
Oro-Dental Manifestations in a Pediatric Patient Affected by Helsmoortel-Van der Aa Syndrome

 , ,
, ,  and
and
Abstract
:1. Introduction
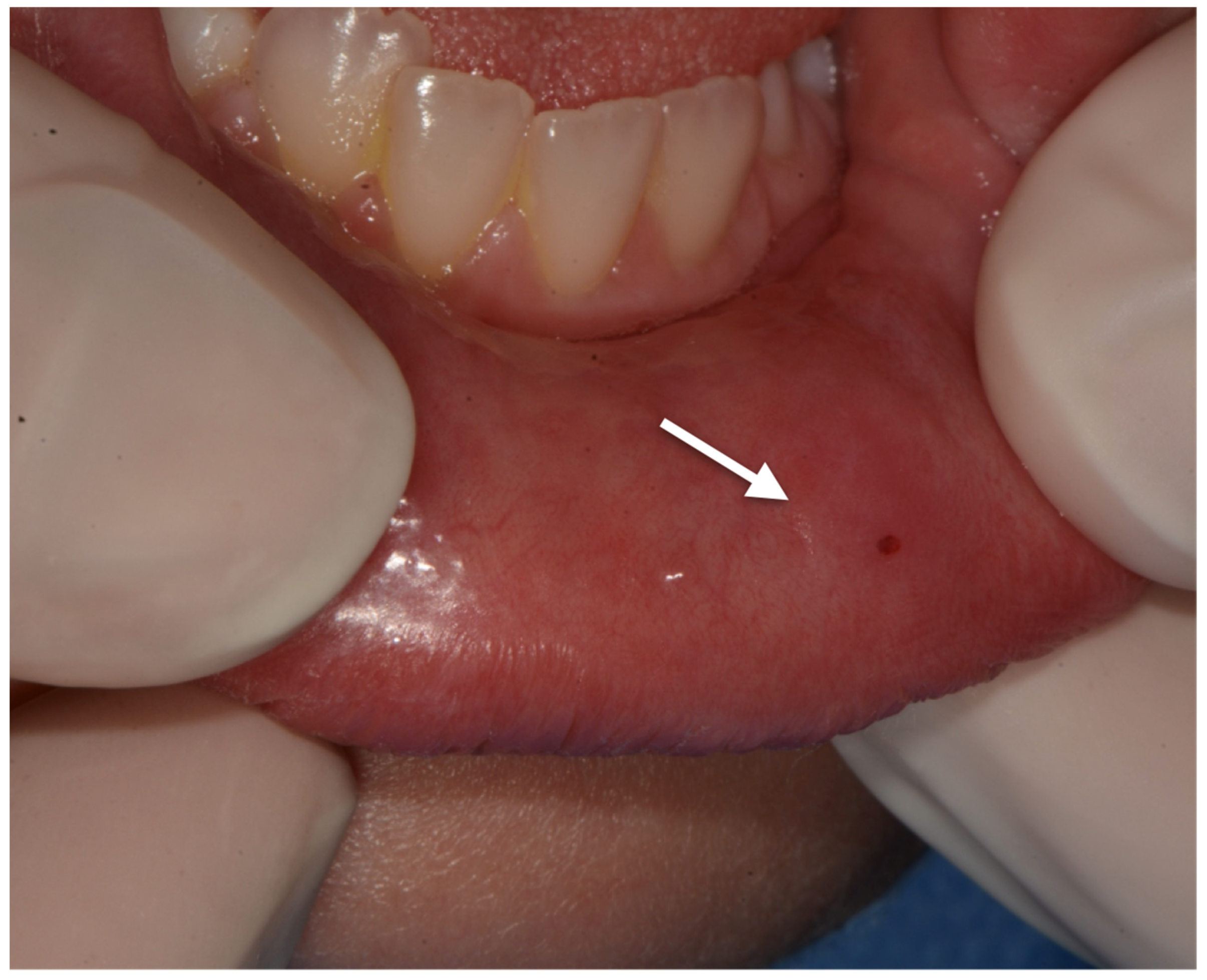
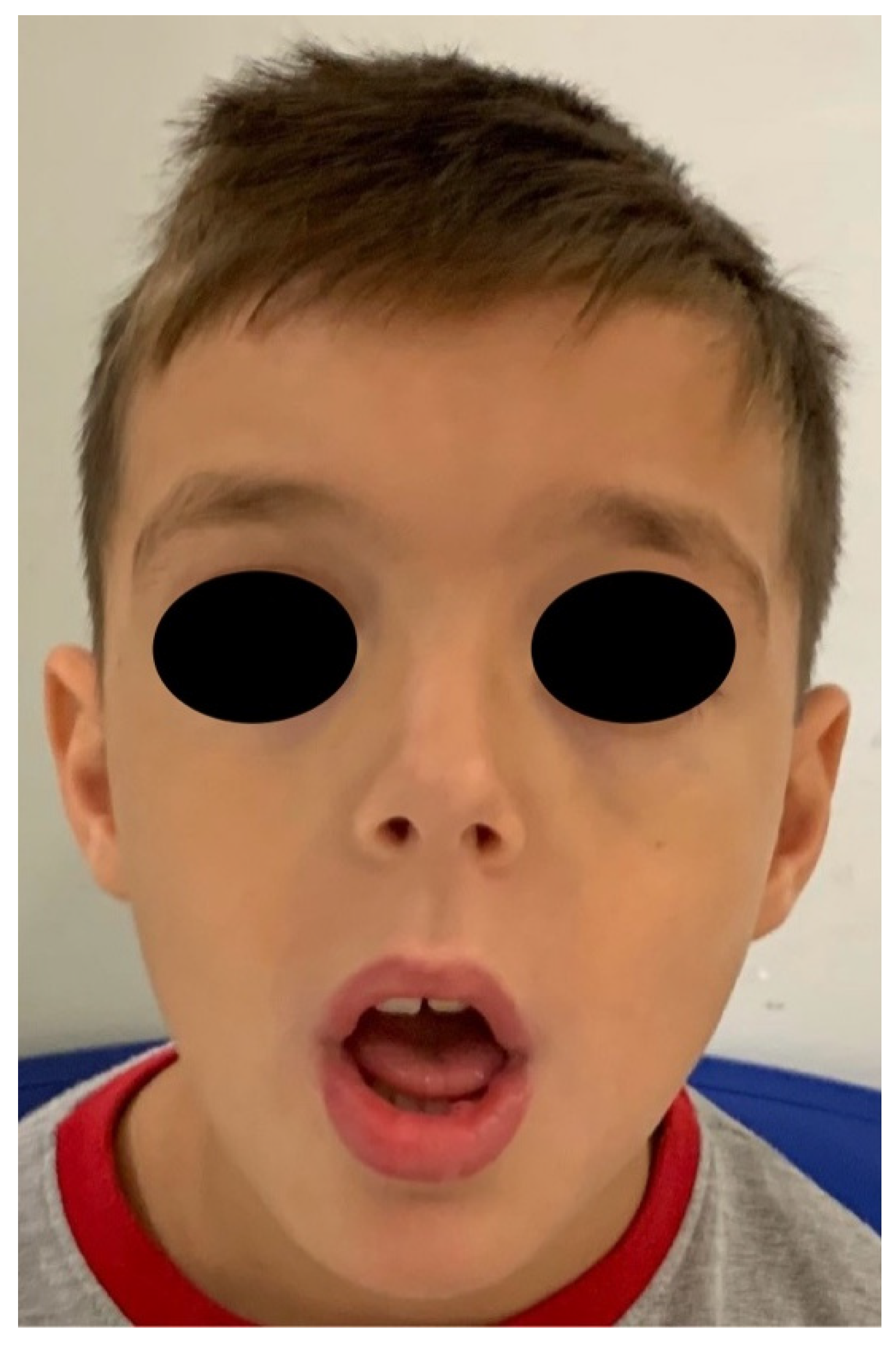
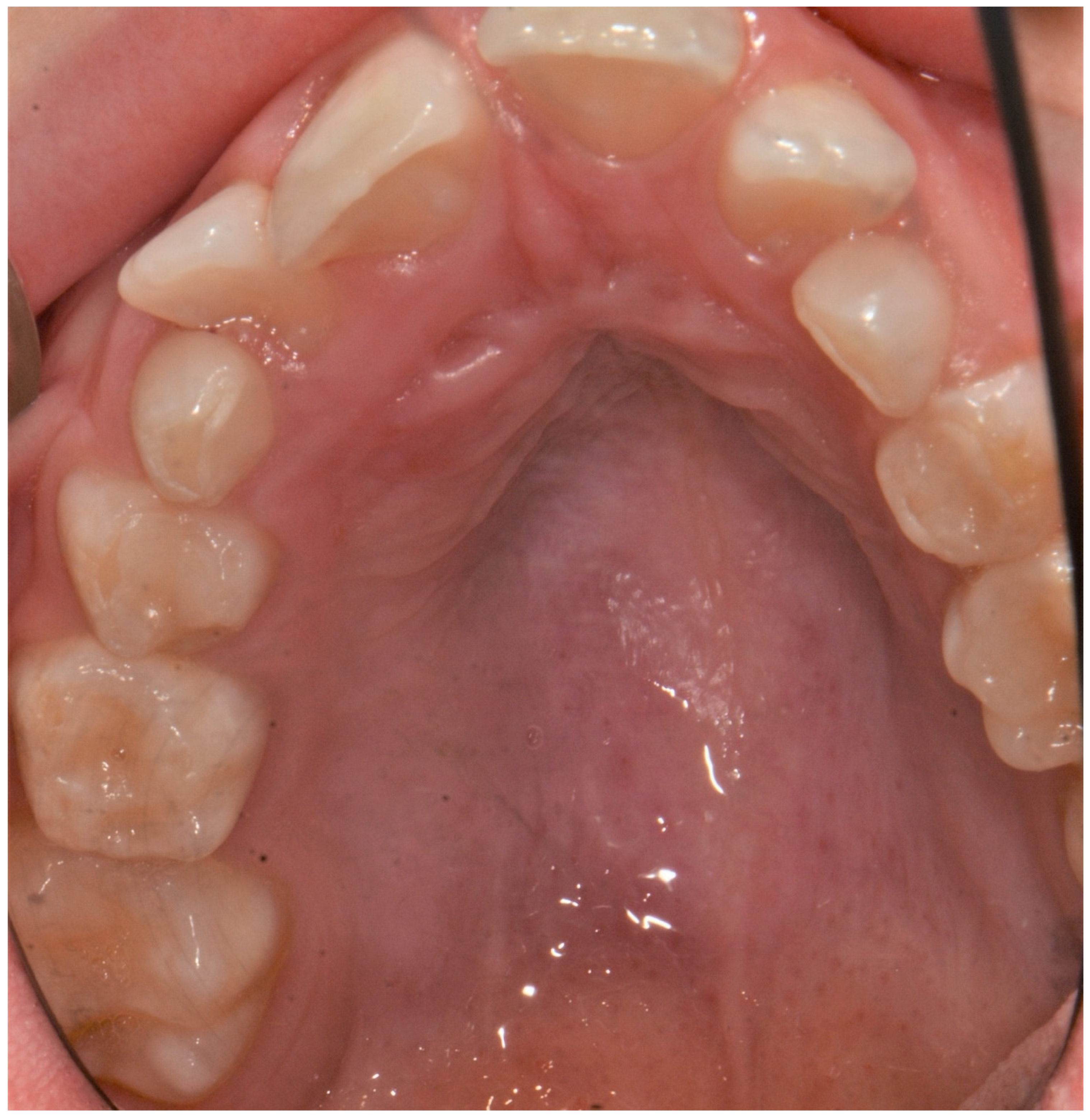
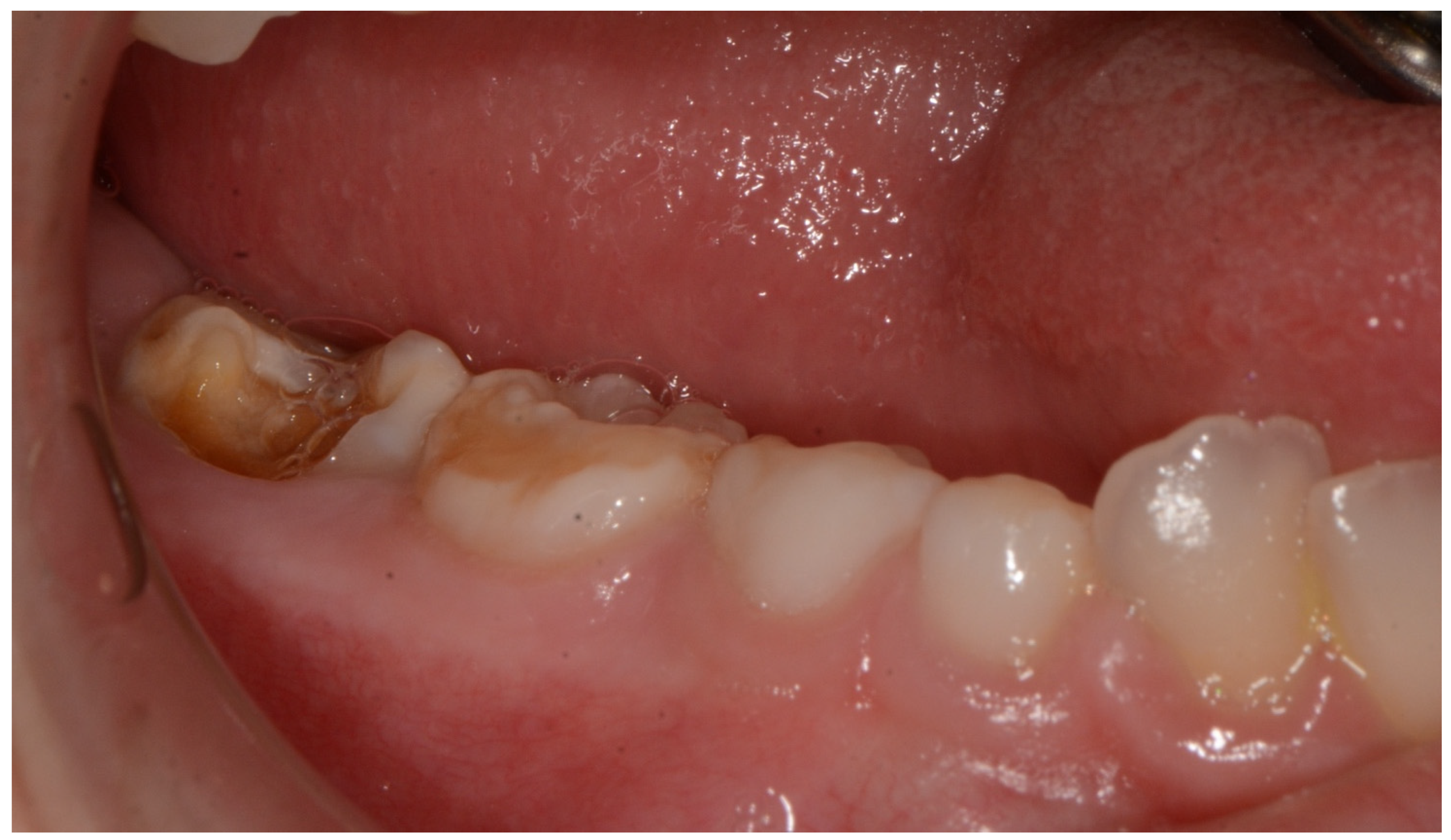
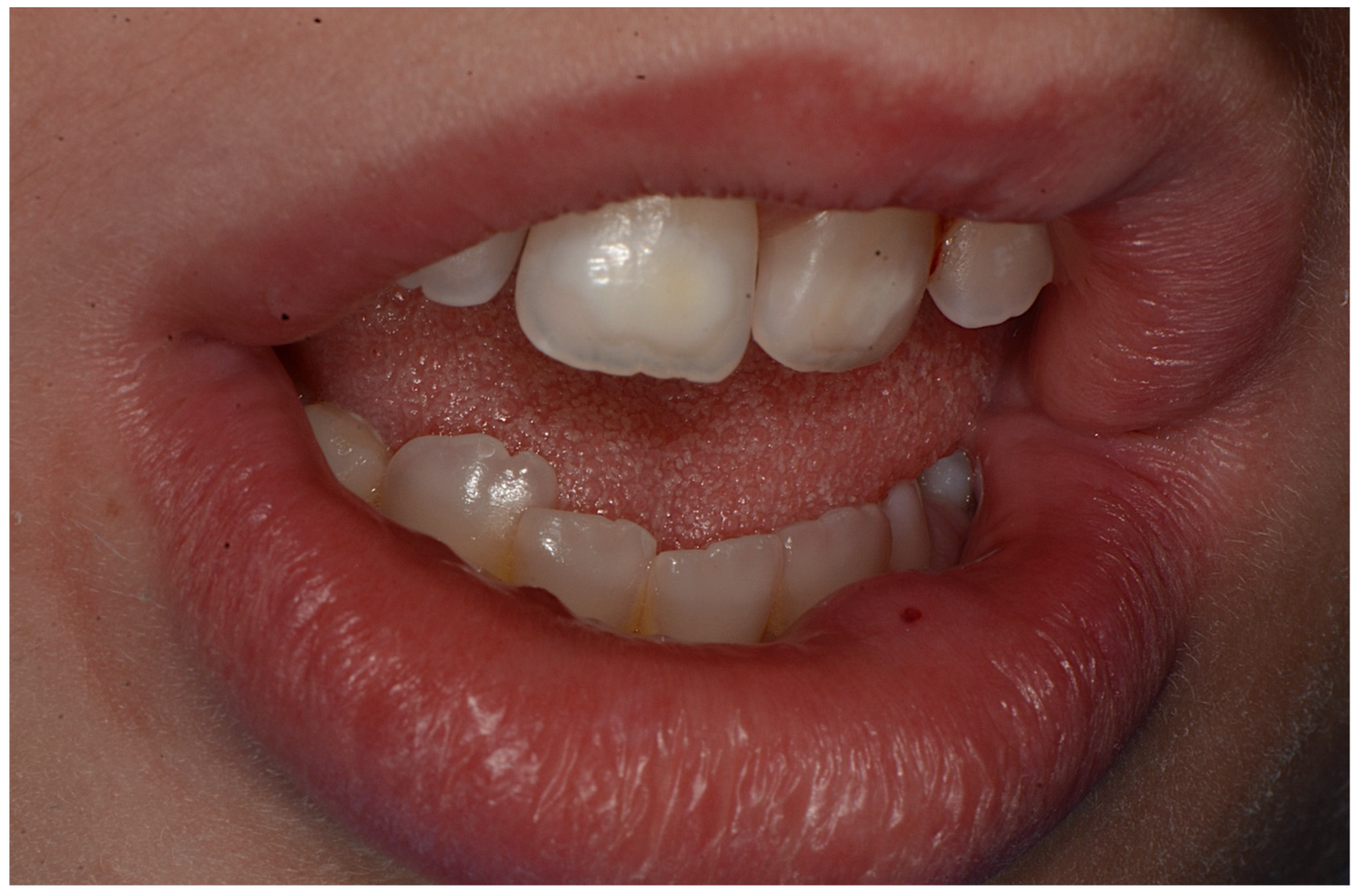
2. Case Report
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Helsmoortel, C.; Vulto-van Silfhout, A.T.; Coe, B.P.; Vandeweyer, G.; Rooms, L.; van den Ende, J.; Schuurs-Hoeijmakers, J.H.; Marcelis, C.L.; Willemsen, M.H.; Vissers, L.E.; et al. A SWI/SNF-related autism syndrome caused by de novo mutations in ADNP. Nat. Genet. 2014, 46, 380–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ADNP Kids. Available online: http://www.adnpkids.com/ (accessed on 4 July 2021).
- ADNP Kids. Research Fundation. Available online: https://www.adnpfoundation.org/ (accessed on 4 July 2021).
