The Value of Case Reports in Systematic Reviews from Rare Diseases. The Example of Enzyme Replacement Therapy (ERT) in Patients with Mucopolysaccharidosis Type II (MPS-II)


Abstract
1. Introduction
2. Methods
2.1. Data Sources, Study Selection, and Quality Assessment from the Previous Meta-Analysis of Case Reports
2.2. Data Search of New Clinical Studies
2.3. Study Selection
- (1)
- Clinical studies (randomized or nonrandomized) and case reports of patients with MPS-II treated with ERT.
- (2)
- The study design evaluates as primary, secondary, or exploratory objective the novelty proposed by the case report.
- (3)
- The results reported included data from new patients where the novelty was the object of research.
- (1)
- Clinical studies or case reports in other diseases or based on treatment without ERT.
- (2)
- Systematic or literature reviews that do not include analyses of new patients.
2.4. Quality Assessment
2.5. Primary Outcome
2.6. Secondary Outcomes
2.7. Statistical Methods
3. Results
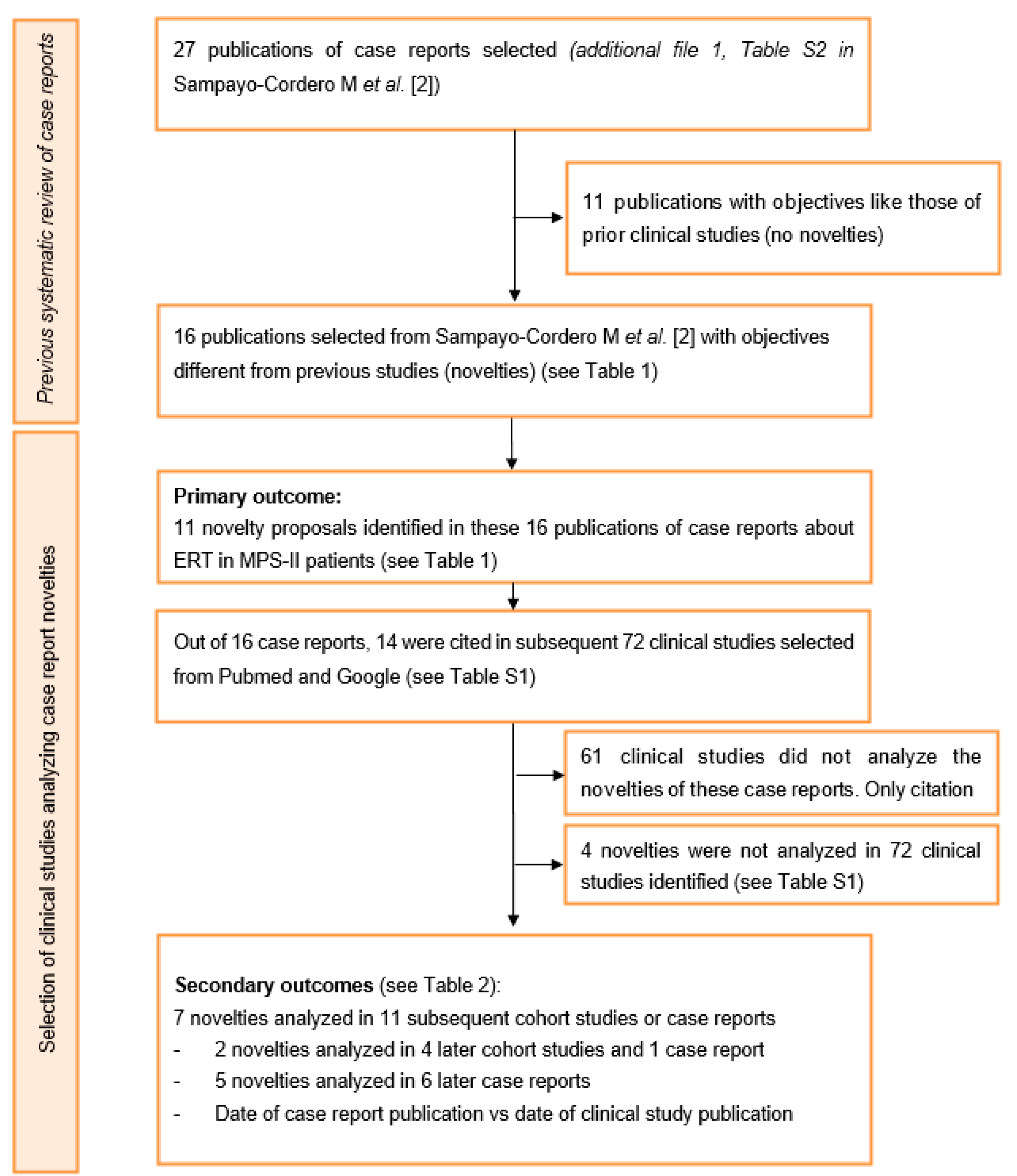
3.1. Data Search Results
3.2. Primary Outcome
3.3. Secondary Outcomes
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Richter, T.; Janoudi, G.; Amegatse, W.; Nester-Parr, S. Characteristics of drugs for ultra-rare diseases versus drugs for other rare diseases in HTA submissions made to the CADTH CDR. Orphanet J. Rare Dis. 2018, 13, 15. [Google Scholar] [CrossRef] [PubMed]
- Sampayo-Cordero, M.; Miguel-Huguet, B.; Pardo-Mateos, A.; Malfettone, A.; Pérez-García, J.; Llombart-Cussac, A.; Cortés, J.; Moltó-Abad, M.; Muñoz-Delgado, C.; Pérez-Quintana, M.; et al. Agreement between results of meta-analyses from case reports and clinical studies, regarding efficacy and safety of idursulfase therapy in patients with mucopolysaccharidosis type II (MPS-II). A new tool for evidence-based medicine in rare diseases. Orphanet J. Rare Dis. 2019, 14, 230. [Google Scholar] [CrossRef] [PubMed]
- Scarpa, M.; Almássy, Z.; Beck, M.; Bodamer, O.; Bruce, I.A.; De Meirleir, L.; Guffon, N.; Guillén-Navarro, E.; Hensman, P.; Jones, S.; et al. Mucopolysaccharidosis type II: European recommendations for the diagnosis and multidisciplinary management of a rare disease. Orphanet J. Rare Dis. 2011, 6, 72. [Google Scholar] [CrossRef] [PubMed]
- Muenzer, J.; Jones, S.A.; Tylki-Szymańska, A.; Harmatz, P.; Mendelsohn, N.J.; Guffon, N.; Giugliani, R.; Burton, B.K.; Scarpa, M.; Beck, M.; et al. Ten years of the Hunter Outcome Survey (HOS): Insights, achievements, and lessons learned from a global patient registry. Orphanet J. Rare Dis. 2017, 12, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Muenzer, J.; Giugliani, R.; Scarpa, M.; Tylki-Szymańska, A.; Jego, V.; Beck, M. Clinical outcomes in idursulfase-treated patients with mucopolysaccharidosis type II: 3-year data from the hunter outcome survey (HOS). Orphanet J. Rare Dis. 2017, 12, 1–11. [Google Scholar] [CrossRef]
- Frieden, T.R. Evidence for Health Decision Making—Beyond Randomized, Controlled Trials. N. Engl. J. Med. 2017, 377, 465–475. [Google Scholar] [CrossRef]
- Stepien, K.M.; Gevorkyan, A.K.; Hendriksz, C.J.; Lobzhanidze, T.V.; Pérez-López, J.; Tol, G.; del Toro Riera, M.; Vashakmadze, N.D.; Lampe, C. Critical clinical situations in adult patients with Mucopolysaccharidoses (MPS). Orphanet J. Rare Dis. 2020, 15, 1–12. [Google Scholar] [CrossRef]
- Lampe, C.; Harmatz, P.R.; Parini, R.; Sharma, R.; Teles, E.L.; Johnson, J.; Sivam, D.; Sisic, Z. Enzyme replacement therapy initiated in adulthood: Findings from the mucopolysaccharidosis VI Clinical Surveillance Program. Mol. Genet. Metab. 2019, 127, 355–360. [Google Scholar] [CrossRef]
- Nissen, T.; Wynn, R. The clinical case report: A review of its merits and limitations. BMC Res. Notes 2014, 7, 264. [Google Scholar] [CrossRef]
- Jackson, D.; Daly, J.; Saltman, D.C. Aggregating case reports: A way for the future of evidence-based health care? Clin. Case Rep. 2014, 2, 23–24. [Google Scholar] [CrossRef]
- Nakamura, T.; Igarashi, H.; Ito, T.; Jensen, R.T. Important of case-reports/series, in rare diseases: Using neuroendocrine tumors as an example. World J. Clin. Cases 2014, 2, 608–613. [Google Scholar] [CrossRef] [PubMed]
- Cochrane Handbook for Systematic Reviews of Interventions|Cochrane Training. Available online: http://training.cochrane.org/handbook (accessed on 4 February 2018).
- Sampayo-Cordero, M.; Miguel-Huguet, B.; Pardo-Mateos, A.; Moltó-Abad, M.; Muñoz-Delgado, C.; Pérez-López, J. Agreement between the results of meta-analyses from case reports and from clinical studies regarding the efficacy of laronidase therapy in patients with mucopolysaccharidosis type I who initiated enzyme replacement therapy in adult age: An example of case reports meta-analyses as an useful tool for evidence-based medicine in rare diseases. Mol. Genet. Metab. 2018, 121, 138–149. [Google Scholar] [CrossRef]
