
Evolution of Singlet Oxygen by Activating Peroxydisulfate and Peroxymonosulfate: A Review


 and
and 
Abstract
1. Introduction
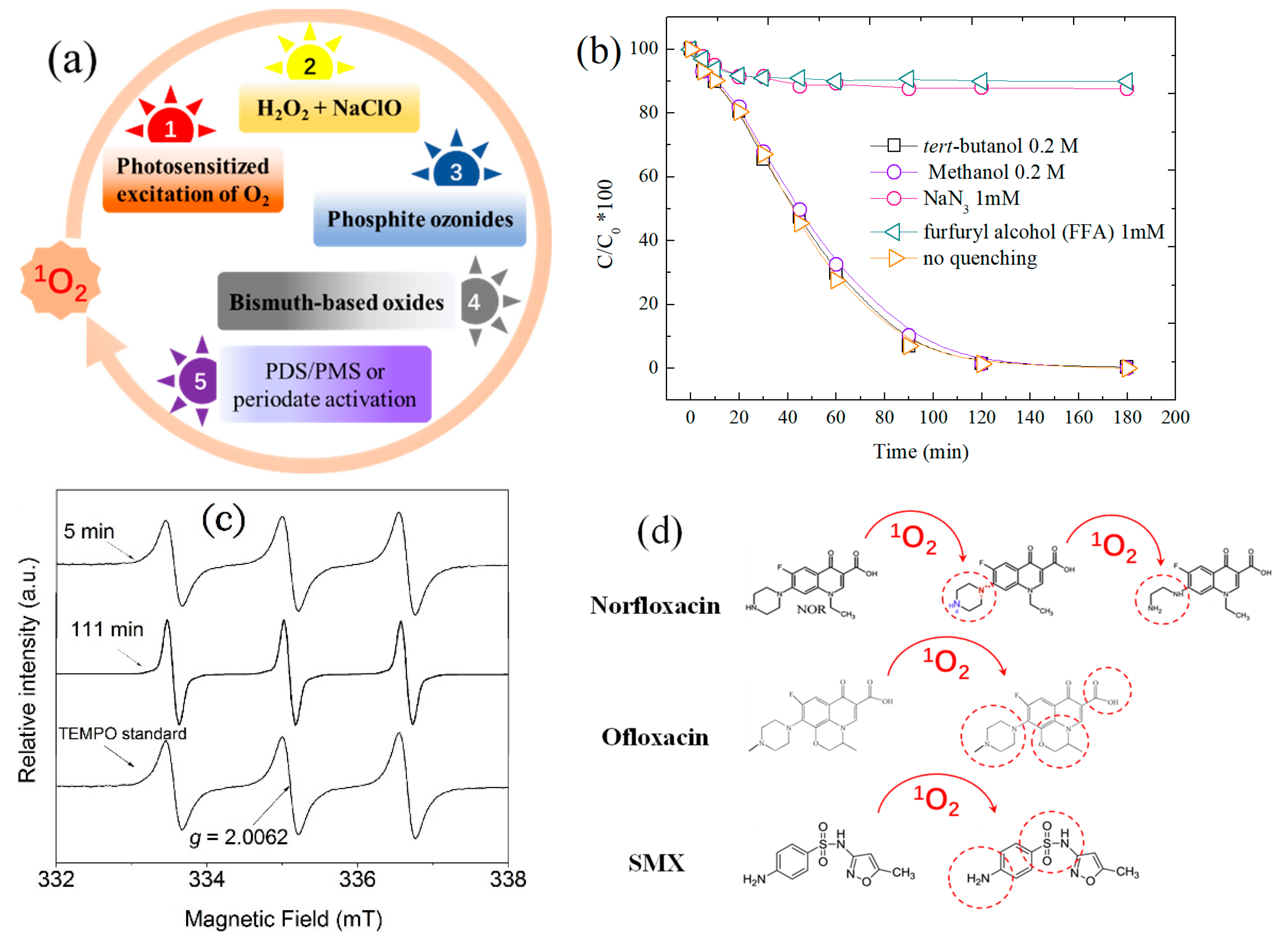
2. Chemical Feature of 1O2 and Its Oxidation
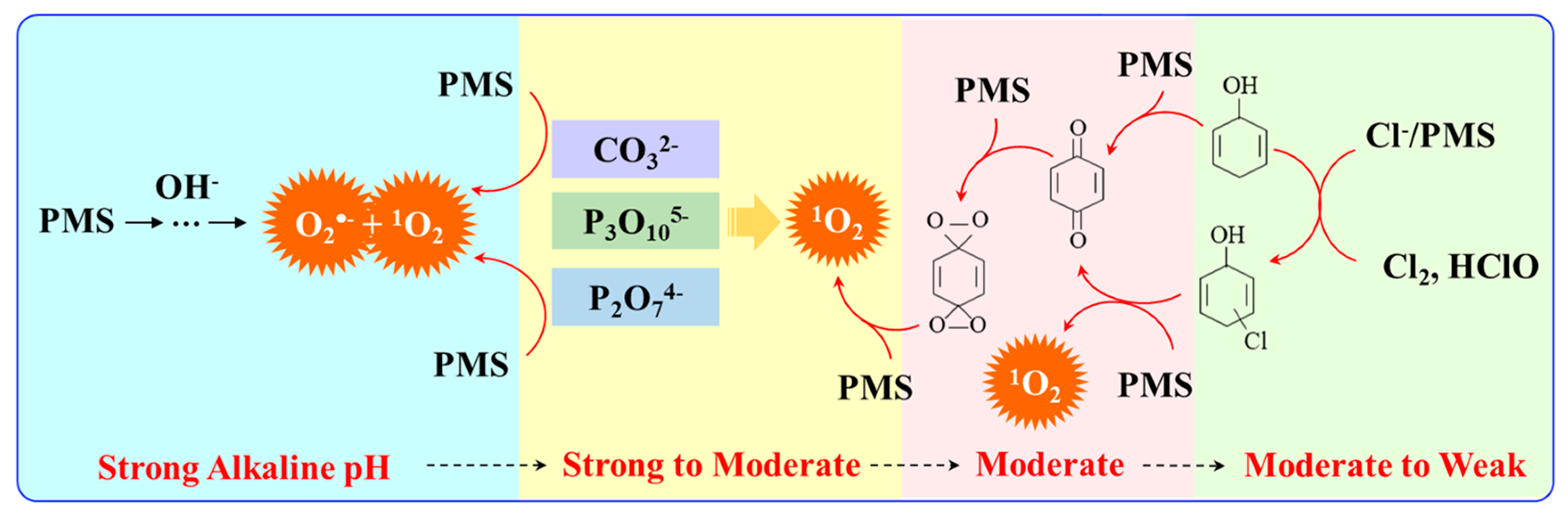
3. Evolution of 1O2 by Activating PDS/PMS in Homogenous Systems




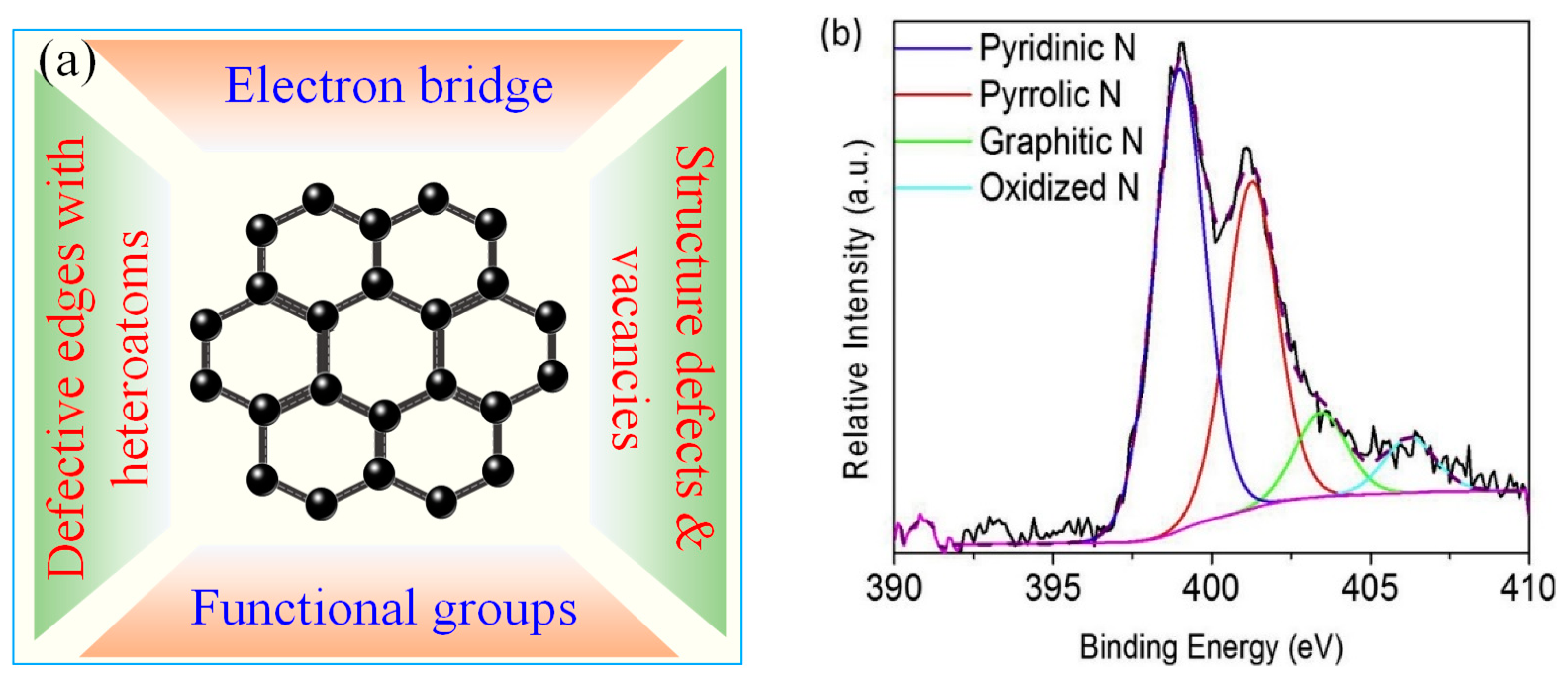
4. Evolution of 1O2 by Activating PDS/PMS with Metal-Free Carbon Catalysts
4.1. Carbon Nanotubes (CNTs)
4.2. GO/rGO
4.3. Biochar
4.4. Other MFCMs
5. Evolution of 1O2 by Activating PDS/PMS with Metal Catalysts and Their Composite
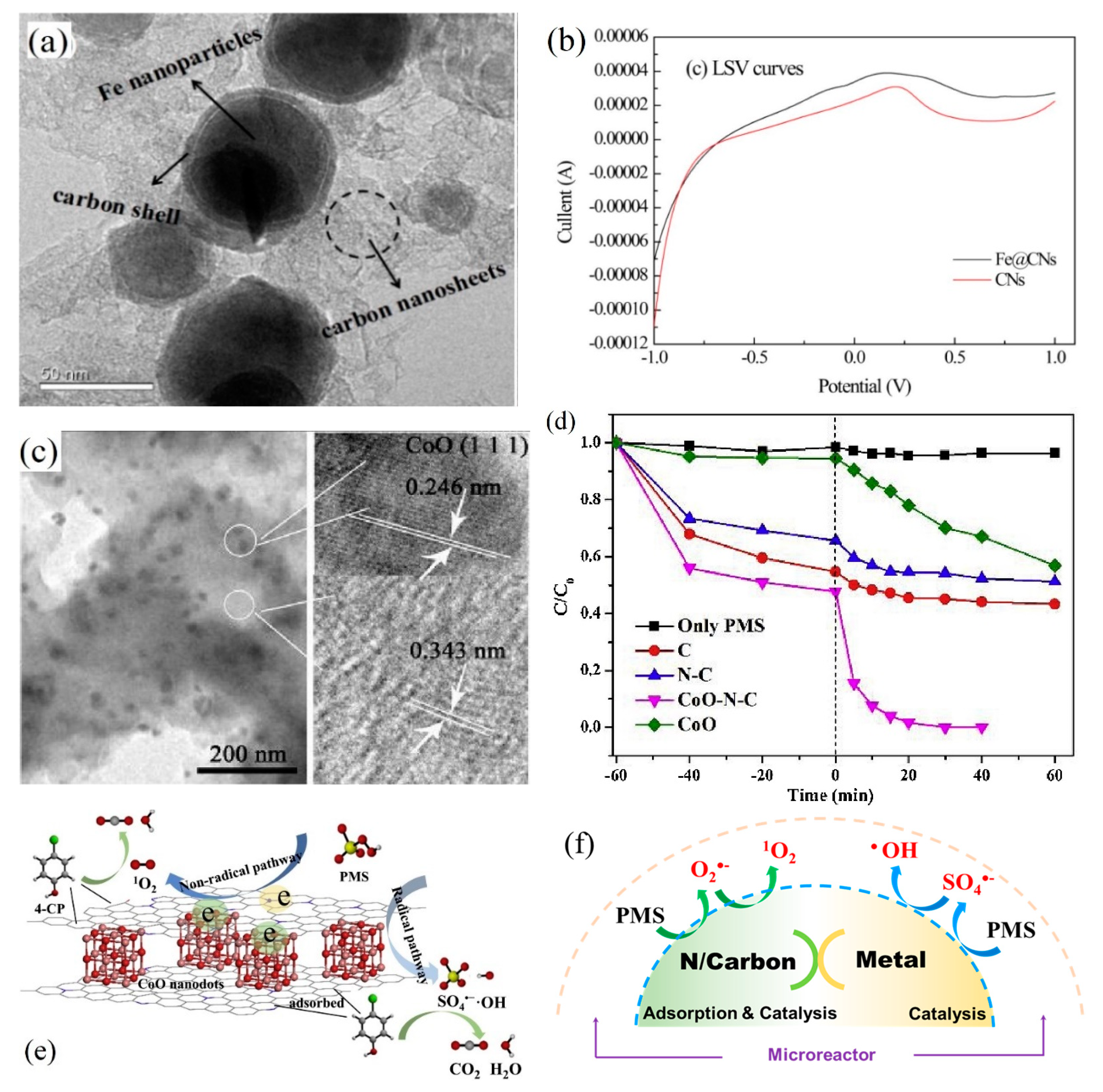
5.1. Iron-Based Catalysts
5.2. Cobalt-Based Catalysts
5.3. Manganese-Based Catalysts
5.4. Copper-Based Catalysts
5.5. Other Metallic Catalysts
6. Implications for In Situ Applications and Future Perspectives
6.1. Implications for In Situ Applications
6.2. Future Perspectives
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Liang, C.; Su, H.-W. Identification of Sulfate and Hydroxyl Radicals in Thermally Activated Persulfate. Ind. Eng. Chem. Res. 2009, 48, 5558–5562. [Google Scholar] [CrossRef]
- Tsitonaki, A.; Petri, B.; Crimi, M.; MosbÆK, H.; Siegrist, R.L.; Bjerg, P.L. In Situ Chemical Oxidation of Contaminated Soil and Groundwater Using Persulfate: A Review. Crit. Rev. Environ. Sci. Technol. 2010, 40, 55–91. [Google Scholar] [CrossRef]
- Wang, J.; Wang, S. Activation of persulfate (PS) and peroxymonosulfate (PMS) and application for the degradation of emerging contaminants. Chem. Eng. J. 2018, 334, 1502–1517. [Google Scholar] [CrossRef]
- Oh, W.-D.; Dong, Z.; Lim, T.-T. Generation of sulfate radical through heterogeneous catalysis for organic contaminants removal: Current development, challenges and prospects. Appl. Catal. B Environ. 2016, 194, 169–201. [Google Scholar] [CrossRef]
- Du, J.; Bao, J.; Liu, Y.; Kim, S.H.; Dionysiou, D.D. Facile preparation of porous Mn/Fe3O4 cubes as peroxymonosulfate activating catalyst for effective bisphenol A degradation. Chem. Eng. J. 2019, 376, 119193. [Google Scholar] [CrossRef]
- Duan, X.; Sun, H.; Shao, Z.; Wang, S. Nonradical reactions in environmental remediation processes: Uncertainty and challenges. Appl. Catal. B Environ. 2018, 224, 973–982. [Google Scholar] [CrossRef]
- Bokare, A.D.; Choi, W. Singlet-Oxygen Generation in Alkaline Periodate Solution. Environ. Sci. Technol. 2015, 49, 14392–14400. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Tian, X.; Nie, Y.; Yang, C.; Zhou, Z.; Wang, Y. Promoted peroxymonosulfate activation into singlet oxygen over perovskite for ofloxacin degradation by controlling the oxygen defect concentration. Chem. Eng. J. 2019, 359, 828–839. [Google Scholar] [CrossRef]
- Liu, S.; Zhao, C.; Wang, Z.; Ding, H.; Deng, H.; Yang, G.; Li, J.; Zheng, H. Urea-assisted one-step fabrication of a novel nitrogen-doped carbon fiber aerogel from cotton as metal-free catalyst in peroxymonosulfate activation for efficient degradation of carbamazepine. Chem. Eng. J. 2020, 386, 124015. [Google Scholar] [CrossRef]
- DeRosa, M.C.; Crutchley, R.J. Photosensitized singlet oxygen and its applications. Coord. Chem. Rev. 2002, 233, 351–371. [Google Scholar]
- Peters, J.W.; Bekowies, P.J.; Winer, A.M.; Pitts, J.N. Potassium perchromate as a source of singlet oxygen. J. Am. Chem. Soc. 1975, 97, 3299–3306. [Google Scholar] [CrossRef]
- Stephenson, L.M.; McClure, D.E. Mechanisms in phosphite ozonide decomposition to phosphate esters and singlet oxygen. J. Am. Chem. Soc. 1973, 95, 3074–3076. [Google Scholar] [CrossRef]
- Foote, C.S.; Wexler, S.; Ando, W.; Higgins, R. Chemistry of singlet oxygen. IV. Oxygenations with hypochlorite-hydrogen peroxide. J. Am. Chem. Soc. 1968, 90, 975–981. [Google Scholar] [CrossRef]
- Du, J.; Xiao, G.; Xi, Y.; Zhu, X.; Su, F.; Kim, S.H. Periodate activation with manganese oxides for sulfanilamide degradation. Water Res. 2020, 169, 115278. [Google Scholar] [CrossRef]
- Yu, K.; Yang, S.; Boyd, S.A.; Chen, H.; Sun, C. Efficient degradation of organic dyes by BiAgxOy. J. Hazard. Mater. 2011, 197, 88–96. [Google Scholar] [CrossRef]
- Yu, J.; Feng, H.; Tang, L.; Pang, Y.; Zeng, G.; Lu, Y.; Dong, H.; Wang, J.; Liu, Y.; Feng, C.; et al. Metal-free carbon materials for persulfate-based advanced oxidation process: Microstructure, property and tailoring. Prog. Mater. Sci. 2020, 111, 100654. [Google Scholar] [CrossRef]
- Ike, I.A.; Linden, K.G.; Orbell, J.D.; Duke, M. Critical review of the science and sustainability of persulphate advanced oxidation processes. Chem. Eng. J. 2018, 338, 651–669. [Google Scholar] [CrossRef]
- Wang, Y.; Indrawirawan, S.; Duan, X.; Sun, H.; Ang, H.M.; Tadé, M.O.; Wang, S. New insights into heterogeneous generation and evolution processes of sulfate radicals for phenol degradation over one-dimensional α-MnO2 nanostructures. Chem. Eng. J. 2015, 266, 12–20. [Google Scholar] [CrossRef]
- Zhu, S.; Li, X.; Kang, J.; Duan, X.; Wang, S. Persulfate activation on crystallographic manganese oxides: Mechanism of singlet oxygen evolution for nonradical selective degradation of aqueous contaminants. Environ. Sci. Technol. 2019, 53, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Cheng, M.; Zhong, H.; Liu, Z.; Liu, Y.; Yang, X.; Liang, Q. Iron-mediated activation of persulfate and peroxymonosulfate in both homogeneous and heterogeneous ways: A review. Chem. Eng. J. 2020, 384, 123265. [Google Scholar] [CrossRef]
- Zhao, Q.; Mao, Q.; Zhou, Y.; Wei, J.; Liu, X.; Yang, J.; Luo, L.; Zhang, J.; Chen, H.; Chen, H.; et al. Metal-free carbon materials-catalyzed sulfate radical-based advanced oxidation processes: A review on heterogeneous catalysts and applications. Chemosphere 2017, 189, 224–238. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer, C.; Schmidt, R. Physical Mechanisms of Generation and Deactivation of Singlet Oxygen. Chem. Rev. 2003, 103, 1685–1757. [Google Scholar] [CrossRef] [PubMed]
