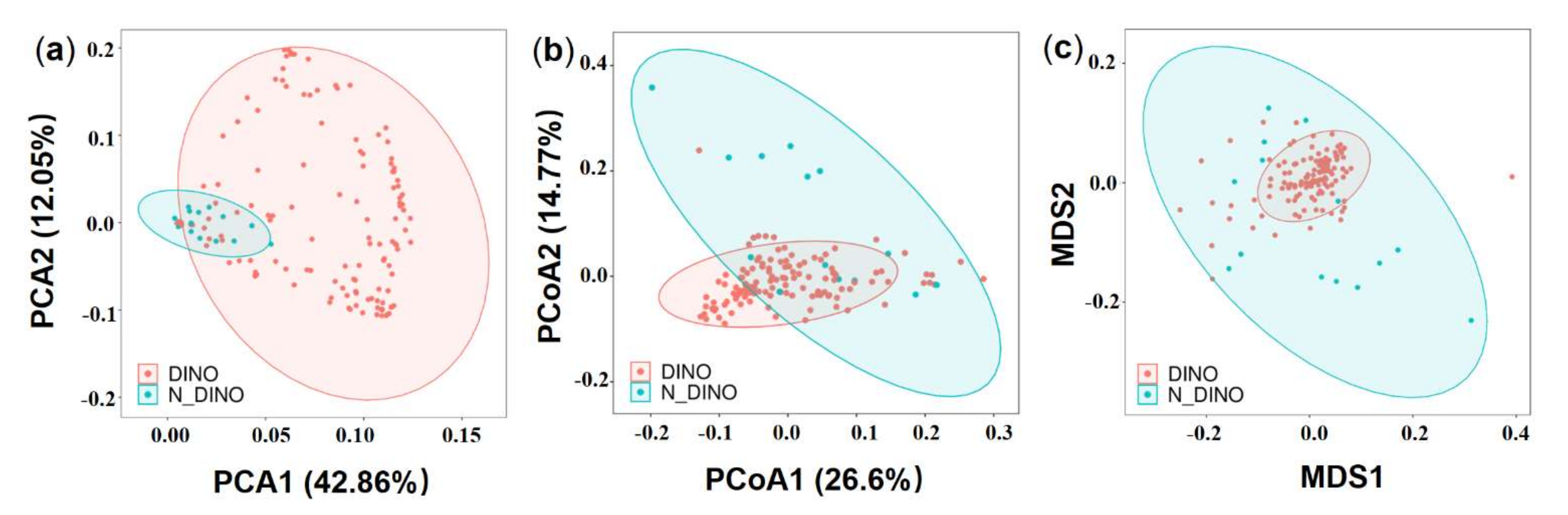
3.1. Global Overview of Bacterial Diversity and Community Composition Associated with Algal Cells
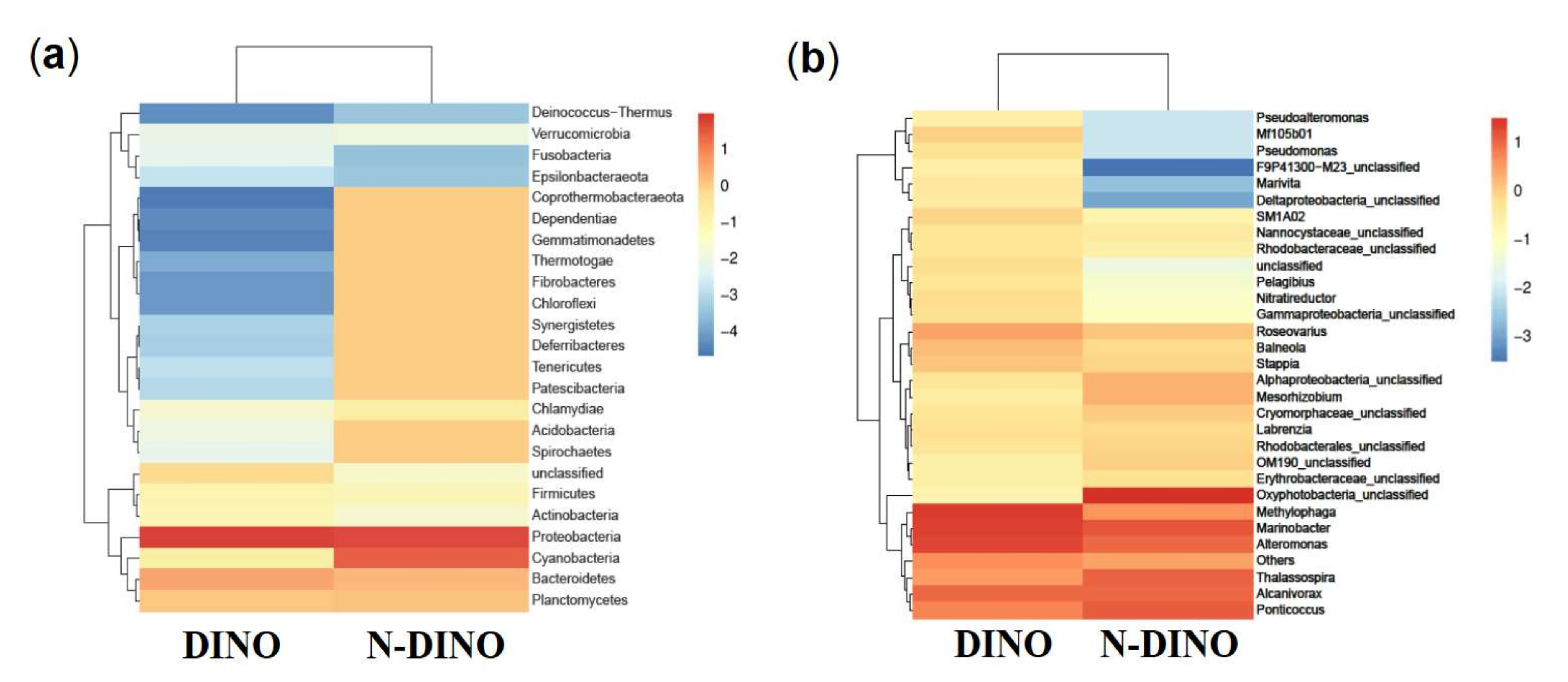
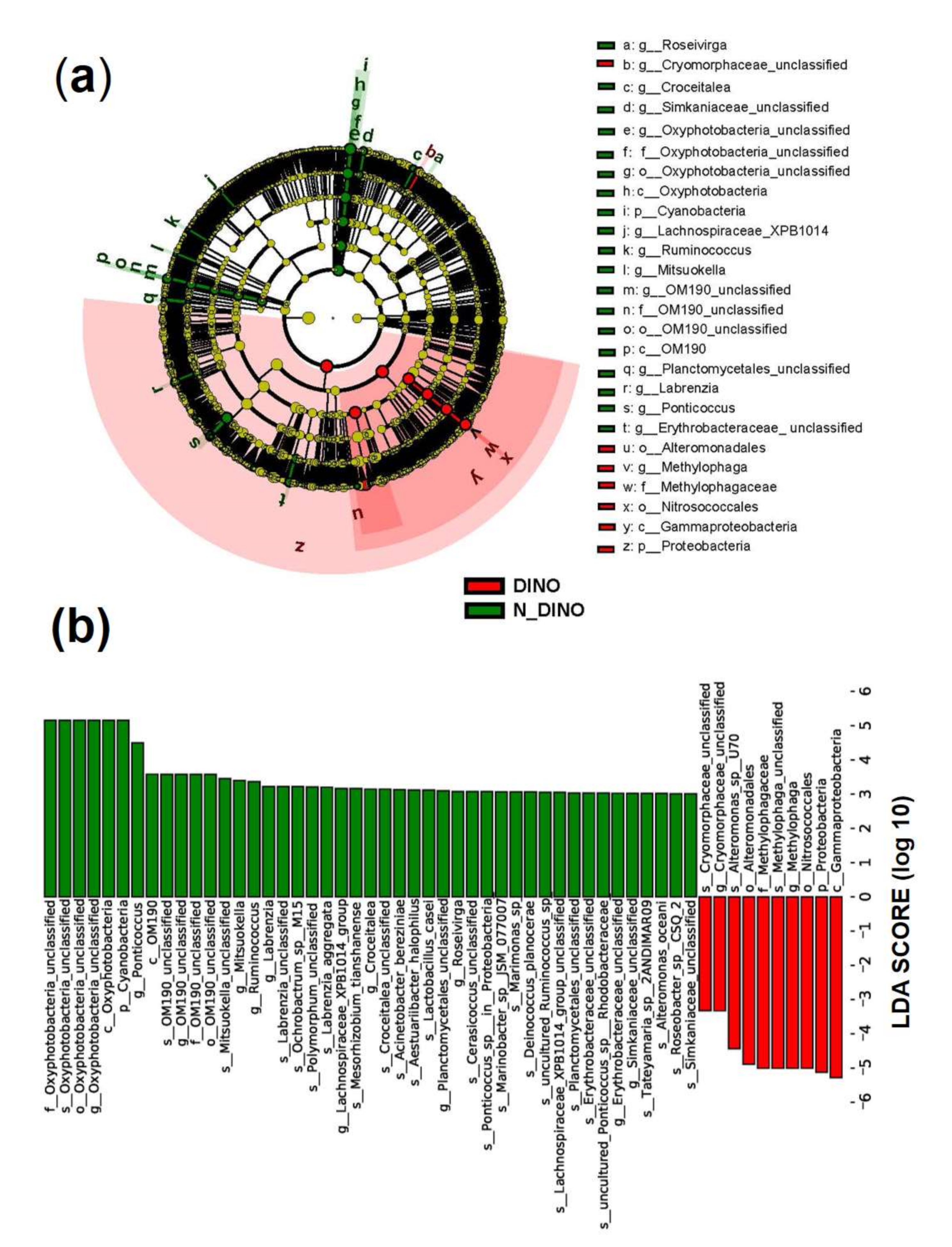
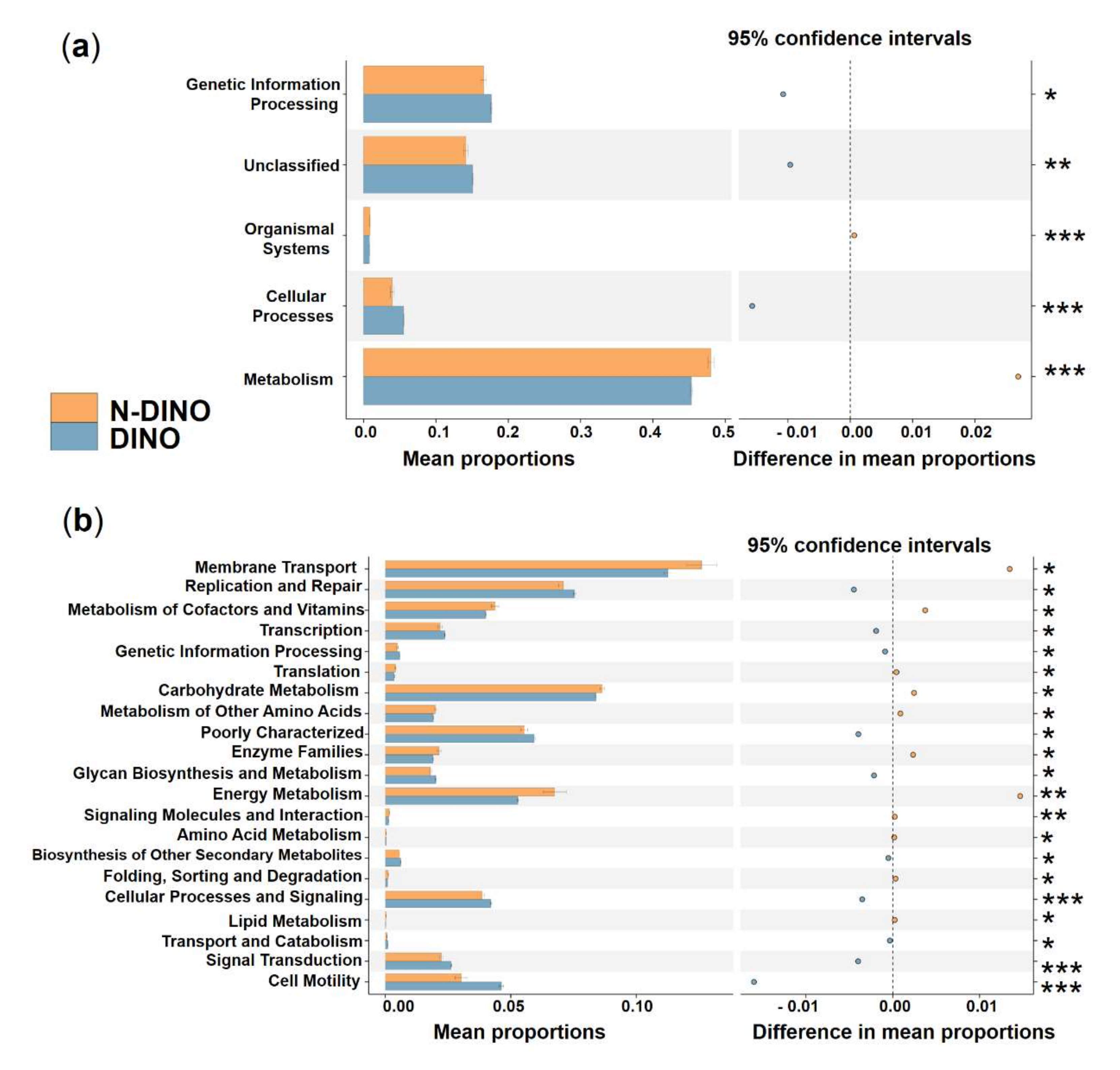
3.2. Species Composition of Bacterial Communities Associated with DINOs and N-DINOs and their Predicted Functions
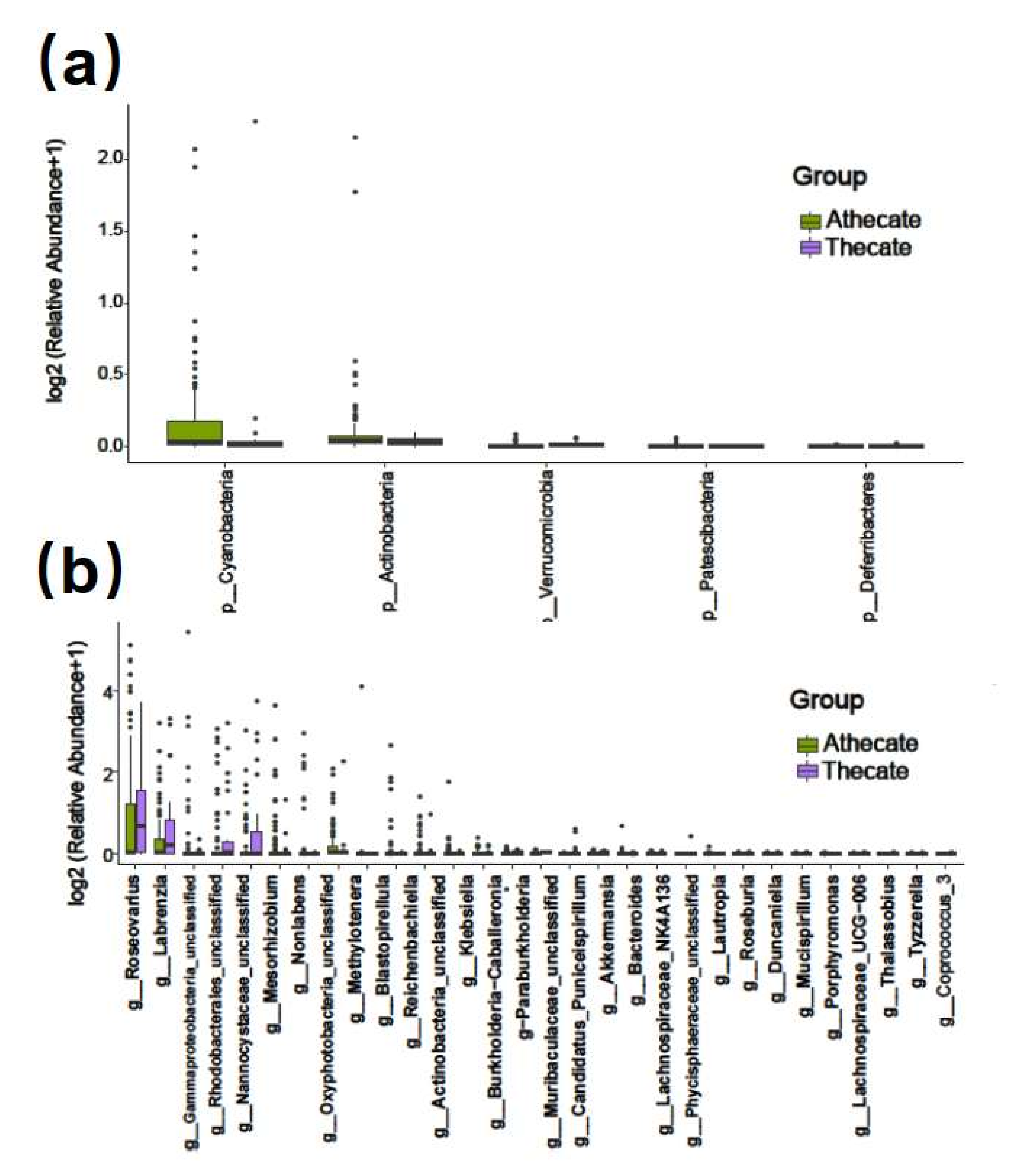
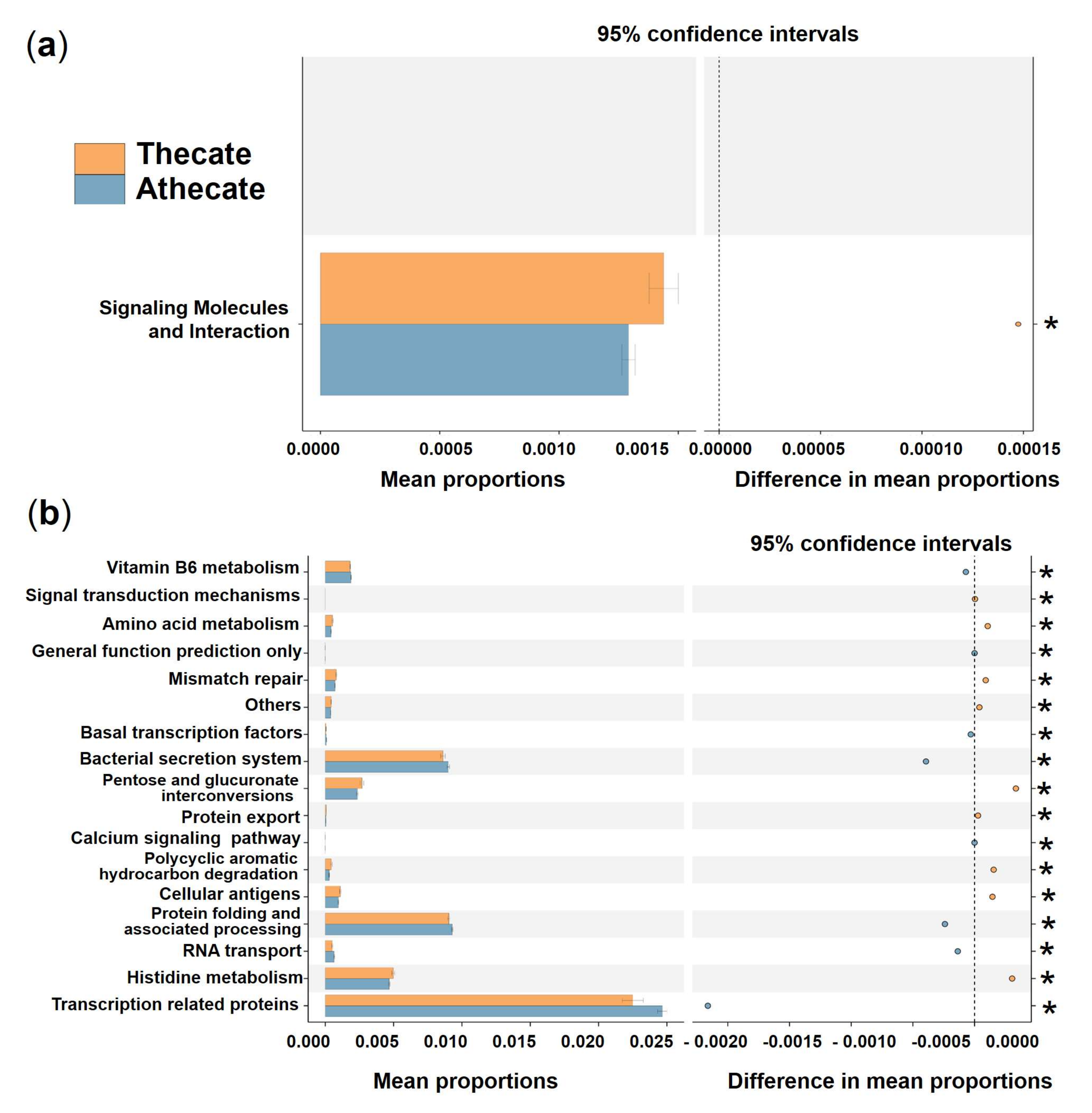
3.3. Comparative Analysis of the Species Composition and Predicted Function of Bacterial Communities between the Thecate and Athecate Groups
3.4. Comparative analysis of the species composition and predicted function of bacterial communities between the Anti and N-Anti groups
4. Discussion
4.1. The Long-Lasting Bacterial Associations to Laboratory-Raised Algal Cultures Hints Bilaterally or/and Unilaterally Beneficial Relationships between Algae and Bacteria
4.2. Bacterial Communities of Dinoflagellates Display Strong Conservation across Strains with an Enrichment of Methylophaga from the Class γ-Proteobacteria and Implies a Potentially Functional Group of Methylotrophs in Methane Consumption
4.3. Athecate Dinoflagellates Provided a Better Niche for Aerobic Cellulolytic Bacteria from the Phylum Actinobacteria and a Possible Reliance on Cellulose Utilization as Energy Source
4.4. Antibiotics Addition Does Not Significantly Affect Bacterial Diversity and Community Composition Associated with Our Microalgal Cultures
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cole, J.J. Interactions between bacteria and algae in aquatic ecosystems. Annu. Rev. Ecol. Syst. 1982, 13, 291–314. [Google Scholar] [CrossRef]
- Amin, S.A.; Hmelo, L.R.; Van Tol, H.M.; Durham, B.P.; Carlson, L.T.; Heal, K.R.; Morales, R.L.; Berthiaume, C.T.; Parker, M.S.; Djunaedi, B.; et al. Interaction and signaling between a cosmopolitan phytoplankton and associated bacteria. Nature 2015, 522, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Seymour, J.R.; Amin, S.A.; Raina, J.B.; Stocker, R. Zooming in on the phycosphere: The ecological interface for phytoplankton-bacteria relationships. Nat. Microbiol. 2017, 2, 17065. [Google Scholar] [CrossRef] [PubMed]