- Pérez-López, J.; Morales-Conejo, M.; López-Rodríguez, M.; Hermida-Ameijeiras, Á.; Moltó-Abad, M. Efficacy of laronidase therapy in patients with mucopolysaccharidosis type I who initiated enzyme replacement therapy in adult age. A systematic review and meta-analysis. Mol. Genet. Metab. 2017, 121, 138–149. [Google Scholar] [CrossRef]
- Pérez-López, J.; Moltó-Abad, M.; Muñoz-Delgado, C.; Morales-Conejo, M.; Ceberio-Hualde, L.; del Toro, M. Efficacy of Idursulfase therapy in patients with Mucopolysaccharidosis type II who initiated enzyme replacement therapy in adult age. A systematic review of the literature. Mol. Genet. Metab. 2018, 124, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Muenzer, J.; Wraith, J.E.; Beck, M.; Giugliani, R.; Harmatz, P.; Eng, C.M.; Vellodi, A.; Martin, R.; Ramaswami, U.; Gucsavas-Calikoglu, M.; et al. A phase II/III clinical study of enzyme replacement therapy with idursulfase in mucopolysaccharidosis II (Hunter syndrome). Genet. Med. 2006, 8, 465–473. [Google Scholar] [CrossRef]
- Muenzer, J.; Beck, M.; Eng, C.M.; Giugliani, R.; Harmatz, P.; Martin, R.; Ramaswami, U.; Vellodi, A.; Wraith, J.E.; Cleary, M.; et al. Long-term, open-labeled extension study of idursulfase in the treatment of Hunter syndrome. Genet. Med. 2011, 13, 95–101. [Google Scholar] [CrossRef]
- Tolar, J.; Grewal, S.S.; Bjoraker, K.J.; Whitley, C.B.; Shapiro, E.G.; Charnas, L.; Orchard, P.J. Combination of enzyme replacement and hematopoietic stem cell transplantation as therapy for Hurler syndrome. Bone Marrow Transpl. 2008, 41, 531–535. [Google Scholar] [CrossRef]
- Wynn, R.F.; Wraith, J.E.; Mercer, J.; O’Meara, A.; Tylee, K.; Thornley, M.; Church, H.J.; Bigger, B.W. Improved Metabolic Correction in Patients with Lysosomal Storage Disease Treated with Hematopoietic Stem Cell Transplant Compared with Enzyme Replacement Therapy. J. Pediatr. 2009, 154, 609–611. [Google Scholar] [CrossRef]
- Eisengart, J.B.; Rudser, K.D.; Tolar, J.; Orchard, P.J.; Kivisto, T.; Ziegler, R.S.; Whitley, C.B.; Shapiro, E.G. Enzyme Replacement is Associated with Better Cognitive Outcomes after Transplant in Hurler Syndrome. J. Pediatr. 2013, 162, 375–380.e1. [Google Scholar] [CrossRef]
- Liberati, A.; Altman, D.G.; Tetzlaff, J.; Mulrow, C.; Gøtzsche, P.C.; Ioannidis, J.P.A.; Clarke, M.; Devereaux, P.J.; Kleijnen, J.; Moher, D. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate healthcare interventions: Explanation and elaboration. BMJ 2009, 339, b2700. [Google Scholar] [CrossRef]
- Stroup, D.F.; Berlin, J.A.; Morton, S.C.; Olkin, I.; Williamson, G.D.; Rennie, D.; Moher, D.; Becker, B.J.; Sipe, T.A.; Thacker, S.B. Meta-analysis of observational studies in epidemiology: A proposal for reporting. Meta-analysis Of Observational Studies in Epidemiology (MOOSE) group. JAMA 2000, 283, 2008–2012. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Whitley, C.B.; Jarnes Utz, J.R. Correlation between urinary GAG and anti-idursulfase ERT neutralizing antibodies during treatment with NICIT immune tolerance regimen: A case report. Mol. Genet. Metab. 2017, 122, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Gkavogiannakis, N.; Aggelides, N.; Makris, M. Case of infusion reaction to idursulafase and successful re-administration with desensitization. Poster Session Group III—Green TPS 39. Allergy 2015, 70, 527–613. [Google Scholar] [CrossRef]
- Volpi, N.; Zampini, L.; Maccari, F.; Santoro, L.; Galeotti, F.; Galeazzi, T.; Gabrielli, O.; Coppa, G.V. Plasmatic kinetics of dermatan sulfate during enzyme replacement therapy with iduronate-2-sulfatase in a mucopolysaccharidosis II Patient. Glycoconj. J. 2013, 30, 727–732. [Google Scholar] [CrossRef]
- Noh, T.K.; Han, J.S.; Won, C.H.; Chang, S.; Choi, J.H.; Moon, K.C.; Lee, M.W.; Yang, J.H.; Soung, J.H. Characteristic “pebbling” skin eruption as a presenting sign of Hunter syndrome. Int. J. Dermatol. 2014, 53, e594–e596. [Google Scholar] [CrossRef]
- Marín, L.L.; Gutiérrez-Solana, L.G.; Fernández, A.T. Hunter Syndrome: Resolution of Extensive Typical Skin Lesions After 9 Months of Enzyme Replacement Therapy with Idursulfase. Pediatr. Dermatol. 2012, 29, 369–370. [Google Scholar] [CrossRef]
- Puiu, M.; Chirita-Emandi, A.; Dumitriu, S.; Arghirescu, S. Hunter syndrome follow-up after 1 year of enzyme-replacement therapy. Case Rep. 2013, 2013, bcr2012007644. [Google Scholar] [CrossRef]
- Wang, R.Y.; Cambray-Forker, E.J.; Ohanian, K.; Karlin, D.S.; Covault, K.K.; Schwartz, P.H.; Abdenur, J.E. Treatment reduces or stabilizes brain imaging abnormalities in patients with MPS I and II. Mol. Genet. Metab. 2009, 98, 406–411. [Google Scholar] [CrossRef]
- Kinoshita, M.; Furujo, M.; Kubo, T. EEG Evaluation of Mucopolysaccharidosis Type II After Enzyme Replacement Therapy. In Proceedings of the 55th Annual Meeting of the Japanese Society of Neurology, Fukuoka, Japan, 22 May 2014. [Google Scholar]