- Min, D.B.; Boff, J.M. Chemistry and Reaction of Singlet Oxygen in Foods. Compr. Rev. Food Sci. Food Saf. 2002, 1, 58–72. [Google Scholar] [CrossRef] [PubMed]
- Neta, P.; Huie, R.E.; Ross, A.B. Rate Constants for Reactions of Inorganic Radicals in Aqueous Solution. J. Phys. Chem. Ref. Data 1988, 17, 1027–1284. [Google Scholar] [CrossRef]
- Asghar, A.; Abdul Raman, A.A.; Wan Daud, W.M.A. Advanced oxidation processes for in-situ production of hydrogen peroxide/hydroxyl radical for textile wastewater treatment: A review. J. Clean. Prod. 2015, 87, 826–838. [Google Scholar] [CrossRef]
- Mostafa, S.; Rosario-Ortiz, F.L. Singlet Oxygen Formation from Wastewater Organic Matter. Environ. Sci. Technol. 2013, 47, 8179–8186. [Google Scholar] [CrossRef]
- Chen, X.; Oh, W.-D.; Lim, T.-T. Graphene-and CNTs-based carbocatalysts in persulfates activation: Material design and catalytic mechanisms. Chem. Eng. J. 2018, 354, 941–976. [Google Scholar] [CrossRef]
- Krieger-Liszkay, A. Singlet oxygen production in photosynthesis. J. Exp. Bot. 2005, 56, 337–346. [Google Scholar] [CrossRef]
- García-Fresnadillo, D. Singlet Oxygen Photosensitizing Materials for Point-of-Use Water Disinfection with Solar Reactors. ChemPhotoChem 2018, 2, 512–534. [Google Scholar] [CrossRef]
- Pibiri, I.; Buscemi, S.; Piccionello, A.P.; Pace, A. Photochemically produced singlet oxygen. ChemPhotoChem 2018, 2, 535–547. [Google Scholar] [CrossRef]
- Chen, L.; Yamane, S.; Mizukado, J.; Suzuki, Y.; Kutsuna, S.; Uchimaru, T.; Suda, H. ESR study of singlet oxygen generation and its behavior during the photo-oxidation of P3HT in solution. Chem. Phys. Lett. 2015, 624, 87–92. [Google Scholar] [CrossRef]
- Rkanofsky, J. Assay for Singlet-Oxygen Generation by Peroxidases Using 1270-nm Chemiluminescence, InSinglet Oxygen, UV-A, and Ozone; Academic Press: San Diego, CA, USA, 2000; Volume 319, pp. 59–67. [Google Scholar]
- Yuan, Y.; Zhang, C.-J.; Xu, S.; Liu, B. A self-reporting AIE probe with a built-in singlet oxygen sensor for targeted photodynamic ablation of cancer cells. Chem. Sci. 2016, 7, 1862–1866. [Google Scholar] [CrossRef]
- Zhou, Y.; Jiang, J.; Gao, Y.; Pang, S.Y.; Yang, Y.; Ma, J.; Gu, J.; Li, J.; Wang, Z.; Wang, L.H.; et al. Activation of peroxymonosulfate by phenols: Important role of quinone intermediates and involvement of singlet oxygen. Water Res. 2017, 125, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Li, C.X.; Wang, Y.J.; Chen, C.B.; Fu, X.Z.; Cui, S.; Lu, J.Y.; Liu, H.Q.; Li, W.W. Interactions between chlorophenols and peroxymonosulfate: pH dependency and reaction pathways. Sci. Total Environ. 2019, 664, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Koppenol, W.H.; Stanbury, D.M.; Bounds, P.L. Electrode potentials of partially reduced oxygen species, from dioxygen to water. Free Radic. Biol. Med. 2010, 49, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Semyachkina-Glushkovskaya, O.V.; Sokolovski, S.G.; Goltsov, A.; Gekaluyk, A.S.; Saranceva, E.I.; Bragina, O.A.; Tuchin, V.V.; Rafailov, E.U. Laser-induced generation of singlet oxygen and its role in the cerebrovascular physiology. Prog. Quantum Electron. 2017, 55, 112–128. [Google Scholar] [CrossRef]
- Niu, X.-Z.; Busetti, F.; Langsa, M.; Croué, J.-P. Roles of singlet oxygen and dissolved organic matter in self-sensitized photo-oxidation of antibiotic norfloxacin under sunlight irradiation. Water Res. 2016, 106, 214–222. [Google Scholar] [CrossRef]
- Montgomery, R.E. Catalysis of peroxymonosulfate reactions by ketones. J. Am. Chem. Soc. 1974, 96, 7820–7821. [Google Scholar] [CrossRef]
- Lange, A.; Brauer, H.D. On the formation of dioxiranes and of singlet oxygen by the ketone-catalysed decomposition of Caro’s acid Journal of the chemical society perkin transactions. J. Chem. Soc. Perkin Trans. 1996, 25, 805–811. [Google Scholar] [CrossRef]
- Zhou, Y.; Jiang, J.; Gao, Y.; Ma, J.; Pang, S.Y.; Li, J.; Lu, X.T.; Yuan, L.P. Activation of Peroxymonosulfate by Benzoquinone: A Novel Nonradical Oxidation Process. Environ. Sci. Technol. 2015, 49, 12941–12950. [Google Scholar] [CrossRef]
- Gallopo, A.R.; Edwards, J.O. Kinetics and mechanism of the oxidation of pyridine by Caro’s acid catalyzed by ketones. J. Org. Chem. 1981, 46, 1684–1688. [Google Scholar] [CrossRef]
- Zhang, H.; Yu, K.; He, J.; Li, N.; You, H.; Jiang, J. Droplet spray ionization mass spectrometry for real-time monitoring of activation of peroxymonosulfate by 1,4-benzoquinone. Microchem. J. 2018, 139, 437–442. [Google Scholar] [CrossRef]
- Li, C.-X.; Chen, C.-B.; Wang, Y.-J.; Fu, X.-Z.; Cui, S.; Lu, J.-Y.; Li, J.; Liu, H.-Q.; Li, W.-W.; Lau, T.-C. Insights on the pH-dependent roles of peroxymonosulfate and chlorine ions in phenol oxidative transformation. Chem. Eng. J. 2019, 362, 570–575. [Google Scholar] [CrossRef]
- Qi, C.; Liu, X.; Ma, J.; Lin, C.; Li, X.; Zhang, H. Activation of peroxymonosulfate by base: Implications for the degradation of organic pollutants. Chemosphere 2016, 151, 280–288. [Google Scholar] [CrossRef]
- Burns, J.M.; Cooper, W.J.; Ferry, J.L.; King, D.W.; DiMento, B.P.; McNeill, K.; Miller, C.J.; Miller, W.L.; Peake, B.M.; Rusak, S.A.; et al. Methods for reactive oxygen species (ROS) detection in aqueous environments. Aquat. Sci. 2012, 74, 683–734. [Google Scholar] [CrossRef]
- Lou, X.; Fang, C.; Geng, Z.; Jin, Y.; Xiao, D.; Wang, Z.; Liu, J.; Guo, Y. Significantly enhanced base activation of peroxymonosulfate by polyphosphates: Kinetics and mechanism. Chemosphere 2017, 173, 529–534. [Google Scholar] [CrossRef]
