
Marine Natural Products in Clinical Use

Abstract
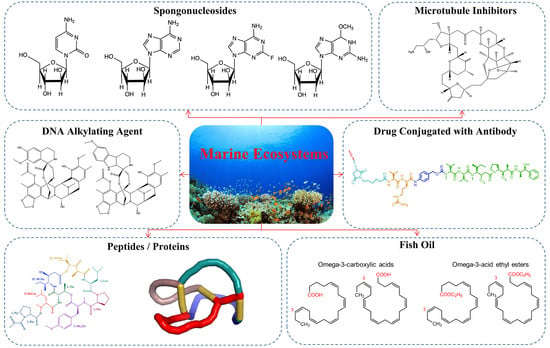
1. Introduction
2. Marine Bioactive Compounds Available on the Market
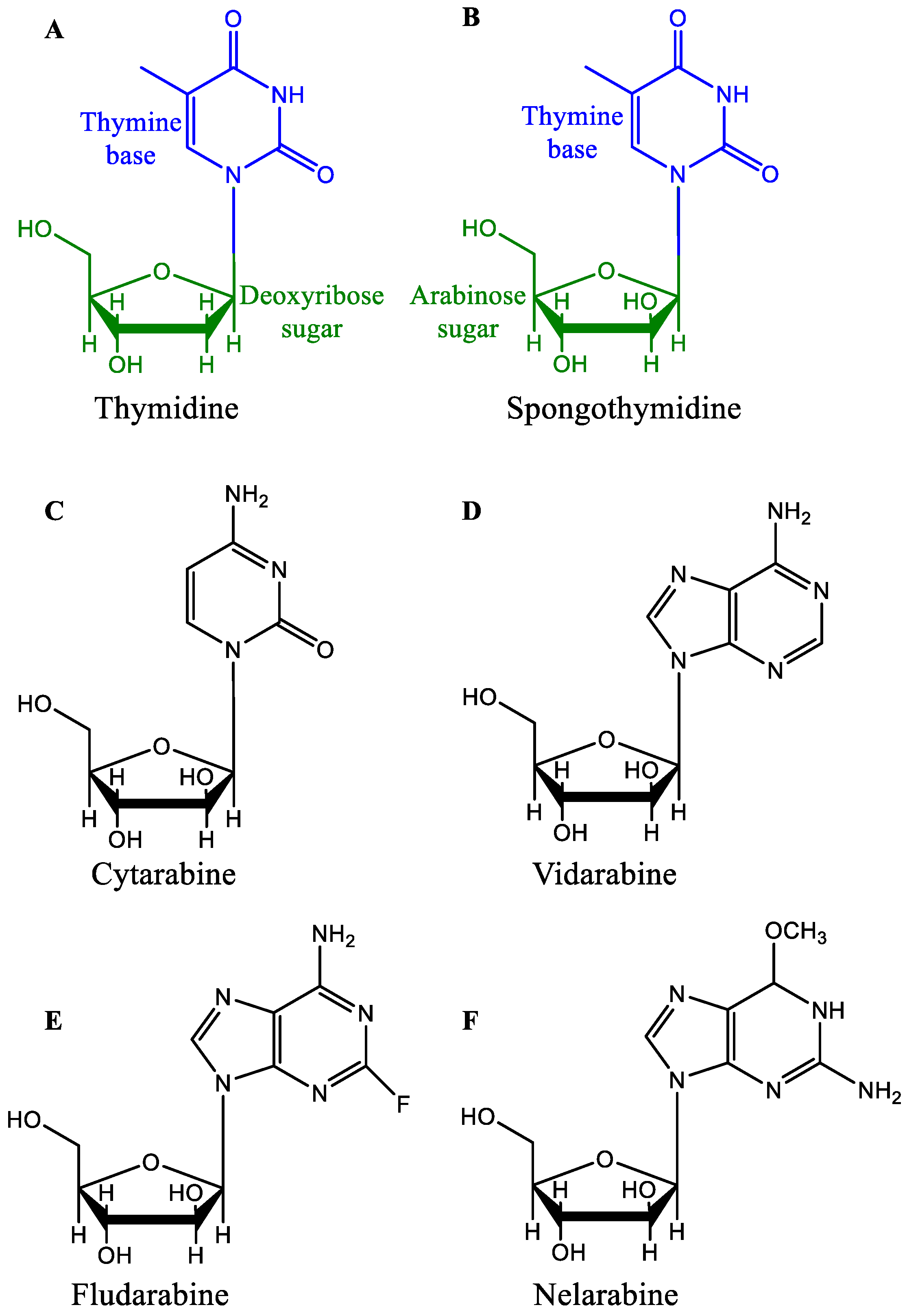
2.1. Nucleoside Analogs
2.1.1. Cytarabine
2.1.2. Vidarabine
2.1.3. Fludarabine
2.1.4. Nelarabine
2.1.5. Histochrome®: Sodium Salt of Echinochrome A—A Common Sea Urchin Pigment
2.2. Microtubule Inhibitors
2.3. DNA Alkyating Agents
2.3.1. Trabectedin
2.3.2. Lurbinectedin
2.4. Antibody-Drug Conjugates
2.4.1. Brentuximab Vedotin
2.4.2. Polatuzumab Vedotin
2.4.3. Enfortumab Vedotin
2.4.4. Belantamab Mafodotin
2.5. Peptides or Proteins Used as Drugs or in Drug Preparations
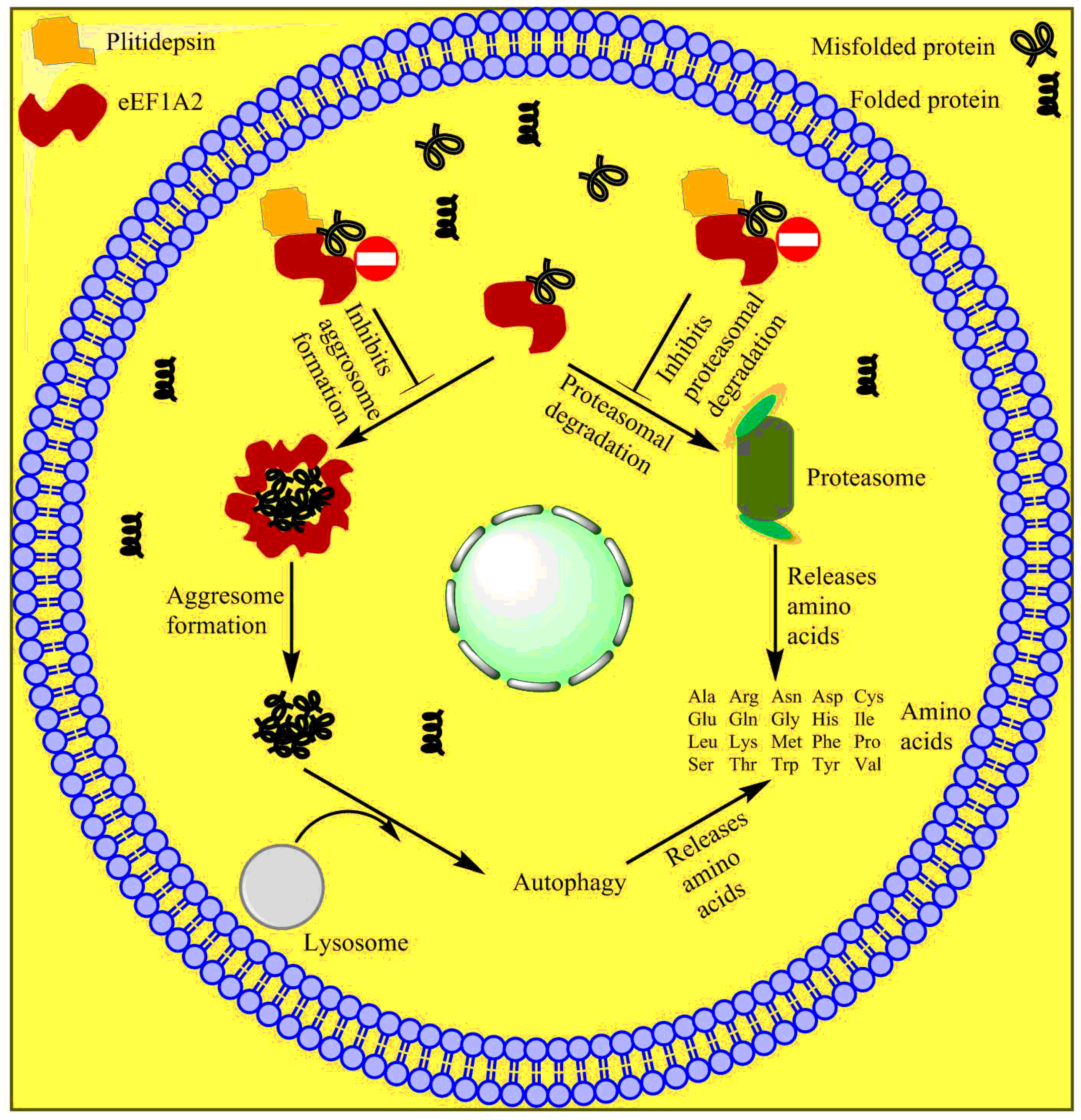
2.5.1. Plitidepsin
2.5.2. Ziconotide
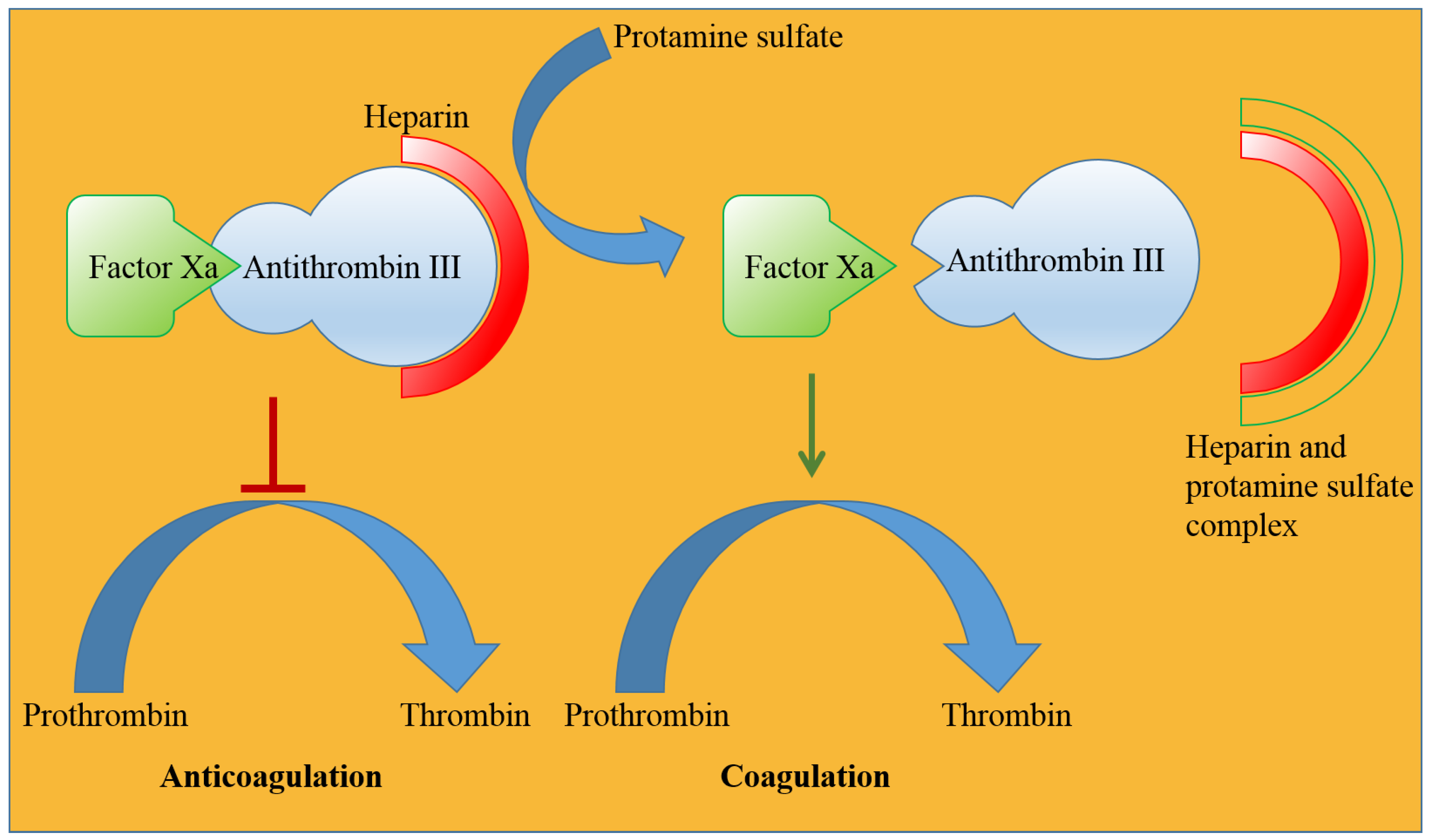
2.5.3. Protamine Sulfate
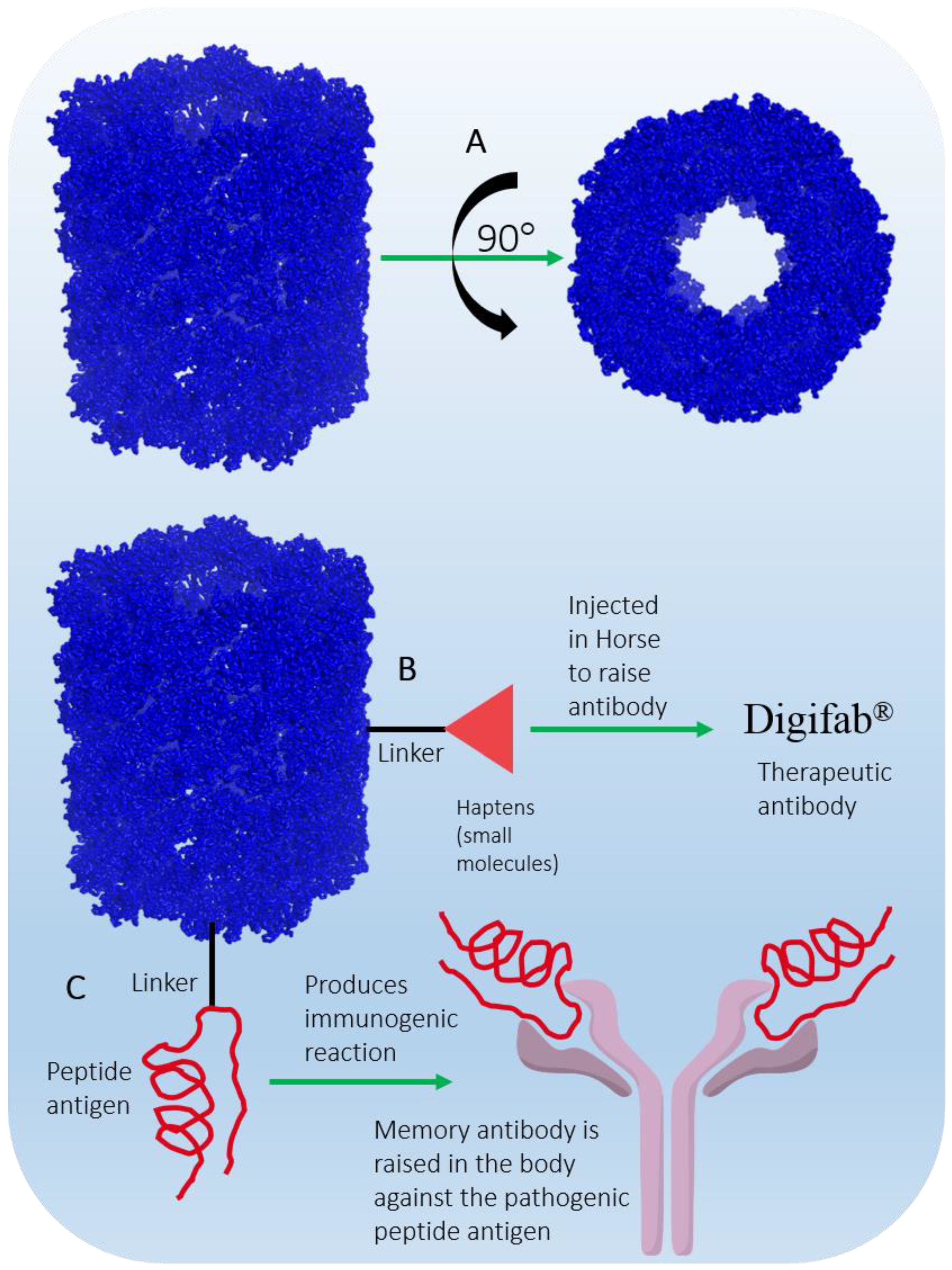
2.5.4. Keyhole Limpet Hemocyanin (KLH)
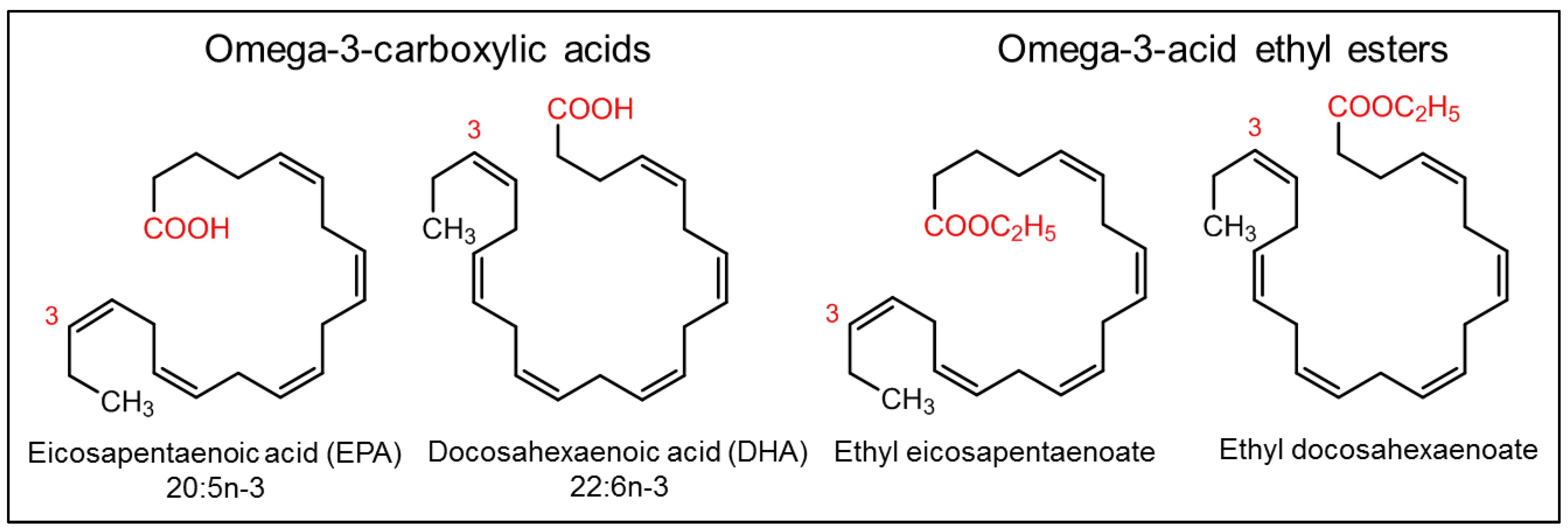
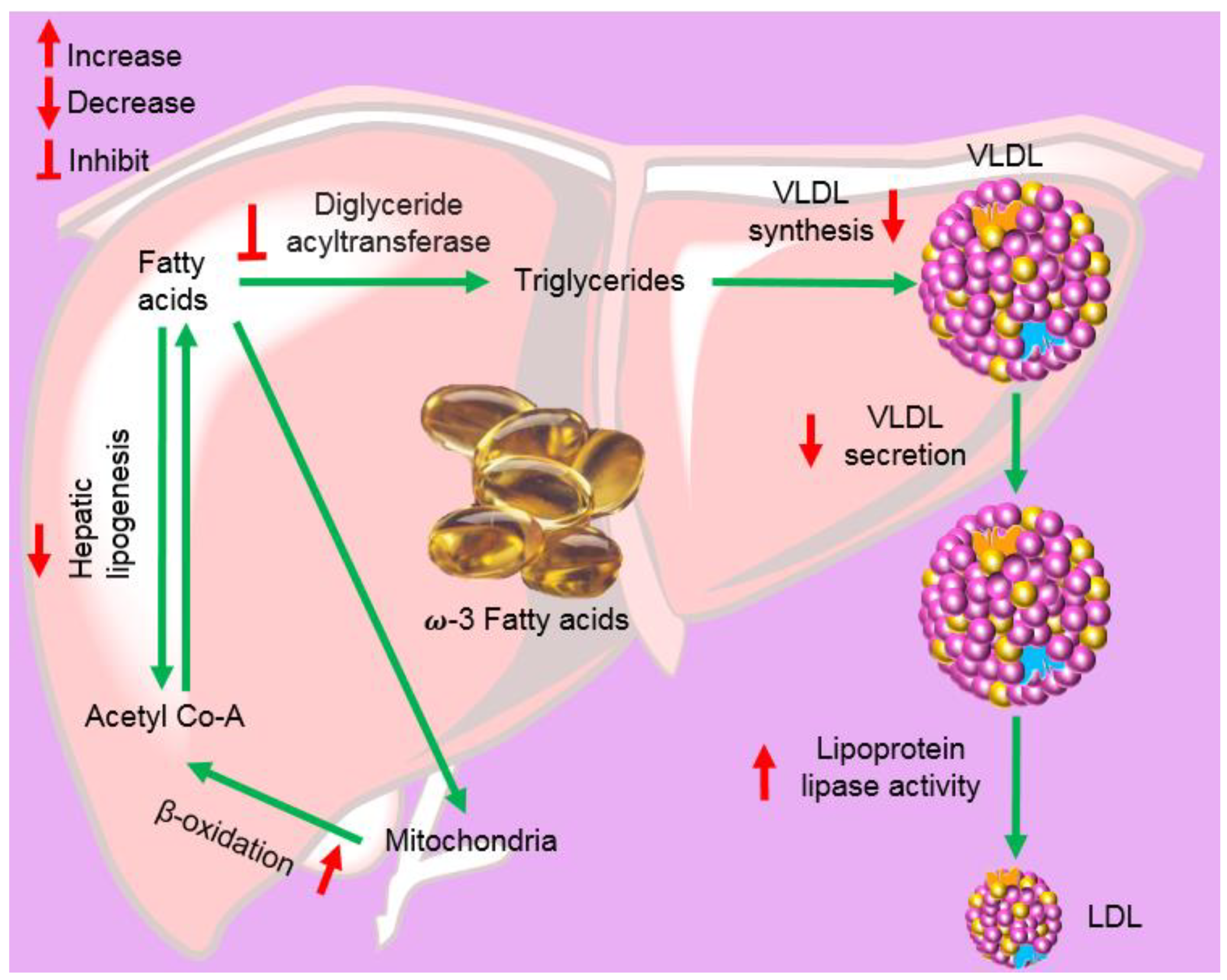
2.6. Fish Oil and Its Components
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bergmann, W.; Feeney, R.J. The isolation of a new thymine pentoside from sponges1. J. Am. Chem. Soc. 1950, 72, 2809–2810. [Google Scholar] [CrossRef]
- Bergmann, W.; Feeney, R.J. Contributions to the study of marine products. XXXII. The nucleosides of sponges I. J. Org. Chem. 1951, 16, 981–987. [Google Scholar] [CrossRef]