- Bonanni, P.; Gubernale, M.; Martinez, F.; Randazzo, G.; Milantoni, L.; Martinuzzi, A.; Boniver, C.; Vecchi, M.; Scarpa, M. Non-convulsive status epilepticus of frontal origin in mucopolysaccharidosis type II successfully treated with ethosuximide: Case Report. Dev. Med. Child. Neurol. 2012, 54, 961–964. [Google Scholar] [CrossRef]
- Sanchez, J.I.; Ascaso, F.J.; Perez, I.; Almenara, C.; Esteban, O.; Martinez, M.; Idoate, A.; Torralba, M.A. Role of SD-OCT in the follow-up of a patient with macular edema associated with mucopoysaccharidosis type II (Hunter syndrome) undergoing idursulfase enzyme replacement therapy. Acta Ophthalmol. 2015, 93. [Google Scholar] [CrossRef]
- Lau, H.A.; Nolan, R.; Narayana, K.; Rucker, J.; Balcer, L.; Galetta, S. Multiple mechanisms of ophthalmologic involvement in attenuated Hunter syndrome: A case report. Mol. Genet. Metab. 2015, 114, S69. [Google Scholar] [CrossRef]
- Fisher, A.; Fernandez, K.; Flores, J.; Deshpande, G.; Croke, B.; Antony, R. 2015 ASPHO Abstracts. Autoimmune thrombocytopenia in a patient with hunter syndrome: Should iduronate-2-sulfatase replacement therapy still be considered? Pediatr. Blood Cancer 2015, 62, S21–S119. [Google Scholar] [CrossRef]
- Uz, B.; Demiroglu, H.; Ozcebe, O.I. Hunter syndrome and new onset idiopathic thrombocytopenic purpura in a young patient. Ann. Hematol. 2012, 91, 303–304. [Google Scholar] [CrossRef] [PubMed]
- Nava, E.; Weber, P.; Gautschi, M.; Nuoffer, J.-M.; Grunt, S. Botulinum Toxin Type A for the Treatment of Equinus Deformity in Patients with Mucopolysaccharidosis Type II. J. Child. Neurol. 2012, 27, 1611–1615. [Google Scholar] [CrossRef]
- Farooq, M.U.; Balmer, S.V.; DeRoos, S.T.; Houtman, K.L.; Chillag, K.L. A novel mutation in the iduronate 2 sulfatase gene resulting in mucopolysaccharidosis type II and chorea: Case report of two siblings. Mov. Disord. 2008, 23, 1487–1488. [Google Scholar] [CrossRef] [PubMed]
- Papadia, F.; Lozupone, M.S.; Gaeta, A.; Capodiferro, D.; Lacalendola, G. Long-term enzyme replacement therapy in a severe case of mucopolysaccharidosis type II (Hunter syndrome). Eur. Rev. Med. Pharm. Sci 2011, 15, 253–258. [Google Scholar]
- Lampe, C.; Atherton, A.; Burton, B.K.; Descartes, M.; Giugliani, R.; Horovitz, D.D.G.; Kyosen, S.O.; Magalhães, T.S.P.C.; Martins, A.M.; Mendelsohn, N.J.; et al. Enzyme Replacement Therapy in Mucopolysaccharidosis II Patients Under 1 Year of Age. In JIMD Reports; Zschocke, J., Gibson, K.M., Brown, G., Morava, E., Peters, V., Eds.; Springer: Heidelberg, Germany, 2014; Volume 14, pp. 99–113. ISBN 978-3-662-43747-6. [Google Scholar]
- Bivina, L.; Boyadjiev, S.A. Mucopolysaccharidosis type II (MPS II): Case report of three affected siblings. Mol. Genet. Metab. 2014, 111, S27. [Google Scholar] [CrossRef]
- Christianto, A.; Watanabe, H.; Nakajima, T.; Inazu, T. Idursulfase enzyme replacement therapy in an adult patient with severe Hunter syndrome having a novel mutation of iduronate-2-sulfatase gene. Clin. Chim. Acta 2013, 423, 66–68. [Google Scholar] [CrossRef]
- Sato, Y.; Fujiwara, M.; Kobayashi, H.; Ida, H. Massive Accumulation of Glycosaminoglycans in the Aortic Valve of a Patient with Hunter Syndrome During Enzyme Replacement Therapy. Pediatr. Cardiol. 2013, 34, 2077–2079. [Google Scholar] [CrossRef]
- Tajima, G.; Sakura, N.; Kosuga, M.; Okuyama, T.; Kobayashi, M. Effects of idursulfase enzyme replacement therapy for Mucopolysaccharidosis type II when started in early infancy: Comparison in two siblings. Mol. Genet. Metab. 2013, 108, 172–177. [Google Scholar] [CrossRef]
- Hoffmann, B.; Schulze-Frenking, G.; Al-Sawaf, S.; Beck, M.; Mayatepek, E. Hunter Disease Before and During Enzyme Replacement Therapy. Pediatr. Neurol. 2011, 45, 181–184. [Google Scholar] [CrossRef] [PubMed]
- Tylki-Szymanska, A.; Jurecka, A.; Zuber, Z.; Rozdzynska, A.; Marucha, J.; Czartoryska, B. Enzyme replacement therapy for mucopolysaccharidosis II from 3 months of age: A 3-year follow-up: Enzyme replacement therapy for mucopolysaccharidosis II. Acta Paediatr. 2012, 101, e42–e47. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Calvo, J.; Bergua Sanclemente, I.; López Moreno, M.J.; Torralba Cabeza, M.Á.; Amores Arriaga, B. Respuesta precoz a idursulfasa en un paciente de 31 años de edad con síndrome de Hunter. Rev. Clín. Española 2011, 211, e42–e45. [Google Scholar] [CrossRef]
- Tchan, M.C.; Devine, K.T.; Sillence, D.O. Three Adult Siblings with Mucopolysaccharidosis Type II (Hunter Syndrome): A Report on Clinical Heterogeneity and 12 Months of Therapy with Idursulfase. In JIMD Reports—Case and Research Reports, 2011/1; JIMD Reports; SSIEM, Ed.; Springer: Berlin/Heidelberg, Germany, 2011; Volume 1, pp. 57–64. ISBN 978-3-642-17707-1. [Google Scholar]