- Cirri, E.; Pohnert, G. Algae-bacteria interactions that balance the planktonic microbiome. New Phytol. 2019, 223, 100–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kouzuma, A.; Watanabe, K. Exploring the potential of algae/bacteria interactions. Curr. Pin. Biotech. 2015, 33, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Croft, M.; Lawrence, A.; Raux-Deery, E.; Warren, M.J.; Smith, A.G. Algae acquire vitamin B12 through a symbiotic relationship with bacteria. Nature 2005, 438, 90–93. [Google Scholar] [CrossRef]
- Droop, M.R. Vitamins, phytoplankton and bacteria: Symbiosis or scavenging? J. Plankton. Res. 2007, 29, 107–113. [Google Scholar] [CrossRef] [Green Version]
- Bolch, C.J.S.; Bejoy, T.A.; Green, D.H. Bacterial associates modify growth dynamics of the dinoflagellate Gymnodinium catenatum. Front. Microbiol. 2017, 8, 670. [Google Scholar] [CrossRef] [Green Version]
- Chai, Z.Y.; Hu, Z.X.; Deng, Y.Y.; Yang, Y.F.; Tang, Y.Z. Interactions between the seaweed Gracilaria and dinoflagellate Akashiwo sanguinea in an indoor co-cultivation system and the interference of bacteria. J. Appl. Phycol. 2021, 33, 3153–3163. [Google Scholar] [CrossRef]
- Bell, W.; Mitchell, R. Chemotactic and growth responses of marine bacteria to algal extracellular products. Biol. Bull. 1972, 143, 265–277. [Google Scholar] [CrossRef]
- Jasti, S.; Sieracki, M.E.; Poulton, N.J.; Giewat, M.W.; Rooney-Varga, J.N. Phylogenetic diversity and specificity of bacteria closely associated with Alexandrium sp. and other phytoplankton. Appl. Environ. Microbiol. 2005, 71, 3483–3494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, F.J.R.; Hoppenrath, M.; Saldarriaga, J.F. Dinoflagellate diversity and distribution. Biodivers. Conserv. 2008, 17, 407–418. [Google Scholar] [CrossRef]