- Mayer, A.M. The Global Marine Pharmaceuticals Pipeline. Available online: https://www.midwestern.edu/departments/marinepharmacology/clinical-pipeline (accessed on 30 June 2022).
- Mayer, A.M.; Glaser, K.B.; Cuevas, C.; Jacobs, R.S.; Kem, W.; Little, R.D.; McIntosh, J.M.; Newman, D.J.; Potts, B.C.; Shuster, D.E. The odyssey of marine pharmaceuticals: A current pipeline perspective. Trends Pharmacol. Sci. 2010, 31, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Kremer, W.B. Drugs five years later: Cytarabine. Ann. Intern. Med. 1975, 82, 684–688. [Google Scholar] [CrossRef]
- Glantz, M.J.; LaFollette, S.; Jaeckle, K.A.; Shapiro, W.; Swinnen, L.; Rozental, J.R.; Phuphanich, S.; Rogers, L.R.; Gutheil, J.C.; Batchelor, T. Randomized trial of a slow-release versus a standard formulation of cytarabine for the intrathecal treatment of lymphomatous meningitis. J. Clin. Oncol. 1999, 17, 3110–3116. [Google Scholar] [CrossRef]
- Buchanan, R.A.; Hess, F. Vidarabine (Vira-A®): Pharmacology and clinical experience. Pharmacol. Ther. 1980, 8, 143–171. [Google Scholar] [CrossRef]
- Rodriguez, G. Fludarabine phosphate. Investig. New Drugs 1994, 12, 75–92. [Google Scholar] [CrossRef]
- Gandhi, V.; Keating, M.J.; Bate, G.; Kirkpatrick, P. Nelarabine. Nat. Rev. Drug Discov. 2006, 5, 17–18. [Google Scholar] [CrossRef]
- Mishchenko, N.P.; Fedoreev, S.A.; Bagirova, V.L. Histochrome: A new original domestic drug. Pharm. Chem. J. 2003, 37, 48–52. [Google Scholar] [CrossRef]
