3.2. Synthesis of Compounds
3.2.1. Synthesis of 2-Hydroxy-5-((Triisopropylsilyl)Oxy)Benzaldehyde (2)
In a 50 mL flask containing 2,5-dihydrobenzaldehyde (1, 20.0 mmol, 2.76 g, 1.1 equiv.), DMAP (3.64 mmol, 444 mg, 0.2 equiv.), and imidazole (54.6 mmol, 3.71 g, 3.0 equiv.) dissolved in dry DCM (20 mL), TIPSCl (18.8 mmol, 3.89 mL, 1.0 equiv.) was added dropwise at 0 °C, and the reaction was stirred overnight at this temperature. After the reaction was completed, additional DCM (20 mL) was added, and the solution was washed with water (10 mL × 3). The organic layer was dried over anhydrous MgSO4, removed under reduced pressure, and the residue was purified with silica gel chromatography (eluent: PE/AcOEt = 50/1) to afford compound 2 as a colorless oil (4.5 g, 77% yield). TLC: Rf = 0.7 (PE/AcOEt = 5:1, UV254). 1H NMR (500 MHz, CDCl3) δ 10.61 (s, 1H), 9.81 (s, 1H), 7.10 (dd, J = 8.9, 3.0 Hz, 1H), 7.01 (d, J = 3.0 Hz, 1H), 6.87 (d, J = 8.9 Hz, 1H), 1.29 − 1.19 (m, 3H), 1.10 (d, J = 7.2 Hz, 18H). 13C NMR (126 MHz, CDCl3) δ 196.1, 156.0, 148.8, 129.7, 122.3, 120.4, 118.4, 17.9, 12.6. LRMS-ESI (m/z): calcd for C16H27O3Si [M+H]+ 295.2, found 295.6.
3.2.2. Synthesis of 3-Bromo-2-Hydroxy-5-((Triisopropylsilyl)Oxy)Benzaldehyde (3)
In a 50 mL flask containing compound 2 (6.0 mmol, 1.75 g, 1.0 equiv.) and anhydrous pyridine (12 mmol, 0.96 mL, 2.0 equiv.) dissolved in DCM (15 mL), bromine (12 mmol, 0.61 mL, 2.0 equiv.) dissolved in DCM was added dropwise at room temperature (25 °C), and the reaction was stirred at this temperature for 3 h. After completion of the reaction, a saturated Na2SO3 solution was added to remove excess bromine, followed by the addition of DCM (15 mL), and the mixture was washed with water (10 mL × 3). The organic layer was dried over anhydrous Na2SO4, removed, and the residue was purified with silica gel chromatography (eluent: PE/AcOEt = 50/1) to afford the target compound 3 as a colorless gel (1.63 g, 73% yield). TLC: Rf = 0.72 (PE/AcOEt = 5:1, UV254). 1H NMR (500 MHz, CDCl3) δ 11.13 (s, 1H), 9.77 (s, 1H), 7.37 (d, J = 2.9 Hz, 1H), 7.01 (d, J = 2.9 Hz, 1H), 1.25 (ddt, J = 13.9, 9.9, 6.6 Hz, 3H), 1.11 (d, J = 7.2 Hz, 18H). 13C NMR (126 MHz, CDCl3) δ 195.6, 152.6, 149.1, 132.4, 122.1, 120.6, 111.0, 17.8, 12.5. LRMS-ESI (m/z): calcd for C16H26BrO3Si [M+H]+ 373.1, 375.1, found 373.1, 375.1.
3.2.3. Synthesis of 3-Bromo-5-Hydroxy-2-Methoxybenzaldehyde (4)
In a 50 mL flask containing compound
3 (8.94 mmol, 3.3 g, 1,0 equiv.) and K
2CO
3 (17.88 mmol, 2.47 g, 2.0 equiv.) dissolved in anhydrous DMF (8 mL, stored with 4 Ǻ MS under N
2), CH
3I (17.88 mmol, 1.11 mL, 2.0 equiv.) was injected via syringe and the reaction was stirred at 10 °C for 4 h. After completion of the reaction, cooled AcOEt (30 mL) and water (5 mL) were added consecutively at this temperature. Then the organic layer was washed with water (10 mL × 5), dried over anhydrous Na
2SO
4, and removed. The crude residue was dissolved in anhydrous THF (10 mL), TBAF (8.94 mmol, 8.94 mL, 1.0 equiv.) was added, and the reaction was stirred at room temperature for 0.5 h. THF was removed under reduced pressure, and AcOEt (30 mL) was added. The organic layer was washed with water (10 mL × 3), dried over Na
2SO
4, and removed. The residue was purified with silica gel chromatography (eluent: PE/AcOEt = 20/1 − 5/1) to afford the target compound
4 [
16] as a white powder (1.75 g, 85% yield). TLC: R
f = 0.3 (PE/AcOEt = 4:1, UV
254).