- Orphanet. Available online: https://www.orpha.net (accessed on 20 August 2021).
- Weerheijm, K.L.; Duggal, M.; Mejàre, I.; Papagiannoulis, L.; Koch, G.; Martens, L.C.; Hallonsten, A.L. Judgement criteria for molar incisor hypomineralisation (MIH) in epidemiologic studies: A summary of the European meeting on MIH held in Athens, 2003. Eur. J. Paediatr. Dent. 2003, 4, 110–113. [Google Scholar] [PubMed]
- Van Dijck, A.; Vulto-van Silfhout, A.T.; Cappuyns, E.; van der Werf, I.M.; Mancini, G.M.; Tzschach, A.; Bernier, R.; Gozes, I.; Eichler, E.E.; Romano, C.; et al. Clinical presentation of a complex neurodevelopmental disorder caused by mutations in ADNP. Biol. Psychiatry 2019, 85, 287–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gozes, I.; Van Dijck, A.; Hacohen-Kleiman, G.; Grigg, I.; Karmon, G.; Giladi, E.; Eger, M.; Gabet, Y.; Pasmanik-Chor, M.; Cappuyns, E.; et al. Premature primary tooth eruption in cognitive/motor-delayed ADNP-mutated children. Transl. Psychiatry 2017, 7, e1043. [Google Scholar] [CrossRef] [PubMed]