- Jeong, H.J.; Kang, H.C.; Lim, A.S.; Jang, S.H.; Lee, K.; Lee, S.Y.; Ok, J.H.; You, J.H.; Kim, J.H.; Lee, K.H.; et al. Feeding diverse prey as an excellent strategy of mixotrophic dinoflagellates for global dominance. Sci. Adv. 2021, 7, eabe4214. [Google Scholar] [CrossRef] [PubMed]
- Hallegraeff, G.M.; Anderson, D.M.; Belin, C.; Bottein, M.Y.D.; Bresnan, E.; Chinain, M.; Zingone, A. Perceived global increase in algal blooms is attributable to intensified monitoring and emerging bloom impacts. Commun. Earth Environ. 2021, 2, 117. [Google Scholar] [CrossRef]
- Buchan, A.; LeCleir, G.R.; Gulvik, C.A.; González, J.M. Master recyclers: Features and functions of bacteria associated with phytoplankton blooms. Nat. Rev. Microbiol. 2014, 12, 686–698. [Google Scholar] [CrossRef]
- Zhou, J.; Richlen, M.L.; Sehein, T.R.; Kulis, D.M.; Anderson, D.M.; Cai, Z. Microbial community structure and associations during a marine dinoflagellate bloom. Front. Microbiol. 2018, 9, 1201. [Google Scholar] [CrossRef]
- Li, D.X.; Zhang, H.; Chen, X.H.; Xie, Z.X.; Zhang, Y.; Zhang, S.F.; Lin, L.; Chen, F.; Wang, D.Z. Metaproteomics reveals major microbial players and their metabolic activities during the blooming period of a marine dinoflagellate Prorocentrum donghaiense. Environ. Microbiol. 2018, 20, 632–644. [Google Scholar] [CrossRef]
- Alavi, M.; Miller, T.; Erlandson, K.; Schneider, R.; Belas, R. Bacterial community associated with Pfiesteria-like dinoflagellate cultures. Environ. Microbiol. 2001, 3, 380–396. [Google Scholar] [CrossRef]
- Hold, G.L.; Smith, E.A.; Birkbeck, T.H.; Gallacher, S. Comparison of paralytic shellfish toxin (PST) production by the dinoflagellates Alexandrium lusitanicum NEPCC 253 and Alexandrium tamarense NEPCC 407 in the presence and absence of bacteria. FEMS Microbiol. Ecol. 2001, 36, 223–234. [Google Scholar] [CrossRef]