- Galán-Gómez, E.; Guerrero-Rico, A.; Cáceres-Marzal, C.; Zambrano-Castaño, M.; Moreno-Tejero, M.-L.; Grande-Tejada, A.-M.; Fernández-Hernández, S.; Vaquerizo-Madrid, J.; Cardesa-García, J.J. Early response to idursulfase treatment in a 3 year-old boy affected of Hunter syndrome. Eur. J. Med Genet. 2008, 51, 268–271. [Google Scholar] [CrossRef] [PubMed]
- Westhoff, M.; Litterst, P. Successful Noninvasive Ventilation and Enzyme Replacement Therapy in an Adult Patient with Morbus Hunter. In JIMD Reports—Case and Research Reports, 2012/2; JIMD Reports; SSIEM, Ed.; Springer: Berlin/Heidelberg, Germany, 2011; Volume 5, pp. 77–82. ISBN 978-3-642-28095-5. [Google Scholar]
- Julien, D.C.; Woolgar, K.; Pollard, L.; Miller, H.; Desai, A.; Lindstrom, K.; Kishnani, P.S. Immune Modulation for Enzyme Replacement Therapy in A Female Patient with Hunter Syndrome. Front. Immunol. 2020, 11, 1000. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, S.; Maganthi, M.; Sanjeev, G. Pebbling of skin: Cutaneous marker of Hunter syndrome. Indian Dermatol. Online J. 2017, 8, 62. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, Y.; Miyazaki, O.; Kosuga, M.; Okuyama, T.; Nosaka, S. Cerebral magnetic resonance findings during enzyme replacement therapy in mucopolysaccharidosis. Pediatr. Radiol. 2017, 47, 1659–1669. [Google Scholar] [CrossRef]
- Manara, R.; Priante, E.; Grimaldi, M.; Santoro, L.; Astarita, L.; Barone, R.; Concolino, D.; Di Rocco, M.; Donati, M.A.; Fecarotta, S.; et al. Brain and spine MRI features of Hunter disease: Frequency, natural evolution and response to therapy. J. Inherit. Metab. Dis. 2011, 34, 763–780. [Google Scholar] [CrossRef]
- Yund, B.; Rudser, K.; Ahmed, A.; Kovac, V.; Nestrasil, I.; Raiman, J.; Mamak, E.; Harmatz, P.; Steiner, R.; Lau, H.; et al. Cognitive, medical, and neuroimaging characteristics of attenuated mucopolysaccharidosis type II. Mol. Genet. Metab. 2015, 114, 170–177. [Google Scholar] [CrossRef]
- Crowe, L.; Yaplito-Lee, J.; Anderson, V.; Peters, H. Cognitive and behaviour profiles of children with mucopolysaccharidosis Type II. Cogn. Neuropsychol. 2017, 34, 347–356. [Google Scholar] [CrossRef]
- Tanaka, A.; Okuyama, T.; Suzuki, Y.; Sakai, N.; Takakura, H.; Sawada, T.; Tanaka, T.; Otomo, T.; Ohashi, T.; Ishige-Wada, M.; et al. Long-term efficacy of hematopoietic stem cell transplantation on brain involvement in patients with mucopolysaccharidosis type II: A nationwide survey in Japan. Mol. Genet. Metab. 2012, 107, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Scarpa, M.; Lourenço, C.M.; Amartino, H. Epilepsy in mucopolysaccharidosis disorders. Mol. Genet. Metab. 2017, 122, 55–61. [Google Scholar] [CrossRef]
- Bonanni, P.; Volzone, A.; Randazzo, G.; Antoniazzi, L.; Rampazzo, A.; Scarpa, M.; Nobili, L. Nocturnal frontal lobe epilepsy in mucopolysaccharidosis. Brain Dev. 2014, 36, 826–829. [Google Scholar] [CrossRef] [PubMed]
- Yamanishi, R.; Nakamura, N.; Tsunoda, K. Recovery of Vision following Enzyme Replacement Therapy in a Patient with Mucopolysaccharidosis Type II, Hunter Syndrome. Case Rep. Ophthalmol. 2019, 10, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Alcántara-Ortigoza, M.A.; García-de Teresa, B.; González-del Angel, A.; Berumen, J.; Guardado-Estrada, M.; Fernández-Hernández, L.; Navarrete-Martínez, J.I.; Maza-Morales, M.; Rius-Domínguez, R. Wide allelic heterogeneity with predominance of large IDS gene complex rearrangements in a sample of Mexican patients with Hunter syndrome: Mutational spectrum in a sample of Mexican MPSII patients. Clin. Genet. 2016, 89, 574–583. [Google Scholar] [CrossRef]
- Nissen, T.; Wynn, R. The recent history of the clinical case report: A narrative review. JRSM Short Rep. 2012, 3, 1–5. [Google Scholar] [CrossRef]
- Bradley, L.A.; Haddow, H.R.M.; Palomaki, G.E. Treatment of mucopolysaccharidosis type II (Hunter syndrome): Results from a systematic evidence review. Genet. Med. 2017, 19, 1187–1201. [Google Scholar] [CrossRef]
- Alegra, T.; Eizerik, D.P.; de Cerqueira, C.C.S.; Pereira, T.V.; Dornelles, A.D.; Schwartz, I.V.D. Eficácia e segurança da terapia com idursulfase em pacientes com mucopolissacaridose tipo II, com e sem comparação com placebo: Revisão sistemática e metanálise. Cad. Saúde Pública 2013, 29, s45–s58. [Google Scholar] [CrossRef]
- da Silva, E.M.; Strufaldi, M.W.L.; Andriolo, R.B.; Silva, L.A. Enzyme replacement therapy with idursulfase for mucopolysaccharidosis type II (Hunter syndrome). Cochrane Database Syst. Rev. 2016, 2, 1465–1858. [Google Scholar] [CrossRef]
- Almeida, P.H.R.F.; Lemos, L.L.P.D.; Alvares, J.; Godman, B.; Bennie, M.; Acurcio, F.D.A.; Junior, A.A.G. Safety of Enzyme Replacement Therapy for Mucopolysaccharidosis II. In Pharmacoepidemiology and Drug Safety; Wiley: Hoboken, NJ, USA, 2018; pp. 222–223. [Google Scholar] [CrossRef]
- Murad, M.H.; Sultan, S.; Haffar, S.; Bazerbachi, F. Methodological quality and synthesis of case series and case reports. BMJ Evid. Based Med. 2018, 23, 60–63. [Google Scholar] [CrossRef]