1H NMR (500 MHz, CDCl
3) δ 10.29 (s, 1H), 7.38 (d,
J = 3.0 Hz, 1H), 7.28 (d,
J = 3.2 Hz, 1H), 5.53 (s, 1H), 3.94 (s, 3H).
13C NMR (126 MHz, (CD
3)
2SO) δ 175.2, 140.4, 138.9, 116.9, 112.8, 104.6, 99.1, 49.8. LRMS-ESI (
m/
z): calcd for C
8H
6BrO
3 [M − H]
− 229.0, 231.0, found 229.0, 231.0.
3.2.4. Synthesis of 5-(3-(Benzyloxy)-4-Formylphenoxy)-3-Bromo-2-Methoxybenzaldehyde (6)
In a 25 mL flask containing compound 4 (1.95 mmol, 448 mg, 1.0 equiv.), 2-(benzyloxy)-4-fluorobenzaldehyde (5, 3.51 mmol, 807.3 mg, 1.8 equiv.), and K2CO3 (5.85 mmol, 806.8 mg, 3.0 equiv.) dissolved in anhydrous DMF (10 mL), the reaction mixture was heated at 100 °C overnight (about 18 h). After completion of the reaction, DMF was removed under reduced pressure, and AcOEt (30 mL) was added. The organic layer was washed with water (10 mL × 3), dried over Na2SO4, and removed, and the residue was purified with silica gel chromatography (eluent: PE/AcOEt = 15/1 − 2/1) to afford the target compound 6 as a colorless gel (0.8 g, 93% yield). TLC: Rf = 0.33 (PE/AcOEt = 4:1, UV254). 1H NMR (500 MHz, CDCl3) δ 10.44 (s, 1H), 10.32 (s, 1H), 7.85 (d, J = 8.7 Hz, 1H), 7.53 (d, J = 3.0 Hz, 1H), 7.45 (d, J = 3.0 Hz, 1H), 7.38 − 7.40 (m, 4H), 7.35 (ddd, J = 8.4, 5.4, 3.3 Hz, 1H), 6.62 (d, J = 2.2 Hz, 1H), 6.55 (dd, J = 8.7, 2.8 Hz, 1H), 5.13 (s, 2H), 4.01 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 188.2, 188.2, 163.1, 162.8, 157.0, 152.1, 135.5, 131.3, 131.0, 130.7, 128.8, 128.5, 127.3, 121.3, 119.1, 118.3, 110.1, 102.9, 70.8, 63.8. LRMS-ESI (m/z): calcd for C22H18BrO5 [M+H]+ 441.0, 443.0, found 441.6, 443.5.