- Seibold, A.; Wichels, A.; Schütt, C. Diversity of endocytic bacteria in the dinoflagellate Noctiluca scintillans. Aquat. Microb. Ecol. 2001, 25, 229–235. [Google Scholar] [CrossRef]
- Ferrier, M.; Martin, J.L.; Rooney-Varga, J.N. Stimulation of Alexandrium fundyense growth by bacterial assemblages from the Bay of Fundy. J. Appl. Microbiol. 2002, 92, 706–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, D.H.; Llewellyn, L.E.; Negri, A.P.; Blackburn, S.I.; Bolch, C.J.S. Phylogenetic and functional diversity of the cultivable bacterial community associated with the paralytic shellfish poisoning dinoflagellate Gymnodinium catenatum. FEMS Microbiol. Ecol. 2004, 47, 345–357. [Google Scholar] [CrossRef]
- Green, D.H.; Hart, M.C.; Blackburn, S.I.; Bolch, C.J.S. Bacterial diversity of Gymnodinium catenatum and the relationship to dinoflagellate toxicity. Aquat. Microb. Ecol. 2010, 61, 73–87. [Google Scholar] [CrossRef]
- Shin, H.; Lee, E.; Shin, J.; Ko, S.R.; Oh, H.S.; Ahn, C.Y.; Cho, S. Elucidation of the bacterial communities associated with the harmful microalgae Alexandrium tamarense and Cochlodinium polykrikoides using nanopore sequencing. Sci. Rep. 2018, 8, 5323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, B.S.; Guo, R.; Lim, W.A.; Ki, J.S. Pyrosequencing reveals specific associations of bacterial clades Roseobacter and Flavobacterium with the harmful dinoflagellate Cochlodinium polykrikoides growing in culture. Mar. Ecol. 2017, 38, e12474. [Google Scholar] [CrossRef]
- Guillard, R.R.L. Culture of phytoplankton for feeding marine invertebrates. In Culture of Marine Invertebrate Animals; Springer: Boston, MA, USA, 1975. [Google Scholar]