- Eisai (Global) Eisai Announces Canadian Approval of Its Anticancer Agent Halaven™. Available online: https://www.eisai.com/news/news201179.html (accessed on 30 June 2022).
- USFDA. Highlights of Prescribing Information: HALAVEN™. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/201532s015lbl.pdf (accessed on 30 June 2022).
- EMA. Yondelis. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/yondelis (accessed on 30 June 2022).
- EMA. Committee for Medicinal Products for Human Use Post-Authorisation Summary of Positive Opinion for Yondelis. Available online: https://www.ema.europa.eu/en/documents/smop/chmp-post-authorisation-summary-positive-opinion-yondelis-24-september-2009_en.pdf (accessed on 30 June 2022).
- Newswire, P.R. Jazz Pharmaceuticals Announces U.S. FDA Accelerated Approval of zepzelca™ (lurbinectedin) for the Treatment of Metastatic Small Cell Lung Cancer. Available online: https://www.prnewswire.com/news-releases/jazz-pharmaceuticals-announces-us-fda-accelerated-approval-of-zepzelca-lurbinectedin-for-the-treatment-of-metastatic-small-cell-lung-cancer-301077082.html (accessed on 30 June 2022).
- USFDA. Highlights of Prescribing Information: ADCETRIS™. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/125388_S056S078lbl.pdf (accessed on 30 June 2022).