- Rao, L.; Yang, Y.; Chen, L.; Liu, X.; Chen, H.; Yao, Y.; Wang, W. Highly efficient removal of organic pollutants via a green catalytic oxidation system based on sodium metaborate and peroxymonosulfate. Chemosphere 2020, 238, 124687. [Google Scholar] [CrossRef] [PubMed]
- Nie, M.; Zhang, W.; Yan, C.; Xu, W.; Wu, L.; Ye, Y.; Hu, Y.; Dong, W. Enhanced removal of organic contaminants in water by the combination of peroxymonosulfate and carbonate. Sci. Total Environ. 2019, 647, 734–743. [Google Scholar] [CrossRef]
- Wang, S.; Wang, J. Treatment of membrane filtration concentrate of coking wastewater using PMS_chloridion oxidation process. Chem. Eng. J. 2019, 122361. [Google Scholar] [CrossRef]
- Yang, F.; Huang, Y.; Fang, C.; Xue, Y.; Ai, L.; Liu, J.; Wang, Z. Peroxymonosulfate/base process in saline wastewater treatment: The fight between alkalinity and chloride ions. Chemosphere 2018, 199, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Xiao, R.; Luo, Z.; Wei, Z.; Luo, S.; Spinney, R.; Yang, W.; Dionysiou, D.D. Activation of peroxymonosulfate/persulfate by nanomaterials for sulfate radical-based advanced oxidation technologies. Curr. Opin. Chem. Eng. 2018, 19, 51–58. [Google Scholar] [CrossRef]
- Duan, X.; Sun, H.; Ao, Z.; Zhou, L.; Wang, G.; Wang, S. Unveiling the active sites of graphene-catalyzed peroxymonosulfate activation. Carbon 2016, 107, 371–378. [Google Scholar] [CrossRef]
- Chen, H.; Carroll, K.C. Metal-free catalysis of persulfate activation and organic-pollutant degradation by nitrogen-doped graphene and aminated graphene. Environ. Pollut. 2016, 215, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Miao, W.; Fang, X.; Tang, Y.; Wu, D.; Mao, S. MOF-derived metal-free N-doped porous carbon mediated peroxydisulfate activation via radical and non-radical pathways: Role of graphitic N and CO. Chem. Eng. J. 2020, 380, 122584. [Google Scholar] [CrossRef]
- Ren, W.; Xiong, L.; Yuan, X.; Yu, Z.; Zhang, H.; Duan, X.; Wang, S. Activation of Peroxydisulfate on Carbon Nanotubes: Electron-Transfer Mechanism. Environ. Sci. Technol. 2019, 53, 14595–14603. [Google Scholar] [CrossRef] [PubMed]
- Yun, E.-T.; Lee, J.H.; Kim, J.; Park, H.-D.; Lee, J. Identifying the Nonradical Mechanism in the Peroxymonosulfate Activation Process: Singlet Oxygenation Versus Mediated Electron Transfer. Environ. Sci. Technol. 2018, 52, 7032–7042. [Google Scholar] [CrossRef]
- Yun, E.-T.; Yoo, H.-Y.; Bae, H.; Kim, H.-I.; Lee, J. Exploring the Role of Persulfate in the Activation Process: Radical Precursor Versus Electron Acceptor. Environ. Sci. Technol. 2017, 51, 10090–10099. [Google Scholar] [CrossRef]
- Ren, W.; Xiong, L.; Nie, G.; Zhang, H.; Duan, X.; Wang, S. Insights into the Electron-Transfer Regime of Peroxydisulfate Activation on Carbon Nanotubes: The Role of Oxygen Functional Groups. Environ. Sci. Technol. 2020, 54, 1267–1275. [Google Scholar] [CrossRef]
- Cheng, X.; Guo, H.; Zhang, Y.; Wu, X.; Liu, Y. Non-photochemical production of singlet oxygen via activation of persulfate by carbon nanotubes. Water Res. 2017, 113, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Kwan, C.; Suvorova, A.; Ang, H.M.; Tadé, M.O.; Wang, S. Catalytic oxidation of organic pollutants on pristine and surface nitrogen-modified carbon nanotubes with sulfate radicals. Appl. Catal. B Environ. 2014, 154, 134–141. [Google Scholar] [CrossRef]
- Ren, W.; Nie, G.; Zhou, P.; Zhang, H.; Duan, X.; Wang, S. The Intrinsic Nature of Persulfate Activation and N-Doping in Carbocatalysis. Environ. Sci. Technol. 2020, 54, 6438–6447. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.; Ao, Z.; Zhou, L.; Sun, H.; Wang, G.; Wang, S. Occurrence of radical and nonradical pathways from carbocatalysts for aqueous and nonaqueous catalytic oxidation. Appl. Catal. Environ. 2016, 188, 98–105. [Google Scholar] [CrossRef]
- Wang, X.; Qin, Y.; Zhu, L.; Tang, H. Nitrogen-Doped Reduced Graphene Oxide as a Bifunctional Material for Removing Bisphenols: Synergistic Effect between Adsorption and Catalysis. Environ. Sci. Technol. 2015, 49, 6855–6864. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhu, Y.; Lyu, L.; Zeng, Q.; Xing, X.; Hu, C. Electronic Structure Modulation of Graphitic Carbon Nitride by Oxygen Doping for Enhanced Catalytic Degradation of Organic Pollutants through Peroxymonosulfate Activation. Environ. Sci. Technol. 2018, 52, 14371–14380. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, L.; Sheng, L.; Zhou, Q.; Wei, T.; Feng, J.; Fan, Z. Edge-Nitrogen-Rich Carbon Dots Pillared Graphene Blocks with Ultrahigh Volumetric/Gravimetric Capacities and Ultralong Life for Sodium-Ion Storage. Adv. Energy Mater. 2018, 8, 1802042. [Google Scholar] [CrossRef]
- Kang, J.; Zhou, L.; Duan, X.; Sun, H.; Wang, S. Catalytic degradation of antibiotics by metal-free catalysis over nitrogen-doped graphene. Catal. Today 2018. [Google Scholar] [CrossRef]
- Wang, S.; Xu, L.; Wang, J. Nitrogen-doped graphene as peroxymonosulfate activator and electron transfer mediator for the enhanced degradation of sulfamethoxazole. Chem. Eng. J. 2019, 375, 122041. [Google Scholar] [CrossRef]
- Sun, P.; Liu, H.; Feng, M.; Guo, L.; Zhai, Z.; Fang, Y.; Zhang, X.; Sharma, V.K. Nitrogen-sulfur co-doped industrial graphene as an efficient peroxymonosulfate activator: Singlet oxygen-dominated catalytic degradation of organic contaminants. Appl. Catal. B Environ. 2019, 251, 335–345. [Google Scholar] [CrossRef]