- Logue, J.B.; Stedmon, C.A.; Kellerman, A.M.; Nielsen, N.J.; Andersson, A.F.; Laudon, H.; Lindström, E.S.; Kritzberg, E.S. Experimental insights into the importance of aquatic bacterial community composition to the degradation of dissolved organic matter. ISME J. 2016, 10, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Gl€ockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Clarke, K.R. Non-parametric multivariate analyses of changes in community structure. Aust. J. Ecol. 1993, 18, 117–143. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polson, S.W. Comparative Analysis of Microbial Community Structure Associated with Acroporid Corals during a Disease Outbreak in the Florida Reef Tract. Ph.D. Thesis, Medical University of South Carolina, Charleston, SC, USA, 2007. [Google Scholar]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Langille, M.G. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef] [Green Version]
- Hattenrath-Lehmann, T.K.; Jankowiak, J.; Koch, F.; Gobler, C.J. Prokaryotic and eukaryotic microbiomes associated with blooms of the ichthyotoxic dinoflagellate Cochlodinium (Margalefidinium) polykrikoides in New York, USA, estuaries. PLoS ONE 2019, 14, e0223067. [Google Scholar] [CrossRef]
- Prieto, A.; Barber-Lluch, E.; Haernandez-Ruiz, M.; Martinez-Garcia, S.; Fernandez, E.; Tiera, E. Assessing the role of phytoplankton-bacterioplankton coupling in response the response of microbial plankton to nutrient additions. J. Plankton Res. 2015, 38, 55–63. [Google Scholar] [CrossRef]
- Hollibaugh, J.T.; Wong, P.S.; Murrell, M.C. Similarity of particleassociated and free-living bacterial communities in northern San Francisco Bay, California. Aquat. Microb. Ecol. 2000, 21, 103–114. [Google Scholar] [CrossRef] [Green Version]
- Neufeld, J.D.; Schäfer, H.; Cox, M.J.; Boden, R.; McDonald, I.R.; Murrell, J.C. Stable-isotope probing implicates Methylophaga spp. and novel Gammaproteobacteria in marine methanol and methylamine metabolism. ISME J. 2007, 1, 480–491. [Google Scholar] [CrossRef] [Green Version]
- Caruana, A.; Steinke, M.; Turner, S.; Malin, G. Concentrations of dimethylsulphoniopropionate and activities of dimethylsulphide-producing enzymes in batch cultures of nine dinoflagellate species. Biogeochemistry 2012, 110, 87–107. [Google Scholar] [CrossRef]