- Amaya, M.; Jimeno, A.; Kamdar, M. Polatuzumab vedotin to treat relapsed or refractory diffuse large B-cell lymphoma, in combination with bendamustine plus rituximab. Drugs Today 2020, 56, 287–294. [Google Scholar] [CrossRef]
- FDA. Padcev®. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-regular-approval-enfortumab-vedotin-ejfv-locally-advanced-or-metastatic-urothelial-cancer (accessed on 30 June 2022).
- FDA. Blenrep®. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-granted-accelerated-approval-belantamab-mafodotin-blmf-multiple-myeloma (accessed on 30 June 2022).
- TGA, A. Australian Public Assessment Report for Plitidepsin. Available online: https://www.tga.gov.au/sites/default/files/auspar-plitidepsin-190513.pdf (accessed on 30 June 2022).
- Bäckryd, E. Do the potential benefits outweigh the risks? An update on the use of ziconotide in clinical practice. Eur. J. Pain 2018, 22, 1193–1202. [Google Scholar] [CrossRef] [PubMed]
- Jorpes, E.; Thaning, T. Neutralisation of action of heparin by protamine. Lancet 1939, 234, 975–976. [Google Scholar] [CrossRef]
- Sokolowska, E.; Kalaska, B.; Miklosz, J.; Mogielnicki, A. The toxicology of heparin reversal with protamine: Past, present and future. Expert Opin. Drug Metab. Toxicol. 2016, 12, 897–909. [Google Scholar] [CrossRef]
- Biosyn. IMMUCOTHEL™. Available online: https://biosyncorp.com/klh/ (accessed on 30 June 2022).
- Biosyn. VACMUNE™. Available online: https://biosyncorp.com/vacmuner/ (accessed on 30 June 2022).