- Chen, X.; Oh, W.-D.; Hu, Z.-T.; Sun, Y.-M.; Webster, R.D.; Li, S.-Z.; Lim, T.-T. Enhancing sulfacetamide degradation by peroxymonosulfate activation with N-doped graphene produced through delicately-controlled nitrogen functionalization via tweaking thermal annealing processes. Appl. Catal. B Environ. 2018, 225, 243–257. [Google Scholar] [CrossRef]
- Guo, Y.; Zeng, Z.; Zhu, Y.; Huang, Z.; Cui, Y.; Yang, J. Catalytic oxidation of aqueous organic contaminants by persulfate activated with sulfur-doped hierarchically porous carbon derived from thiophene. Appl. Catal. B Environ. 2018, 220, 635–644. [Google Scholar] [CrossRef]
- Chen, X.; Duan, X.; Oh, W.-D.; Zhang, P.-H.; Guan, C.-T.; Zhu, Y.-A.; Lim, T.-T. Insights into nitrogen and boron-co-doped graphene toward high-performance peroxymonosulfate activation: Maneuverable N-B bonding configurations and oxidation pathways. Appl. Catal. B Environ. 2019, 253, 419–432. [Google Scholar] [CrossRef]
- Zhang, B.; Wu, T.; Sun, D.; Chen, W.; Li, G.; Li, Y. NH2-MCM-41 supported on nitrogen-doped graphene as bifunctional composites for removing phenol compounds: Synergistic effect between catalytic degradation and adsorption. Carbon 2019, 147, 312–322. [Google Scholar] [CrossRef]
- Deng, B.; Wang, D.; Jiang, Z.; Zhang, J.; Shi, S.; Jiang, Z.-J.; Liu, M. Amine group induced high activity of highly torn amine functionalized nitrogen-doped graphene as the metal-free catalyst for hydrogen evolution reaction. Carbon 2018, 138, 169–178. [Google Scholar] [CrossRef]
- Yu, J.; Zhu, Z.; Zhang, H.; Di, G.; Qiu, Y.; Yin, D.; Wang, S. Hydrochars from pinewood for adsorption and nonradical catalysis of bisphenols. J. Hazard. Mater. 2020, 385, 121548. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.-C.; Jiang, J.; Huang, G.-X.; Yu, H.-Q. Sludge biochar-based catalysts for improved pollutant degradation by activating peroxymonosulfate. J. Mater. Chem. A 2018, 6, 8978–8985. [Google Scholar] [CrossRef]
- Ding, D.; Yang, S.; Qian, X.; Chen, L.; Cai, T. Nitrogen-doping positively whilst sulfur-doping negatively affect the catalytic activity of biochar for the degradation of organic contaminant. Appl. Catal. B Environ. 2020, 263, 118348. [Google Scholar] [CrossRef]
- Wu, D.; Song, W.; Chen, L.; Duan, X.; Xia, Q.; Fan, X.; Li, Y.; Zhang, F.; Peng, W.; Wang, S. High-performance porous graphene from synergetic nitrogen doping and physical activation for advancednonradical oxidation. J. Hazard. Mater. 2020, 381, 121010. [Google Scholar] [CrossRef] [PubMed]
- Yin, R.; Guo, W.; Wang, H.; Du, J.; Wu, Q.; Chang, J.-S.; Ren, N. Singlet oxygen-dominated peroxydisulfate activation by sludge-derived biochar for sulfamethoxazole degradation through a nonradical oxidation pathway: Performance and mechanism. Chem. Eng. J. 2019, 357, 589–599. [Google Scholar] [CrossRef]
- Mian, M.M.; Liu, G. Activation of peroxymonosulfate by chemically modified sludge biochar for the removal of organic pollutants: Understanding the role of active sites and mechanism. Chem. Eng. J. 2020, 392, 123681. [Google Scholar] [CrossRef]
- Hu, W.; Xie, Y.; Lu, S.; Li, P.; Xie, T.; Zhang, Y.; Wang, Y. One-step synthesis of nitrogen-doped sludge carbon as a bifunctional material for the adsorption and catalytic oxidation of organic pollutants. Sci. Total Environ. 2019, 680, 51–60. [Google Scholar] [CrossRef]
- Lee, H.; Kim, H.-i.; Weon, S.; Choi, W.; Hwang, Y.S.; Seo, J.; Lee, C.; Kim, J.-H. Activation of Persulfates by Graphitized Nanodiamonds for Removal of Organic Compounds. Environ. Sci. Technol. 2016, 50, 10134–10142. [Google Scholar] [CrossRef]
- Duan, X.; Ao, Z.; Zhang, H.; Saunders, M.; Sun, H.; Shao, Z.; Wang, S. Nanodiamonds in sp2/sp3configuration for radical to nonradical oxidation: Core-shell layer dependence. Appl. Catal. B Environ. 2018, 222, 176–181. [Google Scholar] [CrossRef]
- Feng, L.; Li, X.; Chen, X.; Huang, Y.; Peng, K.; Huang, Y.; Yan, Y.; Chen, Y. Pig manure-derived nitrogen-doped mesoporous carbon for adsorption and catalytic oxidation of tetracycline. Sci. Total Environ. 2020, 708, 135071. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Zhang, Y.; Zhou, M.; Liang, L.; Li, K. Oxidation of Rhodamine B by persulfate activated with porous carbon aerogel through a non-radical mechanism. J. Hazard. Mater. 2018, 358, 53–61. [Google Scholar] [CrossRef]
- Chen, X.; Oh, W.-D.; Zhang, P.-H.; Webster, R.D.; Lim, T.-T. Surface construction of nitrogen-doped chitosan-derived carbon nanosheets with hierarchically porous structure for enhanced sulfacetamide degradation via peroxymonosulfate activation: Maneuverable porosity and active sites. Chem. Eng. J. 2020, 382, 122908. [Google Scholar] [CrossRef]
- Ahn, Y.-Y.; Bae, H.; Kim, H.-I.; Kim, S.-H.; Kim, J.-H.; Lee, S.-G.; Lee, J. Surface-loaded metal nanoparticles for peroxymonosulfate activation: Efficiency and mechanism reconnaissance. Appl. Catal. B Environ. 2019, 241, 561–569. [Google Scholar] [CrossRef]
- Du, X.; Zhang, Y.; Si, F.; Yao, C.; Du, M.; Hussain, I.; Kim, H.; Huang, S.; Lin, Z.; Hayat, W. Persulfate non-radical activation by nano-CuO for efficient removal of chlorinated organic compounds: Reduced graphene oxide-assisted and CuO (001) facet-dependent. Chem. Eng. J. 2019, 356, 178–189. [Google Scholar] [CrossRef]
- Xiong, X.; Sun, B.; Zhang, J.; Gao, N.; Shen, J.; Li, J.; Guan, X. Activating persulfate by Fe0 coupling with weak magnetic field: Performance and mechanism. Water Res. 2014, 62, 53–62. [Google Scholar] [CrossRef]
- Li, W.; Zhang, Y.; Zhao, P.; Zhou, P.; Liu, Y.; Cheng, X.; Wang, J.; Yang, B.; Guo, H. Enhanced kinetic performance of peroxymonosulfate/ZVI system with the addition of copper ions: Reactivity, mechanism, and degradation pathways. J. Hazard. Mater. 2020, 393, 122399. [Google Scholar] [CrossRef]