- Caruana, A.M.; Malin, G. The variability in DMSP content and DMSP lyase activity in marine dinoflagellates. Prog. Oceanogr. 2014, 120, 410–424. [Google Scholar] [CrossRef]
- Charlson, R.J.; Lovelock, J.E.; Andreae, M.O.; Warren, S.G. Oceanic phytoplankton, atmospheric sulphur, cloud albedo and climate. Nature 1987, 326, 655–661. [Google Scholar] [CrossRef]
- Horn, K. Alga-derived substrates select for distinct betaproteobacterial lineages and contribute to niche separation in limnohabitans strains. Appl. Environ. Microbiol. 2011, 77, 7307–7315. [Google Scholar]
- Boden, R.; Kelly, D.P.; Murrell, J.C.; Schäfer, H. Oxidation of dimethylsulfide to tetrathionate by Methylophaga thiooxidans sp. nov.: A new link in the sulfur cycle. Environ. Microbiol. 2010, 12, 2688–2699. [Google Scholar] [CrossRef]
- Bertram, P.A.; Karrasch, M.; Schmitz, R.A.; Böcher, R.; Albracht, S.P.; Thauer, R.K. Formylmethanofuran dehydrogenases from methanogenic Archaea Substrate specificity, EPR properties and reversible inactivation by cyanide of the molybdenum or tungsten iron-sulfur proteins. Eur. J. Biochem. 1994, 220, 477–484. [Google Scholar] [CrossRef]
- Pomper, B.K.; Vorholt, J.A.; Chistoserdova, L.; Lidstrom, M.E.; Thauer, R.K. A methenyl tetrahydromethanopterin cyclohydrolase and a methenyl tetrahydrofolate cyclohydrolase in Methylobacterium extorquens AM1. Eur. J. Biochem. 1999, 261, 475–480. [Google Scholar] [CrossRef] [Green Version]
- Tortorelli, G.; Oakley, C.A.; Davy, S.K.; van Oppen, M.J.; McFadden, G.I. Cell wall proteomic analysis of the cnidarian photosymbionts Breviolum minutum and Cladocopium goreaui. J. Eukaryot. Microbiol. 2022, 69, e12870. [Google Scholar] [CrossRef]
- Lau, R.K.; Kwok, A.; Chan, W.K.; Zhang, T.Y.; Wong, J.T. Mechanical characterization of cellulosic thecal plates in dinoflagellates by nanoindentation. J. Nanosci. Nanotechno. 2007, 7, 452–457. [Google Scholar] [CrossRef]