- Biosyn. Biosyn Regulatory Documentation. Available online: https://biosyncorp.com/klh/biosyn-regulatory-documentation/ (accessed on 30 June 2022).
- USFDA. Highlights of Prescribing Information: OMTRYG™. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/204977s000lbl.pdf (accessed on 30 June 2022).
- USFDA. OMACOR™. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2004/21–654_Omacor.cfm (accessed on 30 June 2022).
- USFDA. Highlights of Prescribing Information: LOVAZA™. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/021654s043lbl.pdf (accessed on 30 June 2022).
- USFDA. Highlights of Prescribing Information: VASCEPA™. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/202057s019lbl.pdf (accessed on 30 June 2022).
- USFDA. Highlights of Prescribing Information: EPANOVA™. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/205060s000lbl.pdf (accessed on 30 June 2022).
- USFDA. Highlights of Prescribing Information: OMEGAVEN™. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/0210589s000lbledt.pdf (accessed on 30 June 2022).
- Scott, R.B. Cancer chemotherapy—The first twenty-five years. Br. Med. J. 1970, 4, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Chu, M.-Y. Incorporation of arabinosyl cytosine into 2–7S ribonucleic acid and cell death. Biochem. Pharmacol. 1971, 20, 2057–2063. [Google Scholar] [CrossRef]
- Creasey, W.A.; Papac, R.J.; Markiw, M.E.; Calabresi, P.; Welch, A.D. Biochemical and pharmacological studies with 1-β-d-arabinofuranosylcytosine in man. Biochem. Pharmacol. 1966, 15, 1417–1428. [Google Scholar] [CrossRef]
- Lee, W.W.; Benitez, A.; Goodman, L.; Baker, B. Potential anticancer agents. 1 xl. synthesis of the β-anomer of 9-(d-arabinofuranosyl)-adenine. J. Am. Chem. Soc. 1960, 82, 2648–2649. [Google Scholar] [CrossRef]
- Reist, E.J.; Benitez, A.; Goodman, L.; Baker, B.; Lee, W.W. Potential anticancer agents. 1 LXXVI. Synthesis of purine nucleosides of β-D-arabinofuranose. J. Org. Chem. 1962, 27, 3274–3279. [Google Scholar] [CrossRef]
- Reist, E.J.; Benitez, A.; Lee, W.W.; Baker, B.; Goodman, L. Potential anticancer agents. 1 LXXVII. Synthesis of nucleosides of purine-6-thiol (6-Mercaptopurine) containing “Fraudulent” sugars. J. Org. Chem. 1962, 27, 3279–3283. [Google Scholar] [CrossRef]
- Reist, E.J.; Goodman, L. Synthesis of 9-β-D-arabinofuranosylguanine. Biochemistry 1964, 3, 15–18. [Google Scholar] [CrossRef]
- Walwick, E.; Roberts, W.; Dekker, C. Cyclisation during the phosphorylation of uridine and cytidine by polyphosphoric acid-A new route to the O-2, 2’-cyclonucleosides. Proc. Chem. Soc. Lond. 1959, 3, 84. [Google Scholar]
- Brown, D.; Todd, A.; Varadarajan, S. 462. Nucleotides. Part XXXVII. The structure of uridylic acids a and b, and a synthesis of spongouridine (3-β-D-arabofuranosyluracil). J. Chem. Soc. 1956, 2388–2393. [Google Scholar] [CrossRef]
- Cimino, G.; de Rosa, S.; de Stefano, S. Antiviral agents from a gorgonian, eunicella cavolini. Experientia 1984, 40, 339–340. [Google Scholar] [CrossRef]
- Drugs. Cytarabine. Available online: https://www.drugs.com/monograph/cytarabine.html (accessed on 30 June 2022).
- Heuser, M.; Ofran, Y.; Boissel, N.; Mauri, S.B.; Craddock, C.; Janssen, J.; Wierzbowska, A.; Buske, C. Acute myeloid leukaemia in adult patients: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2020, 31, 697–712. [Google Scholar] [CrossRef] [PubMed]
- Chu, M.Y.; Fischer, G. A proposed mechanism of action of 1-β-d-arabinofuranosyl-cytosine as an inhibitor of the growth of leukemic cells. Biochem. Pharmacol. 1962, 11, 423–430. [Google Scholar] [CrossRef]
- Furth, J.; Cohen, S.S. Inhibition of mammalian DNA polymerase by the 5’-triphosphate of 1-β-D-arabinofuranosylcytosine and the 5’-triphosphate of 9-β-D-arabinofuranosyladenine. Cancer Res. 1968, 28, 2061–2067. [Google Scholar]
- Graham, F.; Whitmore, G. The effect of 1-β-D-arabinofuranosylcytosine on growth, viability, and DNA synthesis of mouse L-cells. Cancer Res. 1970, 30, 2627–2635. [Google Scholar]
- Plunkett, W.; Gandhi, V. Evolution of the arabinosides and the pharmacology of fludarabine. Drugs 1994, 47, 30–38. [Google Scholar] [CrossRef]
- Benedict, W.F.; Harris, N.; Karon, M. Kinetics of 1-β-D-arabinofuranosylcytosine-induced chromosome breaks. Cancer Res. 1970, 30, 2477–2483. [Google Scholar]
- Kihlman, B.; Nichols, W.W.; Levan, A. The effect of deoxyadenosine and cytosine arabinoside on the chromosomes of human leukocytes in vitro. Hereditas 1963, 50, 139–143. [Google Scholar] [CrossRef]
- Camiener, G.W.; Smith, C.G. Studies of the enzymatic deamination of cytosine arabinoside—I: Enzyme distribution and species specificity. Biochem. Pharmacol. 1965, 14, 1405–1416. [Google Scholar] [CrossRef]
- Mulligan, L.; Mellett, L. Comparative Metabolism of Cytosine Arabinoside and Inhibition of Deamination by Tetrahydrouridine. Pharmacologist 1968, 10, 167. [Google Scholar]
- Zimm, S.; Collins, J.M.; Miser, J.; Chatterji, D.; Poplack, D.G. Cytosine arabinoside cerebrospinal fluid kinetics. Clin. Pharmacol. Ther. 1984, 35, 826–830. [Google Scholar] [CrossRef]
- Lancet, J.E.; Uy, G.L.; Cortes, J.E.; Newell, L.F.; Lin, T.L.; Ritchie, E.K.; Stuart, R.K.; Strickland, S.A.; Hogge, D.; Solomon, S.R. CPX-351 (cytarabine and daunorubicin) liposome for injection versus conventional cytarabine plus daunorubicin in older patients with newly diagnosed secondary acute myeloid leukemia. J. Clin. Oncol. 2018, 36, 2684–2692. [Google Scholar] [CrossRef]
- Lim, W.-S.; Tardi, P.G.; Dos Santos, N.; Xie, X.; Fan, M.; Liboiron, B.D.; Huang, X.; Harasym, T.O.; Bermudes, D.; Mayer, L.D. Leukemia-selective uptake and cytotoxicity of CPX-351, a synergistic fixed-ratio cytarabine: Daunorubicin formulation, in bone marrow xenografts. Leuk. Res. 2010, 34, 1214–1223. [Google Scholar] [CrossRef]
- Rein, L.A.; Rizzieri, D.A. Clinical potential of elacytarabine in patients with acute myeloid leukemia. Ther. Adv. Hematol. 2014, 5, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Baker, W.J.; Royer, G.L., Jr.; Weiss, R.B. Cytarabine and neurologic toxicity. J. Clin. Oncol. 1991, 9, 679–693. [Google Scholar] [CrossRef] [PubMed]
- Tilly, H.; Castaigne, S.; Bordessoule, D.; Casassus, P.; Le Prise, P.; Tertian, G.; Desablens, B.; Henry-Amar, M.; Degos, L. Low-dose cytarabine versus intensive chemotherapy in the treatment of acute nonlymphocytic leukemia in the elderly. J. Clin. Oncol. 1990, 8, 272–279. [Google Scholar] [CrossRef]