- Gagnier, J.J.; Kienle, G.; Altman, D.G.; Moher, D.; Sox, H.; Riley, D.; The CARE Group; Allaire, A.; Altman, D.G.; Aronson, J.; et al. The CARE guidelines: Consensus-based clinical case reporting guideline development. Case Rep. 2013, 2013, bcr2013201554. [Google Scholar] [CrossRef]
- Luo, M.; Cohen, A.M.; Addepalli, S.; Smalheiser, N.R. Identifying main finding sentences in clinical case reports. Database 2020, 2020. [Google Scholar] [CrossRef] [PubMed]
- Penedones, A.; Alves, C.; Batel Marques, F. A comparison between two recommendations to conduct and report systematic reviews on drug’s safety. Syst. Rev. 2019, 8, 238. [Google Scholar] [CrossRef] [PubMed]
- Piasecki, J.; Waligora, M.; Dranseika, V. Google Search as an Additional Source in Systematic Reviews. Sci. Eng. Ethics 2017, 24, 809–810. [Google Scholar] [CrossRef]
- Lakhani, H.V.; Pillai, S.S.; Zehra, M.; Sharma, I.; Sodhi, K. Systematic Review of Clinical Insights into Novel Coronavirus (CoVID-19) Pandemic: Persisting Challenges in U.S. Rural Population. Int. J. Environ. Res. Public Health 2020, 17, 4279. [Google Scholar] [CrossRef]
- Cook, M.C. Medical case reports in the age of genomic medicine. Clin. Transl. Immunol. 2015, 4, e45. [Google Scholar] [CrossRef]
- Parini, R.; Deodato, F. Intravenous enzyme replacement therapy in mucopolysaccharidoses: clinical effectiveness and limitations. Int. J. Mol. Sci. 2020, 21, 2975. [Google Scholar] [CrossRef]
- Chen, H.H.; Sawamoto, K.; Mason, R.W.; Kobayashi, H.; Yamaguchi, S.; Suzuki, Y.; Orii, K.; Orii, T.; Tomatsu, S. Enzyme replacement therapy for mucopolysaccharidoses; past, present, and future. J. Hum. Genet. 2019, 64, 1153–1171. [Google Scholar] [CrossRef]
- Sawamoto, K.; Stapleton, M.; Alméciga-Díaz, C.J.; Espejo-Mojica, A.J.; Losada, J.C.; Suarez, D.A.; Tomatsu, S. Therapeutic options for mucopolysaccharidoses: current and emerging treatments. Drugs 2019, 79, 1103–1134. [Google Scholar] [CrossRef]
- MPS Consensus Programme Steering Committee; MPS Consensus Programme Co-Chairs; Akyol, M.U.; Alden, T.D.; Amartino, H.; Ashworth, J.; Belani, K.; Berger, K.I.; Borgo, A.; Braunlin, E.; et al. Recommendations for the management of MPS IVA: Systematic evidence- and consensus-based guidance. Orphanet J. Rare Dis. 2019, 14. [Google Scholar] [CrossRef]
- Lagler, F. Innovative Treatments for Mucopolysaccharidoses. J. Child Sci. 2018, 8, e163–e171. [Google Scholar] [CrossRef]
- Lagler, F.B. Current and Emerging Therapies for Mucopolysaccharidoses. In Pediatric Pharmacotherapy; Kiess, W., Schwab, M., van den Anker, J., Eds.; Handbook of Experimental Pharmacology; Springer International Publishing: Cham, Switzerland, 2019; Volume 261, pp. 39–56. ISBN 978-3-030-50493-9. [Google Scholar]
- Singh, A.; Prasad, R.; Mishra, O.P. Spectrum of lysosomal storage disorders at tertiary centre: Retrospective case-record analysis. J. Ped. Genet. 2020, 9, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Kubaski, F.; de Oliveira Poswar, F.; Michelin-Tirelli, K.; Burin, M.G.; Rojas-Málaga, D.; Brusius-Facchin, A.C.; Leistner-Segal, S.; Giugliani, R. Diagnosis of mucopolysaccharidoses. Diagnostics 2020, 10, 172. [Google Scholar] [CrossRef]
- Tjarks, B.J.; Gardner, J.M.; Riddle, N.D. Hamartomas of skin and soft tissue. Semin. Diag. Pathol. 2019, 36, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Almassi, G.H.; Algahim, M. Current and emerging management options for patients with Morquio A syndrome. Ther. Clin. Risk Manag. 2013, 45. [Google Scholar] [CrossRef]
- Valayannopoulos, V.; Wijburg, F.A. Therapy for the mucopolysaccharidoses. Rheumatology 2011, 50, v49–v59. [Google Scholar] [CrossRef] [PubMed]
- D’Aco, K.; Underhill, L.; Rangachari, L.; Arn, P.; Cox, G.F.; Giugliani, R.; Okuyama, T.; Wijburg, F.; Kaplan, P. Diagnosis and treatment trends in mucopolysaccharidosis I: Findings from the MPS I Registry. Eur. J. Ped. 2012, 171, 911–919. [Google Scholar] [CrossRef]
- Zafeiriou, D.I.; Batzios, S.P. Brain and Spinal MR imaging findings in mucopolysaccharidoses: A review. Am. J. Neuroradiol. 2013, 34, 5–13. [Google Scholar] [CrossRef]
- de Ruijter, J.; de Ru, M.H.; Wagemans, T.; IJlst, L.; Lund, A.M.; Orchard, P.J.; Schaefer, G.B.; Wijburg, F.A.; van Vlies, N. Heparan sulfate and dermatan sulfate derived disaccharides are sensitive markers for newborn screening for mucopolysaccharidoses types I, II and III. Mol. Genet. Metab. 2012, 107, 705–710. [Google Scholar] [CrossRef]
- Lin, S.-P.; Lin, H.-Y.; Wang, T.-J.; Chang, C.-Y.; Lin, C.-H.; Huang, S.-F.; Tsai, C.-C.; Liu, H.-L.; Keutzer, J.; Chuang, C.-K. A pilot newborn screening program for Mucopolysaccharidosis type I in Taiwan. Orphanet J. Rare Dis. 2013, 8, 147. [Google Scholar] [CrossRef]