3.2.5. Synthesis of 5-(3-(Benzyloxy)-4-Formylphenoxy)-4′-(Tert-Butoxy)-2-Methoxy-[1,1′-Biphenyl]-3-Carbaldehyde (8)
In a 15 mL Schlenk tube containing compound 6 (0.682 mmol, 300 mg, 1.0 equiv.), 4-tert-butyl-benzoboronic acid (7, 1.36 mmol, 265 mg, 2.0 equiv.), and K2CO3 (2.04 mmol, 282 mg, 3.0 equiv.) dissolved in anhydrous DMF (4 mL) and water (0.5 mL), Pd(OAc)2 (0.136 mmol, 30 mg, 0.2 equiv.) and PPh3 (0.34 mmol, 90 mg, 0.5 equiv.) were added and the solution was bubbled with N2 for 10 min, the mixture was heated at 80 °C in a nitrogen atmosphere overnight. After completion of the reaction, DMF was removed under reduced pressure, and AcOEt (20 mL) was added. The organic layer was washed with water (5 mL × 3), dried over Na2SO4, and removed. The residue was purified with silica gel chromatography (eluent: PE/AcOEt = 50/1 − 10/1) to afford the target compound 8 as a colorless gel (0.3 g, 86% yield). TLC: Rf = 0.3 (PE/DCM = 4:1, then DCM, UV254). 1H NMR (500 MHz, (CD3)2SO) δ 10.32 (s, 1H), 10.29 (s, 1H), 7.73 (d, J = 8.6 Hz, 1H), 7.54 (d, J = 8.6 Hz, 2H), 7.48 − 7.43 (m, 3H), 7.35 (t, J = 7.2 Hz, 2H), 7.33 − 7.30 (m, 2H), 7.09 (d, J = 8.6 Hz, 2H), 6.96 (d, J = 2.2 Hz, 1H), 6.65 (t, J = 9.3 Hz, 1H), 5.26 (s, 2H), 3.52 (s, 3H), 1.34 (s, 9H). 13C NMR (126 MHz, (CD3)2SO) δ 189.5, 187.7, 163.4, 162.4, 157.0, 155.3, 151.1, 137.4, 136.2, 130.7, 130.3, 130.1, 129.5, 128.5, 128.2, 128.1, 127.6, 125.0, 123.4, 120.5, 116.9, 115.1, 109.9, 103.7, 78.4, 70.2, 62.6, 28.6. LRMS-ESI (m/z): calcd for C32H31O6 [M+H]+ 511.2, found 511.5.
3.2.6. Synthesis of 7-(Benzyloxy)-3-(4-(Tert-Butoxy)Phenyl)-2-Methoxydibenzo[b,d]Furan-1,8-Dicarbaldehyde (8a)
In a 10 mL vial containing compound
8 (0.12 mmol, 60 mg, 1.0 equiv.), Pd(OAc)
2 (0.036 mmol, 8 mg, 0.3 equiv.), and AgOAc (0.24 mmol, 40 mg, 2.0 equiv.), PivOH (0.3 mL) was added, and the reaction mixture was heated at 130 °C for 24 h. PivOH was removed under reduced pressure, and AcOEt (10 mL) was added. The organic layer was washed with water (3 mL × 3), dried over Na
2SO
4, and removed, and the residue was purified with silica gel chromatography (eluent: PE/DCM = 10/1 − 1/1.5) to afford the target compound
8a as a white solid (7.8 mg, 13% yield). TLC: R
f = 0.3 (PE/DCM = 4:1, then PE/DCM = 1:1, UV
254).
1H NMR (700 MHz, CDCl
3) δ 10.77 (s, 1H), 10.62 (s, 1H), 9.53 (s, 1H), 7.77 (s, 1H), 7.59 − 7.54 (m, 2H), 7.51 (d,
J = 7.1 Hz, 2H), 7.44 (t,
J = 7.5 Hz, 2H), 7.40 − 7.35 (m, 1H), 7.18 (s, 1H), 7.13 − 7.10 (m, 2H), 5.31 (s, 2H), 3.54 (s, 3H), 1.43 (s, 9H).
13C NMR (176 MHz, CDCl
3) δ 190.9, 189.1, 162.8, 161.9, 158.9, 155.6, 153.5, 135.6, 134.1, 131.5, 129.7, 129.0, 128.8, 128.4, 127.3, 124.9, 124.1, 122.0, 118.9, 117.4, 95.7, 78.9, 70.9, 63.0, 28.9. LRMS-ESI (
m/
z): calcd for C
32H
29O
6 [M+H]
+ 509.2, found 509.4. The crystallization data for this compound were stored in the Cambridge Structural Database (CSD). CCDC 2475075 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via
www.ccdc.cam.ac.uk/data_request/cif, accessed on 20 September 2020.