- Cai, J.; Damaraju, V.L.; Groulx, N.; Mowles, D.; Peng, Y.; Robins, M.J.; Cass, C.E.; Gros, P. Two distinct molecular mechanisms underlying cytarabine resistance in human leukemic cells. Cancer Res. 2008, 68, 2349–2357. [Google Scholar] [CrossRef]
- Funato, T.; Harigae, H.; Abe, S.; Sasaki, T. Assessment of drug resistance in acute myeloid leukemia. Expert Rev. Mol. Diagn. 2004, 4, 705–713. [Google Scholar] [CrossRef]
- Galmarini, C.M.; Thomas, X.; Calvo, F.; Rousselot, P.; Rabilloud, M.; El Jaffari, A.; Cros, E.; Dumontet, C. In vivo mechanisms of resistance to cytarabine in acute myeloid leukaemia. Br. J. Haematol. 2002, 117, 860–868. [Google Scholar] [CrossRef] [PubMed]
- Kojima, H.; Iida, M.; Miyazaki, H.; Koga, T.; Moriyama, H.; Manome, Y. Enhancement of cytarabine sensitivity in squamous cell carcinoma cell line transfected with deoxycytidine kinase. Arch. Otolaryngol.–Head Neck Surg. 2002, 128, 708–713. [Google Scholar] [CrossRef] [PubMed]
- Levin, M.; Stark, M.; Berman, B.; Assaraf, Y.G. Surmounting cytarabine-resistance in acute myeloblastic leukemia cells and specimens with a synergistic combination of hydroxyurea and azidothymidine. Cell Death Dis. 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Ohta, T.; Hori, H.; Ogawa, M.; Miyahara, M.; Kawasaki, H.; Taniguchi, N.; Komada, Y. Impact of cytidine deaminase activity on intrinsic resistance to cytarabine in carcinoma cells. Oncol. Rep. 2004, 12, 1115–1120. [Google Scholar] [CrossRef]
- Privat de Garilhe, M. Effect de deux nucleosides de l’arabinose sur la multiplication des virus de l’herpes et de la vaccine en culture cellulaire. CR Acad. Sci. 1964, 259, 2725–2728. [Google Scholar]
- Sidwell, R.W.; Arnett, G.; Dixon, G.J. Effect of selected biologically active compounds on in vitro cytomegalovirus (CMV) infections. In Proceedings of the 7th Interscience Conference on Antimicrobial Agents and Chemotherapy, Chicago, IL, USA, 28–29 October 1967. [Google Scholar]
- Miller, F.; Dixon, G.; Ehrlich, J.; Sloan, B.J.; McLean, I. Antiviral activity of 9 beta D arabinofuranosyladenîne. I. Cell culture studies. Antimicrob. Agents Chemother. 1969, 8, 136–147. [Google Scholar]
- De Clercq, E.; Li, G. Approved antiviral drugs over the past 50 years. Clin. Microbiol. Rev. 2016, 29, 695–747. [Google Scholar] [CrossRef]
- Plunkett, W.; Chubb, S.; Alexander, L.; Montgomery, J.A. Comparison of the toxicity and metabolism of 9-β-D-arabinofuranosyl-2-fluoroadenine and 9-β-D-arabinofuranosyladenine in human lymphoblastoid cells. Cancer Res. 1980, 40, 2349–2355. [Google Scholar]
- Plunkett, W.; Benjamin, R.S.; Keating, M.J.; Freireich, E.J. Modulation of 9-β-D-arabinofuranosyladenine 5’-triphosphate and deoxyadenosine triphosphate in leukemic cells by 2′-deoxycoformycin during therapy with 9-β-D-arabinofuranosyladenine. Cancer Res. 1982, 42, 2092–2096. [Google Scholar]
- Plunkett, W.; Feun, L.G.; Benjamin, R.S.; Keating, M.; Freireich, E.J. “Modulation of Vidarabine Metabolism by 2’-Deoxycoformycin for Therapy of Acute Leukemia” Conference on 2′-Deoxycoformycin--Current Status and Future Directions, US Department of Health and Human Services, Public Health Service; National Institutes of Health: Washington, DC, USA, 1984; Volume 2, pp. 23–27.
- Montgomery, J.A.; Hewson, K. Synthesis of potential anticancer agents. X. 2-fluoroadenosine1. J. Am. Chem. Soc. 1957, 79, 4559. [Google Scholar] [CrossRef]
- Keating, M.; Kantarjian, H.; Talpaz, M.; Redman, J.; Koller, C.; Barlogie, B.; Velasquez, W.; Plunkett, W.; Freireich, E.J.; McCredie, K. Fludarabine: A new agent with major activity against chronic lymphocytic leukemia. Blood 1989, 74, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Rai, K.R.; Peterson, B.L.; Appelbaum, F.R.; Kolitz, J.; Elias, L.; Shepherd, L.; Hines, J.; Threatte, G.A.; Larson, R.A.; Cheson, B.D. Fludarabine compared with chlorambucil as primary therapy for chronic lymphocytic leukemia. N. Engl. J. Med. 2000, 343, 1750–1757. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, P.; Hagemeister, F.B.; Romaguera, J.E.; Sarris, A.H.; Pate, O.; Younes, A.; Swan, F.; Keating, M.; Cabanillas, F. Fludarabine, mitoxantrone, and dexamethasone: An effective new regimen for indolent lymphoma. J. Clin. Oncol. 1996, 14, 1262–1268. [Google Scholar] [CrossRef] [PubMed]
- Thompson, P.A.; Tam, C.S.; O’Brien, S.M.; Wierda, W.G.; Stingo, F.; Plunkett, W.; Smith, S.C.; Kantarjian, H.M.; Freireich, E.J.; Keating, M.J. Fludarabine, cyclophosphamide, and rituximab treatment achieves long-term disease-free survival in IGHV-mutated chronic lymphocytic leukemia. Blood 2016, 127, 303–309. [Google Scholar] [CrossRef]
- Carpenter, J., Jr.; Vogel, C.; Wang, G.; Raney, M. Phase II evaluation of fludarabine in patients with metastatic breast cancer: A southeastern cancer study group trial. Cancer Treat. Rep. 1986, 70, 1235–1236. [Google Scholar] [PubMed]
- Taylor, S.; Eyre, H. Randomized phase II trials of acivicin (AT-125, NSC 163501) and fludarabine (2-fluoro-ara-AMP, NSC 312887) (2FLAMP) in recurrent malignant gliomas: A SWOG study. Proc. Annu. Meet Am. Soc. Clin. Oncol. 1987, 6, 71. [Google Scholar]
- Taylor, S.A.; Crowley, J.; Vogel, F.S.; Townsend, J.J.; Eyre, H.J.; Jaeckle, K.A.; Hynes, H.E.; Guy, J.T. Phase II evaluation of fludarabine phosphate in patients with central nervous system tumors. Investig. New Drugs 1991, 9, 195–197. [Google Scholar] [CrossRef] [PubMed]
- Weiss, G.; Metch, B.; Von Hoff, D.; Taylor, S.; Saiers, J. Phase II trial of fludarabine phosphate in patients with head and neck cancer: A southwest oncology group study. Cancer Treat. Rep. 1987, 71, 1313–1314. [Google Scholar]
- Casper, E.S.; Mittelman, A.; Kelson, D.; Young, C.W. Phase I clinical trial of fludarabine phosphate (F-ara-AMP). Cancer Chemother. Pharmacol. 1985, 15, 233–235. [Google Scholar] [CrossRef]
- Hutton, J.J.; Von Hoff, D.D.; Kuhn, J.; Phillips, J.; Hersh, M.; Clark, G. Phase I clinical investigation of 9-β-d-arabinofuranosyl-2-fluoroadenine 5’-monophosphate (NSC 312887), a new purine antimetabolite. Cancer Res. 1984, 44, 4183–4186. [Google Scholar]