- Goyal, A. The enigma of molar incisor hypomineralization. Contemp. Clin. Dent. 2021, 12, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Endres, D.; Decher, N.; Röhr, I.; Vowinkel, K.; Domschke, K.; Komlosi, K.; Tzschach, A.; Gläser, B.; Schiele, M.A.; Runge, K.; et al. New Cav1.2 channelopathy with high-functioning autism, affective disorder, severe dental enamel defects, a short QT interval, and a novel CACNA1C loss-of-function mutation. Int. J. Mol. Sci. 2020, 21, 8611. [Google Scholar] [CrossRef] [PubMed]
- Zameer, M.; Peeran, S.A.; Basheer, S.N.; Peeran, S.W.; Birajdar, S.B.; Alzahrani, F.M.; Alkhayrat, A. Molar incisor hypomineralization (MIH) in a child with congenital chronic intestinal pseudoobstruction (CIPO). Case Rep. Dent. 2020, 2020, 8894657. [Google Scholar] [CrossRef] [PubMed]
- Della Vella, F.; Contaldo, M.; Fucile, R.; Panza, F.; Dibello, V.; Kalemaj, Z.; Ninivaggi, R.; Petruzzi, M.; Serpico, R. ORO-dental manifestations in west syndrome. Curr. Top. Med. Chem. 2019, 19, 2824–2828. [Google Scholar] [CrossRef] [PubMed]
- Pascolini, G.; Agolini, E.; Majore, S.; Novelli, A.; Grammatico, P.; Digilio, M.C. Helsmoortel-van der Aa Syndrome as emerging clinical diagnosis in intellectually disabled children with autistic traits and ocular involvement. Eur. J. Paediatr. Neurol. 2018, 22, 552–557. [Google Scholar] [CrossRef] [PubMed]
- Campus, G.; Cagetti, M.G.; Sale, S.; Petruzzi, M.; Solinas, G.; Strohmenger, L.; Lingström, P. Six months of high-dose xylitol in high-risk caries subjects—A 2-year randomised, clinical trial. Clin. Oral Investig. 2013, 17, 785–791. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sex | Oral Manifestations | Age (Years) | ||
|---|---|---|---|---|
| Helsmoortel et al., 2014 | Patient 1 | M | thin upper lip | 4.6 |
| Patient 2 | F | thin upper lip, oligodontia | 6.10 | |