3.2.7. Synthesis of 2-(Benzyloxy)-4-((4′-(Tert-Butoxy)-6-Methoxy-5-(Pivaloyloxy)-[1,1′-Biphenyl]-3-Yl)Oxy)Phenyl Pivalate (9)
In a 25 mL flask containing compound 8 (0.12 mmol, 60 mg, 1.0 equiv.) dissolved in anhydrous DCM (8 mL), m-CPBA (0.48 mmol, 95 mg (85 wt%), 4.0 equiv.) was added in portions at 0 °C, and then the reaction mixture was stirred at room temperature (25 °C) overnight. After the reaction was completed, a saturated Na2S2O3 solution was added to quench excess m-CPBA. Additional DCM (10 mL) was added, and the organic layer was washed with water (3 mL × 3), 10% NaOH solution (3 mL × 3), and removed. The residue was dissolved in MeOH (5 mL), and 2 M NaOH (2 mL, 10 equiv.) was added. The reaction was stirred at room temperature for about 1 h. After the reaction was completed, MeOH was removed, and AcOEt (15 mL) was added. The organic layer was washed with water (3 mL × 3), dried over Na2SO4, and removed. The residue was dried in vacuum overnight. The above crude mixture (0.062 mmol, 30 mg, 1.0 equiv.) dissolved in dry DCM (6 mL) was cooled to 0 °C, and Et3N (0.3 mmol, 42 μL, 5.0 equiv.) was added, followed by adding PivCl (0.28 mmol, 30 μL, 4.0 equiv.) dropwise and DMAP (0.006 mmol, 1.0 mg, 0.1 equiv.). The reaction mixture was further stirred at this temperature for about 3 h. After the reaction was completed, DCM (15 mL) and water (3 mL) were added, and the organic layer was washed with water (3 mL × 3), dried, and removed. The residue was purified with silica gel chromatography (eluent: PE/AcOEt = 50/1 − 40/1) to afford the target compound 9 as a colorless gel (30.1 mg, 38% yield over three steps from 8). TLC: Rf = 0.6 (PE/DCM = 2:1, UV254). 1H NMR (500 MHz, CDCl3) δ 7.44 (d, J = 8.6 Hz, 2H), 7.37 (d, J = 6.4 Hz, 2H), 7.33 (t, J = 7.1 Hz, 2H), 7.30 (d, J = 6.9 Hz, 1H), 7.02 (d, J = 8.6 Hz, 2H), 6.97 (d, J = 8.6 Hz, 1H), 6.88 (d, J = 2.9 Hz, 1H), 6.76 (d, J = 2.7 Hz, 1H), 6.68 (d, J = 2.9 Hz, 1H), 6.62 (dd, J = 8.7, 2.6 Hz, 1H), 4.99 (s, 2H), 3.36 (s, 3H), 1.38 (s, 9H), 1.38 (s, 9H), 1.26 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 176.8, 176.6, 167.3, 155.3, 155.2, 152.8, 151.2, 145.2, 136.6, 136.2, 132.0, 129.6, 128.4, 128.1, 127.8, 123.9, 123.2, 117.9, 112.7, 110.6, 105.2, 78.7, 70.9, 60.7, 39.2, 39.0, 29.0, 27.2, 27.2. LRMS-ESI (m/z): calcd for C40H47O8 [M+H]+ 655.3, found 655.3.
3.2.8. Synthesis of 7-(Benzyloxy)-3-(4-(Tert-Butoxy)Phenyl)-2-Methoxydibenzo[b,d]Furan-1,8-Diyl Bis(2,2-Dimethylpropanoate) (9a)
In a 10 mL vial containing compound 9 (0.035 mmol, 23 mg), Pd(TFA)2 (3.5 mg, 0.01 mmol, 0.3 equiv.), and AgOAc (11.6 mg, 0.07 mmol, 2.0 equiv.), PivOH (0.3 mL) was added, and the mixture was heated at 130 °C for 10 h. PivOH was removed under reduced pressure, and AcOEt (6 mL) was added. The organic layer was washed with water (3 mL × 3), 10% NaOH solution (1 mL × 3), dried over Na2SO4, and removed. The residue was purified with silica gel chromatography (eluent: PE/DCM = 10/1 − 1/1) to afford the target compound 9a as a colorless gel in 53% isolated yield (12 mg) with 13% (3 mg) of 9 recovered. TLC: Rf = 0.5 (PE/DCM = 2:1, UV254). 1H NMR (700 MHz, (CD3)2SO) δ 7.70 (s, 1H), 7.63 (s, 1H), 7.56 (d, J = 8.6 Hz, 2H), 7.44 (d, J = 6.9 Hz, 2H), 7.41 (t, J = 7.6 Hz, 2H), 7.35 (t, J = 7.2 Hz, 1H), 7.32 (s, 1H), 7.09 (d, J = 8.6 Hz, 2H), 5.22 (s, 2H), 3.36 (s, 3H), 1.46 (s, 9H), 1.36 (s, 9H), 1.23 (s, 9H). 13C NMR (126 MHz, (CD3)2SO) δ 175.9, 175.5, 154.9, 154.5, 152.3, 150.7, 145.0, 137.5, 136.6, 136.1, 133.4, 131.5, 129.7, 128.4, 128.2, 127.9, 123.3, 116.9, 114.2, 113.6, 110.2, 98.2, 78.2, 70.6, 60.8, 38.4, 28.6, 26.8, 26.7. LRMS-ESI (m/z): calcd for C40H44O8Na [M+Na]+ 675.3, found 675.3.