- Cheson, B.D.; Bennett, J.M.; Rai, K.R.; Grever, M.R.; Kay, N.E.; Schiffer, C.A.; Oken, M.M.; Keating, M.J.; Boldt, D.H.; Kempin, S.J.; et al. Guidelines for clinical protocols for chronic lymphocytic leukemia: Recommendations of the National Cancer Institute-sponsored working group. Am. J. Hematol. 1988, 29, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Hiddemann, W.; Rottmann, R.; Wörmann, B.; Thiel, A.; Essink, M.; Ottensmeier, C.; Freund, M.; Büchner, T.; van de Loo, J. Treatment of advanced chronic lymphocytic leukemia by fludarabine. Ann. Hematol. 1991, 63, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Hurst, P.G.; Habib, M.P.; Garewal, H.; Bluestein, M.; Paquin, M.; Greenberg, B.R. Pulmonary toxicity associated with fludarabine monophosphate. Investig. New Drugs 1987, 5, 207–210. [Google Scholar] [CrossRef]
- Chun, H.; Leyland-Jones, B.; Caryk, S.; Hoth, D. Central nervous system toxicity of fludarabine phosphate. Cancer Treat. Rep. 1986, 70, 1225–1228. [Google Scholar] [PubMed]
- Warrell, R.P., Jr.; Berman, E. Phase I and II study of fludarabine phosphate in leukemia: Therapeutic efficacy with delayed central nervous system toxicity. J. Clin. Oncol. 1986, 4, 74–79. [Google Scholar] [CrossRef]
- Jensen, K.; L’Aurelle, A.J.; Jacobson, P.A.; Kachler, S.; Kirstein, M.N.; Lamba, J.; Klotz, K.-N. Cytotoxic purine nucleoside analogues bind to A1, A2A, and A3 adenosine receptors. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2012, 385, 519–525. [Google Scholar] [CrossRef]
- Ribeiro, J.; Sebastiao, A.; de Mendonça, A. Adenosine receptors in the nervous system: Pathophysiological implications. Prog. Neurobiol. 2002, 68, 377–392. [Google Scholar] [CrossRef]
- Helman, D.L., Jr.; Byrd, J.C.; Ales, N.C.; Shorr, A.F. Fludarabine-related pulmonary toxicity: A distinct clinical entity in chronic lymphoproliferative syndromes. Chest 2002, 122, 785–790. [Google Scholar] [CrossRef]
- Frederiksen, S. Specificity of adenosine deaminase toward adenosine and 2’-deoxyadenosine analogues. Arch. Biochem. Biophys. 1966, 113, 383–388. [Google Scholar] [CrossRef]
- Avramis, V.I.; Champagne, J.; Sato, J.; Krailo, M.; Ettinger, L.J.; Poplack, D.G.; Finklestein, J.; Reaman, G.; Hammond, G.D.; Holcenberg, J.S. Pharmacology of fludarabine phosphate after a phase I/II trial by a loading bolus and continuous infusion in pediatric patients. Cancer Res. 1990, 50, 7226–7231. [Google Scholar]
- Malspeis, L.; Grever, M.R.; Staubus, A.; Young, D. Pharmacokinetics of 2-F-ara-A (9-beta-D-arabinofuranosyl-2-fluoroadenine) in cancer patients during the phase I clinical investigation of fludarabine phosphate. Semin. Oncol. 1990, 17, 18–32. [Google Scholar] [PubMed]
- Plunkett, W.; Huang, P.; Gandhi, V. Metabolism and action of fludarabine phosphate. Semin. Oncol. 1990, 17, 3–17. [Google Scholar] [PubMed]
- Plunkett, W.; Saunders, P.P. Metabolism and action of purine nucleoside analogs. Pharmacol. Ther. 1991, 49, 239–268. [Google Scholar] [CrossRef]
- Danhauser, L.; Plunkett, W.; Keating, M.; Cabanillas, F. 9-β-D-Arabinofuranosyl-2-fluoroadenine 5’-monophosphate pharmacokinetics in plasma and tumor cells of patients with relapsed leukemia and lymphoma. Cancer Chemother. Pharmacol. 1986, 18, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Chubb, S.; Plunkett, W. Termination of DNA synthesis by 9-beta-D-arabinofuranosyl-2-fluoroadenine. a mechanism for cytotoxicity. J. Biol. Chem. 1990, 265, 16617–16625. [Google Scholar] [CrossRef]
- Spriggs, D.; Robbins, G.; Mitchell, T.; Kufe, D. Incorporation of 9-β-D-arabinofuranosyl-2-fluoroadenine into HL-60 cellular RNA and DNA. Biochem. Pharmacol. 1986, 35, 247–252. [Google Scholar] [CrossRef]
- Krenitsky, T.A.; Koszalka, G.W.; Tuttle, J.V.; Rideout, J.L.; Elion, G.B. An enzymic synthesis of purine D-arabinonucleosides. Carbohydr. Res. 1981, 97, 139–146. [Google Scholar] [CrossRef]
- Lambe, C.U.; Averett, D.R.; Paff, M.T.; Reardon, J.E.; Wilson, J.G.; Krenitsky, T.A. 2-Amino-6-methoxypurine arabinoside: An agent for T-cell malignancies. Cancer Res. 1995, 55, 3352–3356. [Google Scholar]
- Mitchell, B.S.; Mejias, E.; Daddona, P.E.; Kelley, W.N. Purinogenic immunodeficiency diseases: Selective toxicity of deoxyribonucleosides for T cells. Proc. Natl. Acad. Sci. USA 1978, 75, 5011–5014. [Google Scholar] [CrossRef]
- Rodriguez, C.O., Jr.; Stellrecht, C.M.; Gandhi, V. Mechanisms for T-cell selective cytotoxicity of arabinosylguanine. Blood 2003, 102, 1842–1848. [Google Scholar] [CrossRef]
- DeAngelo, D.J.; Yu, D.; Johnson, J.L.; Coutre, S.E.; Stone, R.M.; Stopeck, A.T.; Gockerman, J.P.; Mitchell, B.S.; Appelbaum, F.R.; Larson, R.A. Nelarabine induces complete remissions in adults with relapsed or refractory T-lineage acute lymphoblastic leukemia or lymphoblastic lymphoma: Cancer and leukemia group B study 19801. Blood 2007, 109, 5136–5142. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, V.; Tam, C.; O’Brien, S.; Jewell, R.C.; Rodriguez, C.O., Jr.; Lerner, S.; Plunkett, W.; Keating, M.J. Phase I trial of nelarabine in indolent leukemias. J. Clin. Oncol. 2008, 26, 1098–1105. [Google Scholar] [CrossRef] [PubMed]
- Artyukov, A.A.; Zelepuga, E.A.; Bogdanovich, L.N.; Lupach, N.M.; Novikov, V.L.; Rutckova, T.A.; Kozlovskaya, E.P. Marine polyhydroxynaphthoquinone, echinochrome a: Prevention of atherosclerotic inflammation and probable molecular targets. J. Clin. Med. 2020, 9, 1494. [Google Scholar] [CrossRef]
- Park, J.H.; Lee, N.K.; Lim, H.J.; Mazumder, S.; Kumar Rethineswaran, V.; Kim, Y.J.; Jang, W.B.; Ji, S.T.; Kang, S.; Kim, D.Y.; et al. Therapeutic cell protective role of histochrome under oxidative stress in human cardiac progenitor cells. Mar. Drugs 2019, 17, 368. [Google Scholar] [CrossRef]
- Hirata, Y.; Uemura, D. Halichondrins-antitumor polyether macrolides from a marine sponge. Pure Appl. Chem. 1986, 58, 701–710. [Google Scholar] [CrossRef]
- Bauer, A. Story of eribulin mesylate: Development of the longest drug synthesis. In Synthesis of Heterocycles in Contemporary Medicinal Chemistry; Springer: Berlin/Heidelberg, Germany, 2016; pp. 209–270. [Google Scholar]
- Seshadri, P.; Deb, B.; Kumar, P. Multifarious targets beyond microtubules-role of eribulin in cancer therapy. Front. Biosci. 2021, 13, 157–172. [Google Scholar]
- USFDA. Highlights of Prescribing Information: HALAVEN™. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/201532lbl.pdf (accessed on 30 June 2022).