- Baldo, G.; Mayer, F.Q.; Martinelli, B.Z.; de Carvalho, T.G.; Meyer, F.S.; de Oliveira, P.G.; Meurer, L.; Tavares, Â.; Matte, U.; Giugliani, R. Enzyme replacement therapy started at birth improves outcome in difficult-to-treat organs in mucopolysaccharidosis I mice. Mol. Genet. Metab. 2013, 109, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, T.; Shimizu, H.; Fukuda, T.; Kawagoe, S.; Matsumoto, J.; Shimada, Y.; Kobayashi, H.; Ida, H.; Ohashi, T.; Morimoto, H.; et al. Enzyme replacement therapy (ERT) procedure for mucopolysaccharidosis type II (MPS II) by intraventricular administration (IVA) in murine MPS II. Mol. Genet. Metab. 2012, 107, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Dickson, P.I.; Chen, A.H. Intrathecal enzyme replacement therapy for mucopolysaccharidosis I: Translating success in animal models to patients. Current Pharm. Biotechnol. 2011, 12, 946–955. [Google Scholar] [CrossRef] [PubMed]
- Reichert, R.; Campos, L.G.; Vairo, F.; de Souza, C.F.M.; Pérez, J.A.; Duarte, J.Á.; Leiria, F.A.; Anés, M.; Vedolin, L.M. Neuroimaging findings in patients with mucopolysaccharidosis: What you really need to know. Radiographics 2016, 36, 1448–1462. [Google Scholar] [CrossRef]
- Yang, E.; Prabhu, S.P. Imaging nanifestations of the leukodystrophies, inherited disorders of white matter. Radiol. Clin. N. Am. 2014, 52, 279–319. [Google Scholar] [CrossRef]
- Calleja Gero, M.L.; González Gutiérrez-Solana, L.; López Marín, L.; López Pino, M.A.; Fournier Del Castillo, C.; Duat Rodríguez, A. Neuroimaging findings in patient series with mucopolysaccharidosis. Neurología (English Edition) 2012, 27, 407–413. [Google Scholar] [CrossRef]
- Coppa, G.V.; Buzzega, D.; Zampini, L.; Maccari, F.; Galeazzi, T.; Pederzoli, F.; Gabrielli, O.; Volpi, N. Effect of 6 years of enzyme replacement therapy on plasma and urine glycosaminoglycans in attenuated MPS I patients. Glycobiology 2010, 20, 1259–1273. [Google Scholar] [CrossRef]
- Baldo, G.; Wozniak, D.F.; Ohlemiller, K.K.; Zhang, Y.; Giugliani, R.; Ponder, K.P. Retroviral-vector-mediated gene therapy to mucopolysaccharidosis I mice improves sensorimotor impairments and other behavioral deficits. J. Inherited Metab. Dis. 2013, 36, 499–512. [Google Scholar] [CrossRef]
- Renaud, D. Lysosomal Disorders associated with leukoencephalopathy. Sem. Neurol. 2012, 32, 051–054. [Google Scholar] [CrossRef]
- Baldo, G.; Giugliani, R.; Matte, U. Lysosomal enzymes may cross the blood–brain-barrier by pinocytosis: Implications for Enzyme Replacement Therapy. Med. Hypotheses 2014, 82, 478–480. [Google Scholar] [CrossRef]
- Grosse, S.D.; Lam, W.K.K.; Wiggins, L.D.; Kemper, A.R. Cognitive outcomes and age of detection of severe mucopolysaccharidosis type 1. Genet. Med. 2017, 19, 975–982. [Google Scholar] [CrossRef] [PubMed]
- Arrol, L.P.; Kerrins, A.M.; Yamakawa, Y.; Smith, P.M. Fucosidosis in a domestic shorthair cat. J. Feline Med. Surg. 2011, 13, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.Y.; Chang, Y.S.; Sung, D.K.; Ko, A.; Kim, C.H.; Yoo, D.K.; Lim, K.H.; Sohn, Y.B.; Jin, D.K.; Park, W.S. High-dose enzyme replacement therapy attenuates cerebroventriculomegaly in a mouse model of mucopolysaccharidosis type II. J. Hum. Genet. 2013, 58, 728–733. [Google Scholar] [CrossRef] [PubMed]
- Jelin, A.C.; O’Hare, E.; Blakemore, K.; Jelin, E.B.; Valle, D.; Hoover-Fong, J. Skeletal Dysplasias: Growing Therapy for Growing Bones. Front. Pharmacol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Guillén-Navarro, E.; Blasco, A.J.; Gutierrez-Solana, L.G.; Couce, M.L.; Cancho-Candela, R.; Lázaro, P. Guía de práctica clínica para el tratamiento del síndrome de Hunter. Med. Clín. 2013, 141, 453.e1–453.e13. [Google Scholar] [CrossRef]
- Mendez, D.C.; Stover, A.E.; Rangel, A.D.; Brick, D.J.; Nethercott, H.E.; Torres, M.A.; Khalid, O.; Wong, A.M.; Cooper, J.D.; Jester, J.V.; et al. A novel, long-lived, and highly engraftable immunodeficient mouse model of mucopolysaccharidosis type I. Mol. Ther. Methods Clin. Dev. 2015, 2, 14068. [Google Scholar] [CrossRef]
- Nicolas-Jilwan, M.; AlSayed, M. Mucopolysaccharidoses: Overview of neuroimaging manifestations. Ped. Radiol. 2018, 48, 1503–1520. [Google Scholar] [CrossRef]
- Liang, J.; Singhal, A. Regression of ventriculomegaly following medical management of a patient with Hurler syndrome. J. Neurosurg. Ped. 2016, 17, 537–539. [Google Scholar] [CrossRef]
- Ganesh, A.; Al-Murshedi, F.; Al-Zuhaibi, S.; Al-Thihli, K. Ocular Manifestations of Inborn Errors of Metabolism. In The Eye in Pediatric Systemic Disease; Levin, A.V., Enzenauer, R.W., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 359–460. ISBN 978-3-319-18388-6. [Google Scholar]
- Gera, K. Characterization of a Mucopolysaccharidosis Type I and GalNAc Transferase Deficiency Double Knockout Mouse 2018. Available online: https://lib.dr.iastate.edu/cgi/viewcontent.cgi?article=7589&context=etd. (accessed on 10 September 2020).