3.2.9. Synthesis of Compound 2-(Benzyloxy)-4-((4′-(Tert-Butoxy)-5-((Dimethylcarbamoyl)Oxy)-6-Methoxy-[1,1′-Biphenyl]-3-Yl)Oxy)Phenyl Dimethylcarbamate (10)
Following a similar procedure for the preparation of compound 9, replacing PivCl by (CH3)2NCOCl (5.0 equiv.), and using compound 8 (0.11 mmol, 58.7 mg, 1.0 equiv.), compound 10 (eluent: PE/AcOEt = 10/1 − 5:1) was synthesized as a colorless gel (45 mg, 62% yield over three steps from 8). TLC: Rf = 0.4 (PE/AcOEt = 3:1, UV254). 1H NMR (500 MHz, CDCl3) δ 7.44 (d, J = 8.6 Hz, 2H), 7.39 (d, J = 6.7 Hz, 2H), 7.34 (t, J = 7.3 Hz, 2H), 7.31 − 7.27 (m, 1H), 7.06 (d, J = 8.7 Hz, 1H), 7.01 (d, J = 8.6 Hz, 2H), 6.86 (d, J = 3.0 Hz, 1H), 6.76 (d, J = 2.7 Hz, 1H), 6.75 (d, J = 3.0 Hz, 1H), 6.63 (dd, J = 8.6, 2.7 Hz, 1H), 5.03 (s, 2H), 3.42 (s, 3H), 3.13 (s, 3H), 3.05 (s, 3H), 3.02 (s, 3H), 2.97 (s, 3H), 1.38 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 155.1, 154.9, 154.7, 154.4, 152.9, 151.5, 145.5, 145.4, 137.1, 136.6, 136.3, 132.2, 129.6, 128.4, 127.8, 127.2, 123.7, 123.6, 117.3, 112.8, 111.0, 105.8, 78.6, 70.7, 60.7, 36.9, 36.7, 36.5, 36.5, 28.9. LRMS-ESI (m/z): calcd for C36H41N2O8 [M+H]+ 629.3, found 629.3.
3.2.10. Synthesis of 7-(Benzyloxy)-3-(4-(Tert-Butoxy)Phenyl)-2-Methoxydibenzo[b,d]Furan-1,8-Diyl Bis(Dimethylcarbamate) (10a)
In a 10 mL vial containing compound 10 (0.14 mmol, 86.1 mg), Pd(OAc)2 (0.04 mmol, 9.2 mg, 0.3 equiv.), and AgOAc (45.7 mg, 0.27 mmol, 2.0 equiv.), PivOH (0.3 mL) was added, and the mixture was heated at 130 °C for 10 h. PivOH was removed under reduced pressure, and AcOEt was added. The organic layer was washed with water, dried over Na2SO4, and removed, and the residue was purified with silica gel chromatography (eluent: PE/DCM = 5/1 − 2:1) to afford the target compound 10a as a colorless gel (41.5 mg, 49% yield). TLC: Rf = 0.3 (PE/AcOEt = 3:1, UV254). 1H NMR (500 MHz, CDCl3) δ 7.56 − 7.51 (m, 3H), 7.44 (d, J = 6.9 Hz, 2H), 7.39 (t, J = 7.3 Hz, 2H), 7.35 (s, 1H), 7.33 (t, J = 7.2 Hz, 1H), 7.17 (s, 1H), 7.05 (d, J = 8.6 Hz, 2H), 5.16 (s, 2H), 3.45 (s, 3H), 3.32 (s, 3H), 3.13 (s, 3H), 3.08 (s, 3H), 2.99 (s, 3H), 1.40 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 155.2, 154.9, 154.8, 154.1, 153.1, 150.8, 145.8, 138.3, 137.8, 136.5, 133.6, 132.9, 129.8, 128.5, 128.0, 127.3, 123.8, 118.1, 115.8, 115.2, 109.8, 97.4, 78.6, 71.0, 61.0, 37.1, 36.9, 36.8, 36.6, 28.9. LRMS-ESI (m/z): calcd for C36H39N2O8 [M+H]+ 627.3, found 627.3.