- Yang, S.; Wu, P.; Liu, J.; Chen, M.; Ahmed, Z.; Zhu, N. Efficient removal of bisphenol A by superoxide radical and singlet oxygen generated from peroxymonosulfate activated with Fe0-montmorillonite. Chem. Eng. J. 2018, 350, 484–495. [Google Scholar] [CrossRef]
- Kim, C.; Ahn, J.-Y.; Kim, T.Y.; Shin, W.S.; Hwang, I. Activation of Persulfate by Nanosized Zero-Valent Iron (NZVI): Mechanisms and Transformation Products of NZVI. Environ. Sci. Technol. 2018, 52, 3625–3633. [Google Scholar] [CrossRef] [PubMed]
- Pi, Z.; Li, X.; Wang, D.; Xu, Q.; Tao, Z.; Huang, X.; Yao, F.; Wu, Y.; He, L.; Yang, Q. Persulfate activation by oxidation biochar supported magnetite particles for tetracycline removal: Performance and degradation pathway. J. Clean. Prod. 2019, 235, 1103–1115. [Google Scholar] [CrossRef]
- Feng, M.; Qu, R.; Zhang, X.; Sun, P.; Sui, Y.; Wang, L.; Wang, Z. Degradation of flumequine in aqueous solution by persulfate activated with common methods and polyhydroquinone-coated magnetite/multi-walled carbon nanotubes catalysts. Water Res. 2015, 85, 1–10. [Google Scholar] [CrossRef]
- Liu, L.; Xu, X.; Li, Y.; Su, R.; Li, Q.; Zhou, W.; Gao, B.; Yue, Q. One-step synthesis of “nuclear-shell” structure iron-carbon nanocomposite as a persulfate activator for bisphenol A degradation. Chem. Eng. J. 2020, 382, 122780. [Google Scholar] [CrossRef]
- Zhu, S.; Wang, W.; Xu, Y.; Zhu, Z.; Liu, Z.; Cui, F. Iron sludge-derived magnetic Fe0/Fe3C catalyst for oxidation of ciprofloxacin via peroxymonosulfate activation. Chem. Eng. J. 2019, 365, 99–110. [Google Scholar] [CrossRef]
- Zhang, G.; Ding, Y.; Nie, W.; Tang, H. Efficient degradation of drug ibuprofen through catalytic activation of peroxymonosulfate by Fe3C embedded on carbon. J. Environ. Sci. 2019, 78, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Liu, H.; Yang, C.; Li, X.; Lin, Y.; Yin, K.; Sun, J.; Teng, Q.; Du, C.; Zhong, Y. High-performance porous carbon catalysts doped by iron and nitrogen for degradation of bisphenol F via peroxymonosulfate activation. Chem. Eng. J. 2020, 392, 123683. [Google Scholar] [CrossRef]
- Liu, K.; Yu, M.; Wang, H.; Wang, J.; Liu, W.; Hoffmann, M.R. Multiphase Porous Electrochemical Catalysts Derived from Iron-Based Metal–Organic Framework Compounds. Environ. Sci. Technol. 2019, 53, 6474–6482. [Google Scholar] [CrossRef]
- Liang, S.-X.; Wang, X.; Zhang, W.; Liu, Y.-J.; Wang, W.; Zhang, L.-C. Selective laser melting manufactured porous Fe-based metallic glass matrix composite with remarkable catalytic activity and reusability. Appl. Mater. Today 2020, 19, 100543. [Google Scholar] [CrossRef]
- Liang, S.-X.; Jia, Z.; Zhang, W.C.; Li, X.F.; Wang, W.M.; Lin, H.C.; Zhang, L.C. Ultrafast activation efficiency of three peroxides by Fe78Si9B13 metallic glass under photo-enhanced catalytic oxidation: A comparative study. Appl. Catal. B Environ. 2018, 221, 108–118. [Google Scholar] [CrossRef]
- Xie, M.; Tang, J.; Fang, G.; Zhang, M.; Kong, L.; Zhu, F.; Ma, L.; Zhou, D.; Zhan, J. Biomass Schiff base polymer-derived N-doped porous carbon embedded with CoO nanodots foradsorption and catalytic degradation of chlorophenol by peroxymonosulfate. J. Hazard. Mater. 2020, 384, 121345. [Google Scholar] [CrossRef]
- Anipsitakis, G.P.; Dionysiou, D.D. Radical Generation by the Interaction of Transition Metals with Common Oxidants. Environ. Sci. Technol. 2004, 38, 3705–3712. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; He, D.; Li, X.; Feng, W.; Lyu, C.; Zhang, Y. Mechanism and performance of singlet oxygen dominated peroxymonosulfate activation on CoOOH nanoparticles for 2,4-dichlorophenol degradation in water. J. Hazard. Mater. 2020, 384, 121350. [Google Scholar] [CrossRef]
- Wu, L.; Zhang, Q.; Hong, J.; Dong, Z.; Wang, J. Degradation of bisphenol A by persulfate activation via oxygen vacancy-rich CoFe2O4-x. Chemosphere 2019, 221, 412–422. [Google Scholar] [CrossRef]
- Zhu, C.; Zhang, Y.; Fan, Z.; Liu, F.; Li, A. Carbonate-enhanced catalytic activity and stability of Co3O4 nanowires for 1O2-driven bisphenol A degradation via peroxymonosulfate activation: Critical roles of electron and proton acceptors. J. Hazard. Mater. 2020, 393, 122395. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Zhang, H.; Qian, Z.; Ouyang, T.; Sun, H.; Qin, J.; Tadé, M.O.; Wang, S. Bread-making synthesis of hierarchically Co@C nanoarchitecture in heteroatom doped porous carbons for oxidative degradation of emerging contaminants. Appl. Catal. B Environ. 2018, 225, 76–83. [Google Scholar] [CrossRef]
- Zhang, S.; Gao, H.; Xu, X.; Cao, R.; Yang, H.; Xu, X.; Li, J. MOF-derived CoN/N-C@SiO2yolk-shell nanoreactor with dual active sites for highly efficient catalytic advanced oxidation processes. Chem. Eng. J. 2020, 381, 122670. [Google Scholar] [CrossRef]
- Ding, D.; Yang, S.; Chen, L.; Cai, T. Degradation of norfloxacin by CoFe alloy nanoparticles encapsulated in nitrogen doped graphitic carbon (CoFe@N-GC) activated peroxymonosulfate. Chem. Eng. J. 2020, 392, 123725. [Google Scholar] [CrossRef]
- Liu, C.; Liu, S.; Liu, L.; Tian, X.; Liu, L.; Xia, Y.; Liang, X.; Wang, Y.; Song, Z.; Zhang, Y.; et al. Novel carbon based Fe-Co oxides derived from Prussian blue analogues activating peroxymonosulfate: Refractory drugs degradation without metal leaching. Chem. Eng. J. 2020, 379, 122274. [Google Scholar] [CrossRef]
- Taujale, S.; Baratta, L.R.; Huang, J.; Zhang, H. Interactions in Ternary Mixtures of MnO2, Al2O3, and Natural Organic Matter (NOM) and the Impact on MnO2 Oxidative Reactivity. Environ. Sci. Technol. 2016, 50, 2345–2353. [Google Scholar] [CrossRef]