- Ozkan, M.; Desai, S.G.; Zhang, Y.; Stevenson, D.M.; Beane, J.; White, E.A.; Guerinot, M.L.; Lynd, L.R. Characterization of 13 newly isolated strains of anaerobic, cellulolytic, thermophilic bacteria. J. Ind. Microbiol. Biotechnol. 2001, 27, 275–280. [Google Scholar] [CrossRef]
- Patel, A.K.; Singhania, R.R.; Sim, S.J.; Pandey, A. Thermostable cellulases: Current status and perspectives. Bioresour. Technol. 2019, 279, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Olsen, G.J.; Woese, C.R.; Overbeek, R. The winds of (evolutionary) change: Breathing new life into microbiology. J. Bacteriol. 1994, 176, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Lynd, L.R.; Weimer, P.J.; Van Zyl, W.H.; Pretorius, I.S. Microbial cellulose utilization: Fundamentals and biotechnology. Microbiol. Mol. Biol. R. 2002, 66, 506–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kester, J.C.; Fortune, S.M. Persisters and beyond: Mechanisms of phenotypic drug resistance and drug tolerance in bacteria. Crit. Rev. Biochem. Mol. 2014, 49, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.D. Q&A: Antibiotic resistance: Where does it come from and what can we do about it? BMC Biol. 2010, 8, 123. [Google Scholar]
- Van den Bergh, B.; Fauvart, M.; Michiels, J. Formation, physiology, ecology, evolution and clinical importance of bacterial persisters. FEMS Microbiol. Rev. 2017, 41, 219–251. [Google Scholar] [CrossRef] [PubMed]
- Blair, J.M.A.; Webber, M.A.; Baylay, A.J.; Ogbolu, D.O.; Piddock, L.J.V. Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol. 2015, 13, 42–51. [Google Scholar] [CrossRef]
- Dang, H.; Lovell, C.R. Microbial surface colonization and biofilm development in marine environments. Microbiol. Mol. Biol. 2016, 80, 91–138. [Google Scholar] [CrossRef] [Green Version]