3.2.11. Synthesis of 3′,4′-Bis(Benzyloxy)-5-(3-(Benzyloxy)-4-Formylphenoxy)-2-Methoxy-[1,1′-Biphenyl]-3-Carbaldehyde (11)
Following a similar procedure for the preparation of compound 8, compound 6 (1.33 mmol, 587.6 mg, 1.0 equiv.) and (3,4-bis(benzyloxy)phenyl)boronic acid (1.6 mmol, 533 mg, 1.2 equiv.) were used to afford compound 11 (eluent: PE/AcOEt = 30/1 − 10:1) as a white powder (588 mg, 68%). TLC: Rf = 0.5 (PE/AcOEt = 5:1, UV254). 1H NMR (500 MHz, CDCl3) δ 10.42 (s, 1H), 10.41 (s, 1H), 7.83 (d, J = 8.6 Hz, 1H), 7.48 (t, J = 6.6 Hz, 3H), 7.45 (s, 1H), 7.44 (d, J = 3.0 Hz, 1H), 7.41 − 7.27 (m, 12H), 7.25 (d, J = 2.2 Hz, 1H), 7.23 (d, J = 3.0 Hz, 1H), 7.11 − 6.97 (m, 2H), 6.64 (d, J = 2.2 Hz, 1H), 6.55 (dd, J = 8.6, 2.4 Hz, 1H), 5.23 (s, 2H), 5.22 (s, 2H), 5.12 (s, 2H), 3.41 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 189.4, 188.2, 163.9, 162.8, 157.6, 151.3, 149.2, 148.6, 137.5, 137.0, 137.0, 135.6, 130.9, 130.6, 129.1, 128.8, 128.7, 128.6, 128.5, 128.4, 127.9, 127.9, 127.4, 127.3, 127.3, 122.0, 120.9, 117.5, 115.8, 114.7, 109.7, 102.5, 71.3, 71.2, 70.7, 62.5. LRMS-ESI (m/z): calcd for C42H35O7 [M+H]+ 651.2, found 651.4.
3.2.12. Synthesis of 2-(Benzyloxy)-4-((3′,4′-Bis(Benzyloxy)-6-Methoxy-5-(Pivaloyloxy)-[1,1′-Biphenyl]-3-Yl)Oxy)Phenyl Pivalate (12)
Following similar procedure for the preparation of compound 9, compound 11 (0.32 mmol, 210 mg) was used to afford compound 12 (eluent: PE/AcOEt = 30/1 − 20/1) as a colorless gel (156 mg, 61% yield over three steps from 8). TLC: Rf = 0.75 (PE/AcOEt = 5:1, UV254). 1H NMR (500 MHz, CDCl3) δ 7.47 (t, J = 6.6 Hz, 4H), 7.41 − 7.27 (m, 11H), 7.24 (d, J = 2.2 Hz, 1H), 7.04 (dd, J = 8.3, 2.1 Hz, 1H), 6.98 (s, 1H), 6.96 (s, 1H), 6.83 (d, J = 3.0 Hz, 1H), 6.75 (d, J = 2.7 Hz, 1H), 6.66 (d, J = 2.9 Hz, 1H), 6.60 (dd, J = 8.7, 2.7 Hz, 1H), 5.20 (s, 2H), 5.19 (s, 2H), 4.98 (s, 2H), 3.27 (s, 3H), 1.39 (s, 9H), 1.26 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 176.7, 176.6, 155.2, 152.7, 151.2, 148.8, 148.5, 145.2, 145.1, 137.2, 137.2, 136.3, 136.2, 136.1, 130.5, 128.5, 128.4, 128.4, 128.1, 127.8, 127.8, 127.7, 127.5, 127.3, 123.2, 122.1, 117.9, 116.0, 114.6, 112.5, 110.5, 105.1, 71.3, 71.2, 70.8, 60.5, 39.1, 38.9, 27.2, 27.2. LRMS-ESI (m/z): calcd for C50H51O9 [M+H]+ 795.3, found 795.5.