- Zeng, Z.; Khan, A.; Wang, Z.; Zhao, M.; Mo, W.; Chen, Z. Elimination of atrazine through radical/non-radical combined processes by manganese nano-catalysts/PMS and implications to the structure-performance relationship. Chem. Eng. J. 2020, 397, 125425. [Google Scholar] [CrossRef]
- Huang, J.; Dai, Y.; Singewald, K.; Liu, C.-C.; Saxena, S.; Zhang, H. Effects of MnO2of different structures on activation of peroxymonosulfate. Chem. Eng. J. 2019, 370, 906–915. [Google Scholar] [CrossRef]
- Fan, J.; Qin, H.; Jiang, S. Mn-doped g-C3N4 composite to activate peroxymonosulfate for acetaminophen degradation: The role of superoxide anion and singlet oxygen. Chem. Eng. J. 2019, 359, 723–732. [Google Scholar] [CrossRef]
- Yu, J.; Zeng, T.; Wang, H.; Zhang, H.; Sun, Y.; Chen, L.; Song, S.; Li, L.; Shi, H. Oxygen-defective MnO2−x rattle-type microspheres mediated singlet oxygen oxidation of organics by peroxymonosulfate activation. Chem. Eng. J. 2020, 394, 124458. [Google Scholar] [CrossRef]
- Xing, S.; Li, W.; Liu, B.; Wu, Y.; Gao, Y. Removal of ciprofloxacin by persulfate activation with CuO: A pH-dependent mechanism. Chem. Eng. J. 2020, 382, 122837. [Google Scholar] [CrossRef]
- Wang, S.; Gao, S.; Tian, J.; Wang, Q.; Wang, T.; Hao, X.; Cui, F. A stable and easily prepared copper oxide catalyst for degradation of organic pollutants by peroxymonosulfate activation. J. Hazard. Mater. 2020, 387, 121995. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Sun, Y.; Huang, W.; Xue, C.; Zhu, Y.; Wang, Q.; Liu, D. Innovatively employing magnetic CuO nanosheet to activate peroxymonosulfate for the treatment of high-salinity organic wastewater. J. Environ. Sci. 2020, 88, 46–58. [Google Scholar] [CrossRef]
- Li, Z.; Liu, D.; Huang, W.; Wei, X.; Huang, W. Biochar supported CuO composites used as an efficient peroxymonosulfate activator for highly saline organic wastewater treatment. Sci. Total Environ. 2020, 721, 137764. [Google Scholar] [CrossRef] [PubMed]
- Shahzad, A.; Ali, J.; Ifthikar, J.; Aregay, G.G.; Zhu, J.; Chen, Z.; Chen, Z. Non-radical PMS activation by the nanohybridmaterial with periodic confinement of reduced graphene oxide (rGO) and Cu hydroxides. J. Hazard. Mater. 2020, 392, 122316. [Google Scholar] [CrossRef]
- Dong, X.; Duan, X.; Sun, Z.; Zhang, X.; Li, C.; Yang, S.; Ren, B.; Zheng, S.; Dionysiou, D.D. Natural illite-based ultrafine cobalt oxide with abundant oxygen-vacancies for highly efficient Fenton-like catalysis. Appl. Catal. B Environ. 2020, 261, 118214. [Google Scholar] [CrossRef]
- Yu, R.; Zhao, J.; Zhao, Z.; Cui, F. Copper substituted zinc ferrite with abundant oxygen vacancies for enhanced ciprofloxacin degradation via peroxymonosulfate activation. J. Hazard. Mater. 2020, 390, 121998. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Liu, D.; Zhao, Y.; Li, S.; Wei, X.; Meng, F.; Huang, W.; Lei, Z. Singlet oxygen dominated peroxymonosulfate activation by CuO-CeO2for organic pollutants degradation: Performance and mechanism. Chemosphere 2019, 233, 549–558. [Google Scholar] [CrossRef]
- Jawad, A.; Zhan, K.; Wang, H.; Shahzad, A.; Zeng, Z.; Wang, J.; Zhou, X.; Ullah, H.; Chen, Z.; Chen, Z. Tuning of Persulfate Activation from a Free Radical to a Nonradical Pathway through the Incorporation of Non-Redox Magnesium Oxide. Environ. Sci. Technol. 2020, 54, 2476–2488. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Bi, S.; Zhao, G.; Chen, Y.; Hu, Y. Enhanced degradation of triclosan by cobalt manganese spinel-type oxide activated peroxymonosulfate oxidation process via sulfate radicals and singlet oxygen: Mechanisms and intermediates identification. Sci. Total Environ. 2020, 711, 134715. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Yan, Z.; Chen, C.; Chen, J. Spinels: Controlled preparation, oxygen reduction/evolution reaction application, and beyond. Chem. Rev. 2017, 117, 10121–10211. [Google Scholar] [CrossRef]
- Zhao, Y.; An, H.; Dong, G.; Feng, J.; Wei, T.; Ren, Y.; Ma, J. Oxygen vacancies induced heterogeneous catalysis of peroxymonosulfate by Ni-doped AgFeO2materials: Evolution of reactive oxygen species and mechanism. Chem. Eng. J. 2020, 388, 124371. [Google Scholar] [CrossRef]
- Deng, J.; Cheng, Y.-Q.; Lu, Y.-A.; Crittenden, J.C.; Zhou, S.-Q.; Gao, N.-Y.; Li, J. Mesoporous manganese Cobaltite nanocages as effective and reusable heterogeneous peroxymonosulfate activators for Carbamazepine degradation. Chem. Eng. J. 2017, 330, 505–517. [Google Scholar] [CrossRef]
- Li, C.; Wu, J.; Peng, W.; Fang, Z.; Liu, J. Peroxymonosulfate activation for efficient sulfamethoxazole degradation by Fe3O4/β-FeOOH nanocomposites: Coexistence of radical and non-radical reactions. Chem. Eng. J. 2019, 356, 904–914. [Google Scholar] [CrossRef]
- Lu, S.; Wang, G.; Chen, S.; Yu, H.; Ye, F.; Quan, X. Heterogeneous activation of peroxymonosulfate by LaCo1-xCuxO3 perovskites for degradation of organic pollutants. J. Hazard. Mater. 2018, 353, 401–409. [Google Scholar] [CrossRef]
- Ahn, Y.-Y.; Yun, E.-T.; Seo, J.-W.; Lee, C.; Kim, S.H.; Kim, J.-H.; Lee, J. Activation of Peroxymonosulfate by Surface-Loaded Noble Metal Nanoparticles for Oxidative Degradation of Organic Compound. Environ. Sci. Technol. 2016, 50, 10187–10197. [Google Scholar] [CrossRef]
- Wang, Y.; Cao, D.; Liu, M.; Zhao, X. Insights into heterogeneous catalytic activation of peroxymonosulfate by Pd/g-C3N4: The role of superoxide radical and singlet oxygen. Catal. Commun. 2017, 102, 85–88. [Google Scholar] [CrossRef]