- Biegala, I.C.; Kennaway, G.; Alverca, E.; Lennon, J.F.; Vaulot, D.; Simon, N. Identification of bacteria associated with dinoflagellates (Dinophyceae) Alexandrium spp. using tyramide signal amplification-fluorescent in situ hybridization and confocal microscopy. J. Phycol. 2002, 38, 404–411. [Google Scholar] [CrossRef] [Green Version]
- Maas, E.W.; Latter, R.M.; Thiele, J.; Waite, A.M.; Brooks, H.J.L. Effect of multiple antibiotic treatments on a paralytic shellfish toxin-producing culture of the dinoflagellate Alexandrium minutum. Aquat. Microb. Ecol. 2007, 48, 255–260. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deng, Y.; Wang, K.; Hu, Z.; Tang, Y.-Z. Abundant Species Diversity and Essential Functions of Bacterial Communities Associated with Dinoflagellates as Revealed from Metabarcoding Sequencing for Laboratory-Raised Clonal Cultures. Int. J. Environ. Res. Public Health 2022, 19, 4446. https://doi.org/10.3390/ijerph19084446
Deng Y, Wang K, Hu Z, Tang Y-Z. Abundant Species Diversity and Essential Functions of Bacterial Communities Associated with Dinoflagellates as Revealed from Metabarcoding Sequencing for Laboratory-Raised Clonal Cultures. International Journal of Environmental Research and Public Health. 2022; 19(8):4446. https://doi.org/10.3390/ijerph19084446
Chicago/Turabian StyleDeng, Yunyan, Kui Wang, Zhangxi Hu, and Ying-Zhong Tang. 2022. "Abundant Species Diversity and Essential Functions of Bacterial Communities Associated with Dinoflagellates as Revealed from Metabarcoding Sequencing for Laboratory-Raised Clonal Cultures" International Journal of Environmental Research and Public Health 19, no. 8: 4446. https://doi.org/10.3390/ijerph19084446
APA StyleDeng, Y., Wang, K., Hu, Z., & Tang, Y.-Z. (2022). Abundant Species Diversity and Essential Functions of Bacterial Communities Associated with Dinoflagellates as Revealed from Metabarcoding Sequencing for Laboratory-Raised Clonal Cultures. International Journal of Environmental Research and Public Health, 19(8), 4446. https://doi.org/10.3390/ijerph19084446