3.2.13. Synthesis of 7-(Benzyloxy)-3-(3,4-Bis(Benzyloxy)Phenyl)-2-Methoxydibenzo[b,d]Furan-1,8-Diyl Bis(2,2-Dimethylpropanoate) (12a)
Following similar procedure for the preparation of compound 9a, compound 12 (0.055 mmol, 44 mg) was used to afford compound 12a (eluent: PE/DCM = 10/1 − 1/1.5) as a colorless gel (18 mg, 41%, 51% brsm). TLC: Rf = 0.4 (PE/DCM = 4:1, then PE/DCM = 1:1, UV254). 1H NMR (500 MHz, CD2Cl2) δ 7.49 (t, J = 6.6 Hz, 4H), 7.47 − 7.42 (m, 2H), 7.42 (s, 1H), 7.41 − 7.38 (m, 5H), 7.37 (s, 1H), 7.36 (d, J = 1.5 Hz, 2H), 7.35 − 7.30 (m, 3H), 7.24 (s, 1H), 7.18 (dd, J = 8.2, 2.2 Hz, 1H), 7.06 (d, J = 8.4 Hz, 1H), 5.19 (s, 4H), 5.15 (s, 2H), 3.31 (s, 3H), 1.52 (s, 9H), 1.29 (s, 9H). 13C NMR (126 MHz, CD2Cl2) δ 177.1, 176.6, 155.6, 153.6, 151.4, 149.3, 149.1, 145.9, 138.7, 137.9, 137.9, 137.7, 136.7, 134.3, 131.6, 129.1, 129.1, 129.0, 128.8, 128.5, 128.4, 128.4, 128.2, 128.1, 122.9, 118.1, 116.6, 115.6, 115.3, 114.9, 110.5, 97.9, 71.8, 71.8, 71.7, 61.4, 39.9, 39.4, 27.7, 27.4. LRMS-ESI (m/z): calcd for C50H49O9 [M+H]+ 793.3, 793.4.
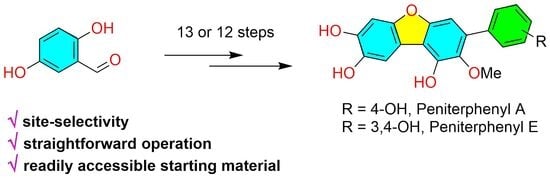
3.2.14. Synthesis of Peniterphenyl A
In a 10 mL vial, compound
9a (0.031 mmol, 20 mg, 1.0 equiv.) was dissolved in DCM (0.5 mL), and TFA (0.1 mL) was added dropwise, and the mixture was stirred at room temperature for 1 h. After the reaction was completed, DCM was removed under reduced pressure. Additional DCM (1.0 mL) was added, and the solvent was removed under reduced pressure; this procedure was repeated twice. The residue was dissolved in MeOH/THF mixture (1.0 mL,
v/
v = 1/1), followed by the addition of 2 M NaOH (0.62 mmol, 0.3 mL, 20 equiv.). The mixture was heated at 50 °C for 1 h. MeOH and THF were removed and the residue was dissolved in AcOEt (10 mL). The organic layer was washed with water (3 mL × 3) and removed. The residue was dissolved in MeOH (1.5 mL), and 10% Pd/C (4 mg, 20 wt%) and HCO
2NH
4 (0.62 mmol, 40 mg, 20 equiv.) were added, and the reaction mixture was heated at 85 °C for 2 h. MeOH was removed, and the residue was redissolved in AcOEt (10 mL). The organic layer was washed with water (3 mL × 3), dried over Na
2SO
4, and removed. The residue was dissolved in MeOH and purified by semi-preparative HPLC (RP-C18, MeOH/H
2O = 60/40, flow rate = 3.0 mL/min, λ = 254 nm) to afford peniterphenyl A (t
R = 16 min) as a colorless gel (6.0 mg, 59% yield over three steps from
9a). Its
1H/
13C NMR signals were carefully assigned and listed in
Table S1. The NMR data (
Table S1, Figures S1 and S2) and the HRMS result (
Figure S5) for synthesized peniterphenyl A were in excellent agreement with the natural one, indicating our successful synthesis of this natural product. TLC: R
f = 0.2 (PE/AcOEt = 1:1.5, UV
254).