- Valayannopoulos, V. Enzyme replacement therapy in lysosomal storage diseases. In Rare Diseases; Özgüç, M., Ed.; Advances in Predictive, Preventive and Personalised Medicine; Springer: Dordrecht, The Netherlands, 2015; Volume 6, pp. 91–107. ISBN 978-94-017-9213-4. [Google Scholar]
- Kubaski, F.; de Oliveira Poswar, F.; Michelin-Tirelli, K.; da Matte, U.S.; Horovitz, D.D.; Barth, A.L.; Baldo, G.; Vairo, F.; Giugliani, R. Mucopolysaccharidosis Type I. Diagnostics 2020, 10, 161. [Google Scholar] [CrossRef]
- Kubaski, F. Diagnosis and Therapies for Mucopolysaccharidoses. Doctoral Dissertation. 2017. Available online: https://udspace.udel.edu/bitstream/handle/19716/21745/Kubaski_udel_0060D_12805.pdf?sequence=1&isAllowed=y (accessed on 27 July 2020).
- Zhao, X.; Huang, Y.; Li, S.; Lin, W.; Zhou, Z.; Liu, L. Quantitative determination of urinary mucopolysaccharide and its clinical application. Chin. J. Children’s Health 2010, 11, 885–889. Available online: http://www.cqvip.com/qk/98529a/201011/35809101.html. (accessed on 10 September 2020).
- Zheng, M.; Tan, J.; Pan, L.; Cai, R. Analysis of clinical features of mucopolysaccharidosis type II. Chinese J. Eugenics Genet. 2013, 110–111. Available online: http://www.cqvip.com/qk/98444x/201301/44774375.html (accessed on 10 September 2020).
- Wang, L.; Sun, J. The role of prenatal fetal heart monitoring in reducing fetal and neonatal injury. Chinese J. Eugenics Genet. 2013, 62–63. Available online: http://www.cqvip.com/qk/98444x/201301/44774346.html (accessed on 10 September 2020).
- van der Lee, J.H.; Morton, J.; Adams, H.R.; Clarke, L.; Ebbink, B.J.; Escolar, M.L.; Giugliani, R.; Harmatz, P.; Hogan, M.; Jones, S.; et al. Cognitive endpoints for therapy development for neuronopathic mucopolysaccharidoses: Results of a consensus procedure. Mol. Genet. Metab. 2017, 121, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Janzen, D.; Delaney, K.A.; Shapiro, E.G. Cognitive and adaptive measurement endpoints for clinical trials in mucopolysaccharidoses types I, II, and III: A review of the literature. Mol. Genet. Metab. 2017, 121, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Kasteleijn- Nolst Trenite, D.; Genton, P.; Brandt, C.; Reed, R.C. The ‘Photosensitivity Model’ is ( also) a model for focal (partial) seizures. Epilepsy Res. 2017, 133, 113–120. [Google Scholar] [CrossRef]
- Kasteleijn-Nolst Trenite, D. Rebuttal to the manuscript by R.J. Porter. Epilepsy Res. 2017, 133, 123–125. [Google Scholar] [CrossRef] [PubMed]
- Brandl, U. Ethosuximid bei strukturellen und läsionellen Epilepsien. Z. Epileptol. 2014, 27, 132–138. [Google Scholar] [CrossRef]
- Scarpa, M.; Cinzia, B. Coinvolgimento neurologico nelle MPS. La Rivista delle Malattie Rare. 2013. Nr. 2. Available online: https://malattierare.aou.udine.it/news/articoli/coinvolgimento-neurologico-nelle-mps (accessed on 10 September 2020).
- Porter, R.J. The photosensitivity model is not a model for partial (focal) seizures—REBUTTAL. Epilepsy Res. 2017, 133, 121–122. [Google Scholar] [CrossRef]
- Huang, H. Clinical analysis of status epilepticus secondary to acute cerebral hemorrhage. J. Guangxi Med. Univ. 2013, 5, 777–779. Available online: http://www.cqvip.com/qk/91948a/2013005/47755900.html. (accessed on 10 September 2020).
- Chen, Z.; Chen, M.; He, J.; Jia, W.; Qin, X. Segmentation of diabetic macular edema for retinal OCT images. In Proceedings of the Optics in Health Care and Biomedical Optics VIII; Luo, Q., Li, X., Tang, Y., Gu, Y., Eds.; SPIE: Beijing, China, 2018; p. 32. [Google Scholar]
- Emel, N. Evaluation of hematologic findings in mucopolysaccharidosis cases. Acta Med. Medit. 2020, 797–800. [Google Scholar] [CrossRef]
- Panigrahi, I.; Dhanorkar, M.; Didel, S.; Koganti, R.A. Hunter syndrome with persistent thrombocytopenia. BMJ Case Rep. 2019, 12, e226518. [Google Scholar] [CrossRef] [PubMed]
- Kuzenkova, L.; Namazova-Baranova, L.; Gevorkyan, A.; Vashakmadze, N.; Podkletnova, T.; Studenikin, V.; Lazurenko, S. The modern view of mucopolysaccharidosis type II in children: a multidisciplinary approach to the problem. Effect. Pharmacother. Pediatrics 2012. Available online: https://umedp.ru/articles/sovremennyy_vzglyad_na_mukopolisakharidoz_ii_tipa_u_detey_multidistsiplinarnyy_podkhod_k_probleme.html (accessed on 10 September 2020).