| Patient 3 | F | N.D. | 8 | |
| Patient 4 | M | small mouth with thin lips, small teeth and minor tongue tie | 4.11 | |
| Patients 5 | M | normal palate, normal teeth | 8.7 | |
| Patient 6 | M | largemouth widely spaced teeth | 8.5 | |
| Patient 7 | F | thin upper lip | 5 | |
| Patient 8 | M | high narrow palate | 10.8 | |
| Patient 9 | F | N.D. | 10.6 | |
| Patient 10 | M | N.D. | 5.6 | |
| Pascolini et al., 2018 | Patient n.2 | F | small mouth, dental anomalies with multiple caries. | 3.9 |
| Van Dijck A et al., 2019 | 78 patients | M:F = 44:34 | 8.2 (mean) | |
| Present study 2021 | 1 patient | M | high narrow palate, crowding, molar incisor hypomineralization (MIH), second class | 9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petruzzi, M.; Stella, A.; Capra, V.; Contaldo, M.; della Vella, F. Oro-Dental Manifestations in a Pediatric Patient Affected by Helsmoortel-Van der Aa Syndrome. Int. J. Environ. Res. Public Health 2021, 18, 8957. https://doi.org/10.3390/ijerph18178957
Petruzzi M, Stella A, Capra V, Contaldo M, della Vella F. Oro-Dental Manifestations in a Pediatric Patient Affected by Helsmoortel-Van der Aa Syndrome. International Journal of Environmental Research and Public Health. 2021; 18(17):8957. https://doi.org/10.3390/ijerph18178957
Chicago/Turabian StylePetruzzi, Massimo, Alessandro Stella, Valeria Capra, Maria Contaldo, and Fedora della Vella. 2021. "Oro-Dental Manifestations in a Pediatric Patient Affected by Helsmoortel-Van der Aa Syndrome" International Journal of Environmental Research and Public Health 18, no. 17: 8957. https://doi.org/10.3390/ijerph18178957
APA StylePetruzzi, M., Stella, A., Capra, V., Contaldo, M., & della Vella, F. (2021). Oro-Dental Manifestations in a Pediatric Patient Affected by Helsmoortel-Van der Aa Syndrome. International Journal of Environmental Research and Public Health, 18(17), 8957. https://doi.org/10.3390/ijerph18178957