- Luo, R.; Li, M.; Wang, C.; Zhang, M.; Nasir Khan, M.A.; Sun, X.; Shen, J.; Han, W.; Wang, L.; Li, J. Singlet oxygen-dominated non-radical oxidation process for efficient degradation of bisphenol Aunder high salinity condition. Water Res. 2019, 148, 416–424. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Cai, J.; Wang, X.; Li, Y.; Wu, Z.; Wu, W.D.; Chen, X.D.; Sun, J.; Sun, S.P.; Wang, Z. A Bimetallic Fe-Mn Oxide-Activated Oxone for In Situ Chemical Oxidation (ISCO) of Trichloroethylene in Groundwater: Efficiency, Sustained Activity, and Mechanism Investigation. Environ. Sci. Technol. 2020, 54, 3714–3724. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Teel, A.L.; Watts, R.J. Activationof Peroxymonosulfate by Subsurface Minerals. J. Contam. Hydrol. 2016, 191, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, M.; Teel, A.L.; Watts, R.J. Persulfate activation by subsurface minerals. J. Contam. 2010, 115, 34–45. [Google Scholar] [CrossRef]
- Sra, K.S.; Thomson, N.R.; Barker, J.F. Persulfate injection into a gasoline source zone. J. Contam. Hydrol. 2013, 150, 35–44. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
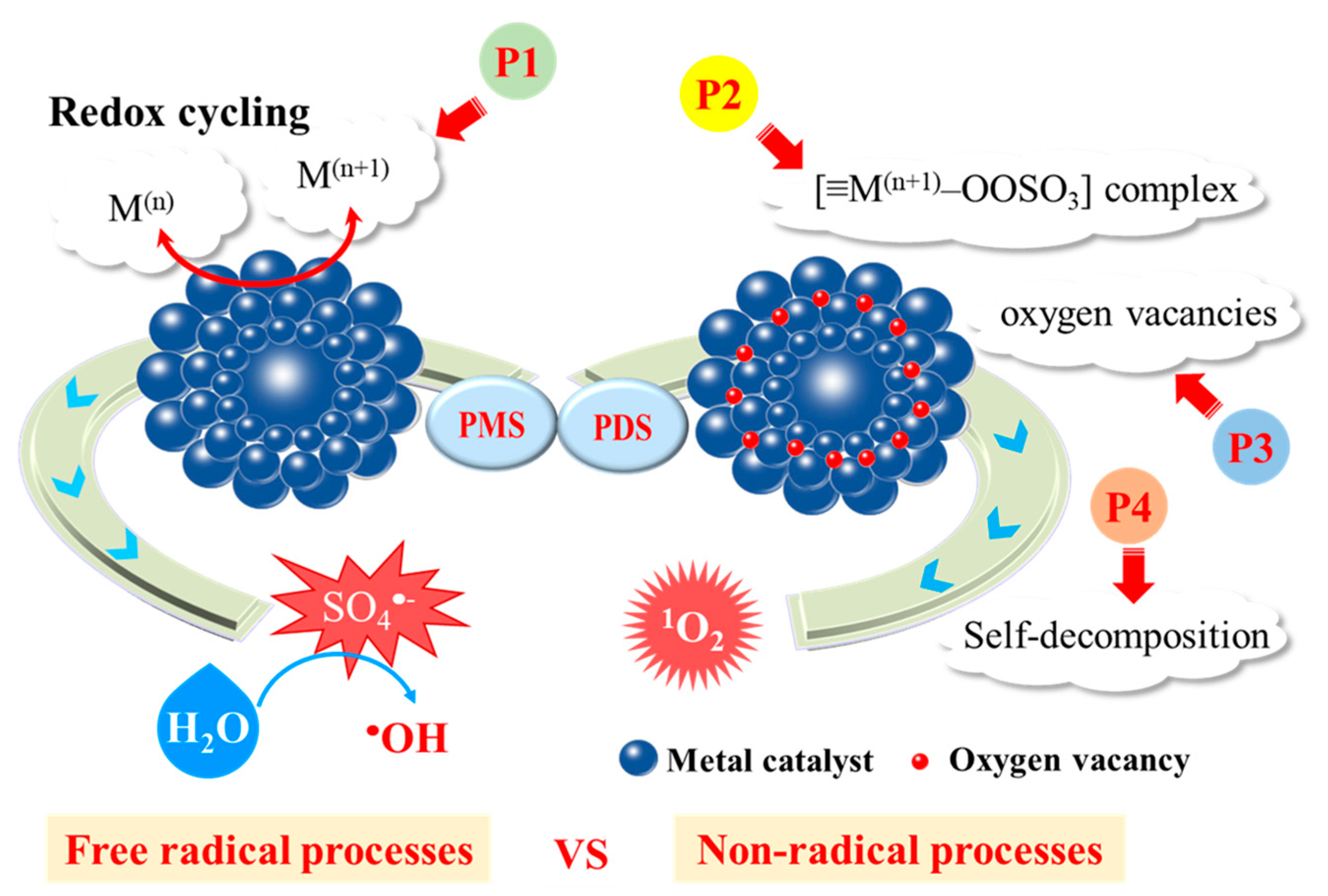{kind=link}
| Systems | Target/Pollutant | Degradation Rate and Time | Radicals Involve | Active Catalytic SitesOr Activation Mechanism | References |
|---|---|---|---|---|---|
| phenols/PMS | sulfamethoxazole | 100% in 60 min at pH 10 | 1O2 | quinone intermediates formed from phenolic parents | Zhou et al. [34] |
| PMS | chlorophenols | 65% of 4-CP, 70% of 2,4-DCP and 95% of 2,4,6-TCP in 60 min at pH 9 | 1O2, SO4•−, •OH | chlorophenols | Li et al. [35] |
| ketones/PMS | / | / | 1O2, | ketone carbonyl | Lange and Brauer [40] |
| benzoquinone/PMS | sulfamethoxazole | 86% in 3 min at pH 10 | 1O2 | quinone carbonyl | Zhou et al. [41] |
| benzoquinone/PMS | / | / | 1O2 | quinone carbonyl | Zhang et al. [43] |
| Cl−/phenol/PMS | phenols | 100% in 60 min at pH 10 | 1O2 | Cl− interact with PMS to form chlorophenols and benzoquinone | Li et al. [44] |
| base/PMS | acid orange 7 | 90% in 60 min | 1O2, O2•− | O2•− intermediates for 1O2 generation under too much pH conditions | Qi et al. [45] |
| base/polyphosphates/PMS | acid orange 7 | 100% in 500 s | 1O2, O2•− | remains unclear | Luo et al. [47] |
| BO2−/PMS | acid red 1 | 97.8% in 10 min | 1O2 | nucleophilic mechanisms | Rao et al. [48] |
| CO32−/PMS | acid orange 7 | 100% in 40 min | 1O2, O2•− | PMS self-decomposition, O2•− intermediates by base-catalyzed hydrolysis for 1O2 generation | Nie et al. [49] |
| Cl−/PMS | 2,4-dimethylphenol | 100% in 20 min | SO4•−, •OH, 1O2, Cl• | / | Wang et al. [50] |
| Cl−/PMS | methylene blue | 100% in 120 min | 1O2, Cl• | PMS self-decomposition, reactive chlorine | Yang et al. [51] |
| Catalyst | Oxidant | Target Pollutant | Degradation Rate and Time | Radicals Mechanism | Active Catalytic Sites | References | |
|---|---|---|---|---|---|---|---|
| reduced graphene oxide | PMS | phenol | 100% | 45 min | 1O2 | sp2 carbon, defective edges | Duan et al. [53] |
| single-wall carbon nanotubes | PDS | 4-chlorophenol | 100% | 60 min | 1O2 | sp2 carbon, nonradical electron-transfer | Yun et al. [58] |
| carbon nanotubes | PDS | phenol | 100% | 30 min | / | nonradical electron-transfer | Ren et al. [59] |
| carbon nanotubes | PDS | 2,4-dichlorophenol | 95.9% | 30 min | 1O2, O2•− | sp2 carbon | Cheng et al. [60] |
| nitrogen-doped carbon nanotubes | PMS | phenol | 95.6% | 10 min | / | nonradical electron-transfer | Ren et al. [62] |
| N-doping reduced graphene oxide | PMS | Sulfachloro-pyridazine | 100% | 9 min | SO4•−, •OH, 1O2 | hydroxyl group, sp2 carbon, pyridine N, pyrrole N, graphite N | Kang et al. [67] |
| N-S co-doped graphene | PMS | methyl p-hydroxy-benzoate | 100% | 30 min | SO4•−, •OH, 1O2 | sp2 carbon, pyrrolic N, pyridinic N, graphitic N, thiophenic S | Sun et al. [69] |
| amino-functionalized mesoporous silica anchoring N-doped graphene oxide | PMS | bisphenol A | 100% | 600 min | 1O2 | sp2 carbon, phenolic hydroxyl group, amino group | Zhang et al. [73] |
| sludge-biochar (600 °C) | PMS | bisphenol | 100% | 30 min | 1O2 | ketone structure inside the biochar | Huang et al. [76] |
| biochar doped with N and S | PMS | metolachlor | about 80% | 120 min | •OH, 1O2 | surface ketone of biochar | Ding et al. [77] |
| porous nitrogen-doped reduced graphene oxide | PMS | phenol | 100% | 25 min | •OH, 1O2, O2•− | structure vacancies with two nitrogen atoms of graphene structure | Wu et al. [78] |
| sludge-derived biochar | PDS | sulfamethoxazole | 94.6% | 180 min | 1O2 | sp2 carbon, Fe(II), N atoms | Yin et al. [79] |
| nitrogen-doped sludge carbon | PMS | methylene blue | 98.7% | 20 min | SO4•−, •OH, 1O2 | not mentioned | Hu et al. [81] |
| graphited nanodiamond | PDS | phenol | 100% | 10 min | SO4•−, •OH, 1O2 | nonradical electron-transfer | Lee et al. [82] |
| nano diamonds with a core/shell structure | PMS | phenol | 100% | 90 min | / | sp2/sp3 carbon of graphite structure | Duan er al. [83] |
| porous carbon aerogel | PDS | rhodamine B | 100% | 60 min | 1O2, O2•− | sp2 carbon, defective edges and the carbon edges of carbon aerogel | Jiang et al. [85] |
| N-doped chitosan-derived carbon nanosheets | PMS | sulfacetamide | 100% | 10 min | 1O2, O2•− | sp2 carbon, graphitic N | Chen et al. [86] |
| Oxidant | Catalyst | Target/Pollutant | Degradation Rate and Time | Radicals Involve | Active Catalytic SitesOr Activation Mechanism | References | |
|---|---|---|---|---|---|---|---|
| PDS | β-MnO2 | phenol | over 99% | 180 min | 1O2, O2•− | metastable manganese intermediates for O2•− generation | Zhu et al. [19] |
| sheet-like CuO | 2,4,6-trichlorophenol | 90% | 180 min | •OH, non-radical | facet (001) of CuO, electron-defective center | Du et al. [88] | |
| CoFe2O4-x | bisphenol A | 98% | 60 min | SO4•−, •OH, 1O2 | Fe(III)/Fe(II), Co(III)/Co(II), oxygen vacancies, | Wu et al. [105] | |