1H NMR (700 MHz, (CD
3)
2SO) δ 9.64 (s, 1H), 9.54 (s, 1H), 9.29 (s, 1H), 9.03 (s, 1H), 7.43 (d,
J = 8.6 Hz, 2H), 7.39 (s, 1H), 6.96 (s, 1H), 6.89 (s, 1H), 6.84 (d,
J = 8.6 Hz, 2H), 3.33 (s, 3H).
13C NMR (176 MHz, (CD
3)
2SO) δ 157.1, 153.1, 150.0, 145.9, 144.8, 142.5, 140.4, 132.4, 130.4, 129.3, 115.6, 114.7, 113.0, 107.6, 102.8, 98.7, 60.7. HRMS-ESI (
m/
z): calcd for C
19H
13O
6 [M − H]
– 337.0718, found 337.0726.
3.2.15. Synthesis of Peniterphenyl E
In a 15 mL Schlenk tube under a nitrogen atmosphere, compound
12a (0.038 mmol, 30 mg, 1.0 equiv.) was dissolved in dry THF (3 mL). The mixture was cooled to 0 °C, and LiAlH
4 (0.38 mmol, 15.0 mg, 10 equiv.) was added in portions. The reaction was stirred at this temperature for 1 h. Upon completion, a saturated NH
4Cl solution was added dropwise. AcOEt (10 mL) was then added, and the mixture was washed with saturated NH
4Cl (2 mL × 2) solution and H
2O (2 mL × 2). The organic layer was removed under reduced pressure, and the residue was subjected to short silica gel chromatography (eluent: PE/AcOEt = 10/1). The crude product was dissolved in MeOH (2 mL), followed by the addition of HCO
2NH
4 (1.52 mmol, 96 mg, 40 equiv.) and 10% Pd/C (6 mg, 20 wt%). The reaction mixture was heated at 85 °C for 1 h. After completion, the solution was directly purified by semi-preparative HPLC (RP-C18, MeOH/H
2O = 50/50, flow rate = 3.0 mL/min, λ = 254 nm) to afford peniterphenyl E (t
R = 14 min) as a white gel (4.0 mg, 30% yield over two steps from
12a). Its
1H/
13C NMR signals were carefully assigned and listed in
Table S2. The NMR data (
Table S2, Figures S3 and S4) and the HRMS result (
Figure S6) for synthesized peniterphenyl E were in excellent agreement with the natural one, indicating our successful synthesis of this natural product. TLC: R
f = 0.2 (PE/AcOEt = 1:3, UV
254).
1H NMR (700 MHz, CD
3OD) δ 7.47 (s, 1H), 7.12 (d,
J = 2.2 Hz, 1H), 6.97 (dd,
J = 8.2, 2.2 Hz, 1H), 6.93 (s, 1H), 6.85 (s, 1H), 6.83 (d,
J = 8.2 Hz, 1H), 3.43 (s, 3H).
13C NMR (176 MHz, CD
3OD) δ 154.9, 152.0, 146.5, 146.1, 145.9, 145.8, 143.0, 141.2, 133.9, 131.9, 121.8, 117.4, 116.6, 116.2, 113.8, 108.6, 103.7, 98.9, 61.0. HRMS-ESI (
m/
z): calcd for C
19H
13O
7 [M − H]
– 353.0667, found 353.0672.

 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