- Pawliuk, C.; Widger, K.; Dewan, T.; Brander, G.; Brown, H.L.; Hermansen, A.-M.; Grégoire, M.-C.; Steele, R.; Siden, H. (Hal) Scoping review of symptoms in children with rare, progressive, life-threatening disorders. BMJ Support. Palliat. Care 2020, 10, 91–104. [Google Scholar] [CrossRef]
- Guo, Y.-B.; Pan, H.-D.; Guo, C.-M.; Li, Y.-M.; Chen, L.-M. Identification of a novel mutation of IDS gene from a Chinese pedigree with MPS II. Hereditas (Beijing) 2009, 31, 1101–1106. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, M.F.; Lacerda, L.; Alves, S. Glycosaminoglycan storage disorders: A review. Biochem. Res. Int. 2012, 2012, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Noh, H.; Lee, J.I. Current and potential therapeutic strategies for mucopolysaccharidoses. J. Clin. Pharm. Ther. 2014, 39, 215–224. [Google Scholar] [CrossRef]
- Tanaka, N.; Kida, S.; Kinoshita, M.; Morimoto, H.; Shibasaki, T.; Tachibana, K.; Yamamoto, R. Evaluation of cerebrospinal fluid heparan sulfate as a biomarker of neuropathology in a murine model of mucopolysaccharidosis type II using high-sensitivity LC/MS/MS. Mol. Genet. Metab. 2018, 125, 53–58. [Google Scholar] [CrossRef]
- Zuber, Z.; Jurecka, A.; Jurkiewicz, E.; Kiec-Wilk, B.; Tylki-Szymanska, A. Cervical Spine MRI Findings in Patients with Mucopolysaccharidosis Type II. Pediatr. Neurosurg. 2015, 50, 26–30. [Google Scholar] [CrossRef]
- Rout-Pitt, N.B. The Effects of Undegraded Glycosaminoglycans from Mucopolysaccharidoses on Osteoblast Differentiation and Mineralisation In Vitro; University of Adelaide, School of Medicine: Adelaide, Australia, 2015. [Google Scholar]


{kind=link}
| Novelties Proposed in Case Reports for Treatment of MPS-II Patient with ERT * | Case Reports Selected |
|---|---|
| New therapeutic strategies to increase the immunotolerance for ERT | |
| (1) Immune tolerance regimen and desensitization procedures | Kim et al. 2014; Gkavogiannakis N et al. 2015 [23,24] |
| (2) New therapeutic strategies based on the evaluation of plasmatic dermatan sulfate | Volpi et al. 2013 [25] |
| Different outcomes and therapeutic situations | |
| (3) Effects in pebbling skin lesions | NoH et al. 2014; Marín LL et al. 2012 [26,27] |
| (4) Effects in hyperactivity, aggressive behavior, language functioning, and social interaction | Puiu M et al. 2013 [28] |
| (5) Evaluation of the effects of ERT in central nervous system development | Wang RY et al. 2009 [29] |
| (6) Evaluation of the effects of ERT in epileptogenic symptoms | Kinoshita M et al. 2014; Bonanni P et al. 2012 [30,31] |
| (7) ERT effects in vision | Sanchez JI et al. 2015; Lau HA et al. 2015 [32,33] |
| (8) ERT effect in autoimmune anemia, thrombocytopenia, or thrombocytopenic purpura | Fisher et al. 2015; Uz B et al. 2012 [34,35] |
| (9) Botulinum Toxin for the treatment of equinus deformity with an ERT | Nava E et al. 2012 [36] |
| (10) ERT effects in involuntary movements (chorea) | Farooq MU et al. 2008 [37] |
| (11) Early ERT effects in bone abnormalities | Papadia F et al. 2011 [38] |
| Novelties Proposed in Case Reports for ERT in MPS-II Patients (Citation) * | Next Studies, Article Type (Citation) | Time Until Clinical Study (Case Reports not Considered) |
|---|---|---|
| (1) Immune tolerance regimen and desensitization procedures [23,24] | Case report (Julien DC et al., 2020) [50] | No clinical study after 6 years |
| (2) New therapeutic strategies based on the evaluation of plasmatic dermatan sulfate [25] | No new citations | No clinical study after 7 years |
| (3) Effects in pebbling skin lesions [26,27] | Case report (Srinivas SM et al., 2017) [51] | No clinical study after 8 years |
| (4) Effects in hyperactivity, aggressive behavior, language functioning, and social interaction [28] | No clinical study after 7 years | No clinical study after 7 years |
| (5) Evaluation of the effects of ERT in central nervous system development [29] | Cohort studies Matsubara et al., 2017; Manara et al., 2011 **; Yund et al., 2015; Tanaka A et al., 2012 (3 case reports) Crowe et al., 2017 [52,53,54,55,56] | 2 years/Not randomized |
| (6) Evaluation of the effects of ERT in epileptogenic symptoms [30,31] | Case report Scarpa et al., 2017 Bonanni et al., 2014 [57,58] | No clinical study after 8 years |
| (7) ERT effects in vision [32,33] | Case report (Yamanishi R et al., 2019) [59] | No clinical study after 5 years |
| (8) ERT effect in autoimmune anemia, thrombocytopenia, or thrombocytopenic purpura [34,35] | Case report (Alcántara-Ortigoza et al., 2016) [60] | No clinical study after 8 years |
| (9) Botulinum Toxin for the treatment of equinus deformity with an ERT [36] | No new citations | No clinical study after 8 years |
| (10) ERT effects in involuntary movements (chorea) [37] | No new citations | No prospective study after 12 years |
| (11) Early ERT effects in bone abnormalities [38] | Cohort study (Manara R et al., 2011 **) [53] | 4 months/Not randomized |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sampayo-Cordero, M.; Miguel-Huguet, B.; Malfettone, A.; Pérez-García, J.M.; Llombart-Cussac, A.; Cortés, J.; Pardo, A.; Pérez-López, J. The Value of Case Reports in Systematic Reviews from Rare Diseases. The Example of Enzyme Replacement Therapy (ERT) in Patients with Mucopolysaccharidosis Type II (MPS-II). Int. J. Environ. Res. Public Health 2020, 17, 6590. https://doi.org/10.3390/ijerph17186590
Sampayo-Cordero M, Miguel-Huguet B, Malfettone A, Pérez-García JM, Llombart-Cussac A, Cortés J, Pardo A, Pérez-López J. The Value of Case Reports in Systematic Reviews from Rare Diseases. The Example of Enzyme Replacement Therapy (ERT) in Patients with Mucopolysaccharidosis Type II (MPS-II). International Journal of Environmental Research and Public Health. 2020; 17(18):6590. https://doi.org/10.3390/ijerph17186590
Chicago/Turabian StyleSampayo-Cordero, Miguel, Bernat Miguel-Huguet, Andrea Malfettone, José Manuel Pérez-García, Antonio Llombart-Cussac, Javier Cortés, Almudena Pardo, and Jordi Pérez-López. 2020. "The Value of Case Reports in Systematic Reviews from Rare Diseases. The Example of Enzyme Replacement Therapy (ERT) in Patients with Mucopolysaccharidosis Type II (MPS-II)" International Journal of Environmental Research and Public Health 17, no. 18: 6590. https://doi.org/10.3390/ijerph17186590
APA StyleSampayo-Cordero, M., Miguel-Huguet, B., Malfettone, A., Pérez-García, J. M., Llombart-Cussac, A., Cortés, J., Pardo, A., & Pérez-López, J. (2020). The Value of Case Reports in Systematic Reviews from Rare Diseases. The Example of Enzyme Replacement Therapy (ERT) in Patients with Mucopolysaccharidosis Type II (MPS-II). International Journal of Environmental Research and Public Health, 17(18), 6590. https://doi.org/10.3390/ijerph17186590