| CuO | ciprofloxacin | 100% | 30 min | 1O2, O2•−, SO4•− •OH | Cu(II)/Cu(III) for O2•− and 1O2, Cu(I)/Cu(II) for SO4•−, •OH | Xing et al. [116] | |
| PMS | LaNiO3 | ofloxacin | 93% | 10 min | 1O2, SO4•−, •OH | Ni(III)/Ni(II), oxygen vacancies | Gao et al. [8] |
| nZVI/Cu2+ | rhodamine B | 99.3% | 60 min | 1O2, O2•−, SO4•−, •OH | Fe(III)/Fe(II), Cu(II)/Cu(I) | Li et al. [90] | |
| Fe0-Mt | bisphenol A | 99.3% at pH 3 | 120 min | 1O2, O2•−, SO4•−, •OH | Fe0, released Fe2+ | Yang et al. [91] | |
| CoOOH | 2,4-dichlorophenol | 100% | 120 min | 1O2, O2•−, SO4•− •OH | Co(III)/Co(II), -OH | Zhang et al. [104] | |
| Co3O4/CO32− | bisphenol A | 100% | 10 min | 1O2, O2•−, SO4•− •OH | Co(III)/Co(II), OH−, CO32− | Zhu et al. [106] | |
| Mn oxides in different structure | atrazine | 100% | 100 min | 1O2, SO4•−, •OH | Mn(IV)/Mn(III), Mn(III)/Mn(II) | Zeng et al. [112] | |
| δ-MnO2 | bisphenol A | 42% | 10 min | 1O2, SO4•−, •OH | δ-MnO2 direct oxidation, Mn(IV)/Mn(III) | Huang et al. [113] | |
| Mn-g-C3N4 | acetaminophen | 100% | 15 min | 1O2, O2•− | Mn(III)/Mn(II) in the N-pot | Fan et al. [114] | |
| Oxygen-defective MnO2 | bisphenol A | 100% | 60 min | 1O2, SO4•−, •OH | oxygen vacancies | Yu et al. [115] | |
| CuO | bisphenol A | 100% | 60 min | 1O2, O2•− | Cu(II)-(O)-OSO3− formed on surface of CuO for O2•− generation | Wang et al. [117] | |
| magnetic CuO nanosheet | AO7 in high salinity wastewater | 95.8% | 30 min | 1O2, SO4•−, •OH | synergistic effect of Cu(I)/Cu(II) and Fe(II)/Fe(III) | Li et al. [118] | |
| copper substituted zinc ferrate | ciprofloxacin | 96.6% | 15 min | 1O2, O2•−, SO4•−, •OH | Fe(III)/Fe(II), Cu(II)/ Cu(I), oxygen vacancies | Yu et al. [122] | |
| CuO-CeO2 | rhodamine B | 100% | 60 min | 1O2, O2•−, SO4•−, •OH | Ce(IV)/Ce(III), Cu(II)/Cu(I), oxygen vacancies, electron transfer | Li et al. [123] | |
| CuOMgO/Fe3O4 | 4-chlorophenol | 100% | 40 min | 1O2, O2•− | [≡Cu(III)–OOSO3] intermediates for O2•− generation | Jawad et al. [124] | |
| Co2Mn1O4 | triclosan phenol | 96.4% | 30 min | 1O2, SO4•− | oxygen vacancies, Co(II)/Co(III), Mn(III)/Mn(II)/Mn(IV) | Chen et al. [125] | |
| LaCo0.4Cu0.6O3 | phenol | 100% | 12 min | 1O2, SO4•−, •OH | Co(II)/Co(III), Cu(II)/Cu(I), oxygen vacancies | Lu et al. [130] | |
| Oxidant | Catalyst | Target/Pollutant | Degradation Rate and Time | Radicals Involve | Active Catalytic SitesOr Activation Mechanism | References | |
|---|---|---|---|---|---|---|---|
| PDS | oxidation biochar supported magnetite particles | tetracycline | 92.3% | 120 min | SO4•−, •OH, 1O2 | Fe(II)/Fe(III), sp2-hybrid C atom with defective structures, ketone groups mediated electron transfer | Pi et al. [93] |
| Nuclear-shell structure iron-carbon | bisphenol A | 96% | 30 min | SO4•−, •OH, 1O2, O2•− | Fe0 Fe(II)/Fe(III), electron transfer for O2•− formation to produce 1O2 | Liu et al. [95] | |
| PMS | sludge-derived magnetic Fe0/Fe3C | ciprofloxacin | 99% | 20 min | SO4•−, •OH, 1O2, O2•− | Fe0, Fe(II)/Fe(III), electron transfer for O2•− formation to produce 1O2 | Zhu et al. [96] |
| Fe3C embedded on carbon | ibuprofen | 100% | 30 min | SO4•−, •OH, 1O2 | Fe(II)/Fe(III), N-doped carbon area, enhanced electron transfer process due to the carbon shell | Zhang et al. [97] | |
| iron and nitrogen co-doped porous carbon | bisphenol F | 97.1% | 90 min | SO4•−, •OH, 1O2, O2•− | pyridine N, graphite N, adjacent C region of Fe-doping | Wu et al. [98] | |
| N-doped porous carbon embedded with CoO nanodots | chlorophenol | 100% | 30 min | SO4•−, •OH, 1O2 | Co(II)/Co(III), increase defects sites of C by CoO doping, enhanced electron transfer by N doping | Xie et al. [102] | |
| core-shell Co@C nanoparticles with nitrogen and sulfur | p-hydroxybenzoic acid | 100% | 45 min | SO4•−, •OH, 1O2 | sp2 carbon, defect sites procuced by Co and N doping, Co(II)/Co(III) | Tian et al. [107] | |
| CoFe alloy nanoparticles encapsulated in nitrogen doped graphitic carbon | norfloxacin | 94.4% | 20 min | SO4•−, •OH, 1O2 | Co(II)/Co(III), Fe(II)/Fe(III), neighboring C atoms of graphitic N, self-decomposition of PMS | Ding et al. [109] | |
| carbon-based Fe-Co oxide derived from Prussian blue | 4-aminobenzoic acid ethyl ester | 95.5% | 60 min | SO4•−, •OH, 1O2 | Co(II)/Co(III), Fe(II)/Fe(III), sp2 hybridized carbon, pyridinic-N and pyrrolic-N | Liu et al. [110] | |
| Pd nanoparticles anchored C3N4 | bisphenol A | 91% | 60 min | SO4•−, •OH, 1O2 | Pd0/Pd(II), electron transfer for 1O2 production | Wang et al. [132] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, G.; Xu, T.; Faheem, M.; Xi, Y.; Zhou, T.; Moryani, H.T.; Bao, J.; Du, J. Evolution of Singlet Oxygen by Activating Peroxydisulfate and Peroxymonosulfate: A Review. Int. J. Environ. Res. Public Health 2021, 18, 3344. https://doi.org/10.3390/ijerph18073344
Xiao G, Xu T, Faheem M, Xi Y, Zhou T, Moryani HT, Bao J, Du J. Evolution of Singlet Oxygen by Activating Peroxydisulfate and Peroxymonosulfate: A Review. International Journal of Environmental Research and Public Health. 2021; 18(7):3344. https://doi.org/10.3390/ijerph18073344
Chicago/Turabian StyleXiao, Guangfeng, Tiantian Xu, Muhammad Faheem, Yanxing Xi, Ting Zhou, Haseeb Tufail Moryani, Jianguo Bao, and Jiangkun Du. 2021. "Evolution of Singlet Oxygen by Activating Peroxydisulfate and Peroxymonosulfate: A Review" International Journal of Environmental Research and Public Health 18, no. 7: 3344. https://doi.org/10.3390/ijerph18073344
APA StyleXiao, G., Xu, T., Faheem, M., Xi, Y., Zhou, T., Moryani, H. T., Bao, J., & Du, J. (2021). Evolution of Singlet Oxygen by Activating Peroxydisulfate and Peroxymonosulfate: A Review. International Journal of Environmental Research and Public Health, 18(7), 3344. https://doi.org/10.3390/ijerph18073344