- Polastro, L.; Aftimos, P.G.; Awada, A. Eribulin mesylate in the management of metastatic breast cancer and other solid cancers: A drug review. Expert Rev. Anticancer. Ther. 2014, 14, 649–665. [Google Scholar] [CrossRef]
- Towle, M.J.; Salvato, K.A.; Budrow, J.; Wels, B.F.; Kuznetsov, G.; Aalfs, K.K.; Welsh, S.; Zheng, W.; Seletsky, B.M.; Palme, M.H.; et al. In vitro and in vivo anticancer activities of synthetic macrocyclic ketone analogues of halichondrin B. Cancer Res. 2001, 61, 1013–1021. [Google Scholar]
- Vahdat, L.T.; Pruitt, B.; Fabian, C.J.; Rivera, R.R.; Smith, D.A.; Tan-Chiu, E.; Wright, J.; Tan, A.R.; DaCosta, N.A.; Chuang, E.; et al. Phase II study of eribulin mesylate, a halichondrin B analog, in patients with metastatic breast cancer previously treated with an anthracycline and a taxane. J. Clin. Oncol. 2009, 27, 2954–2961. [Google Scholar] [CrossRef]
- Shablak, A. Eribulin for advanced breast cancer: A drug evaluation. J. Breast Cancer 2013, 16, 12–15. [Google Scholar] [CrossRef]
- Cortes, J.; O’Shaughnessy, J.; Loesch, D.; Blum, J.L.; Vahdat, L.T.; Petrakova, K.; Chollet, P.; Manikas, A.; Diéras, V.; Delozier, T.; et al. Investigators (EMBRACE), Eribulin monotherapy versus treatment of physician’s choice in patients with metastatic breast cancer (EMBRACE): A phase 3 open-label randomised study. Lancet 2011, 377, 914–923. [Google Scholar] [CrossRef]
- Swami, U.; Shah, U.; Goel, S. Eribulin in cancer treatment. Mar. Drugs 2015, 13, 5016–5058. [Google Scholar] [CrossRef] [PubMed]
- Yuan, P.; Xu, B. Clinical utility of eribulin mesylate in the treatment of breast cancer: A Chinese perspective. Breast Cancer: Targets Ther. 2021, 13, 135–150. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.; Calvo, J.A.; Samson, L.D. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat. Rev. Cancer 2012, 12, 104–120. [Google Scholar] [CrossRef] [PubMed]
- Ganjoo, K.N.; Patel, S.R. Trabectedin: An anticancer drug from the sea. Expert Opin. Pharmacother. 2009, 10, 2735–2743. [Google Scholar] [CrossRef]
- Guan, Y.; Sakai, R.; Rinehart, K.; Wang, A. Molecular and crystal structures of ecteinascidins: Potent antitumor compounds from the caribbean tunicate ecteinascidia turbinata. J. Biomol. Struct. Dyn. 1993, 10, 793–818. [Google Scholar]
- Corey, E.J.; Gin, D.Y.; Kania, R.S. Enantioselective total synthesis of ecteinascidin 743. J. Am. Chem. Soc. 1996, 118, 9202–9203. [Google Scholar] [CrossRef]
- He, W.; Zhang, Z.; Ma, D. A scalable total synthesis of the antitumor agents ET-743 and lurbinectedin. Angew. Chem. Int. Ed. 2019, 58, 3972–3975. [Google Scholar] [CrossRef]
- EMA. Public Summary of Opinion on Orphan Designation: Trabectedin for the Treatment of Ovarian Cancer. Available online: https://www.ema.europa.eu/en/documents/orphan-designation/eu/3/03/171-public-summary-positive-opinion-orphan-designation-trabectedin-treatment-ovarian-cancer_en.pdf (accessed on 30 June 2022).
- De Sanctis, R.; Marrari, A.; Santoro, A. Trabectedin for the treatment of soft tissue sarcomas. Expert Opin. Pharmacother. 2016, 17, 1569–1577. [Google Scholar] [CrossRef]
- Zijoo, R.; von Mehren, M. Efficacy of trabectedin for the treatment of liposarcoma. Expert Opin. Pharmacother. 2016, 17, 1953–1962. [Google Scholar] [CrossRef]
- USFDA. Highlights of Prescribing Information: YONDELIS™. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/207953s000lbl.pdf (accessed on 30 June 2022).
- Gordon, E.M.; Sankhala, K.K.; Chawla, N.; Chawla, S.P. Trabectedin for soft tissue sarcoma: Current status and future perspectives. Adv. Ther. 2016, 33, 1055–1071. [Google Scholar] [CrossRef] [PubMed]
- Grosso, F.; D’Ambrosio, L.; Zucchetti, M.; Ibrahim, T.; Tamberi, S.; Matteo, C.; Rulli, E.; Comandini, D.; Palmerini, E.; Baldi, G.G.; et al. Pharmacokinetics, safety, and activity of trabectedin as first-line treatment in elderly patients who are affected by advanced sarcoma and are unfit to receive standard chemotherapy: A phase 2 study (TR1US study) from the Italian sarcoma group. Cancer 2020, 126, 4726–4734. [Google Scholar] [CrossRef] [PubMed]
- Beumer, J.H.; Rademaker-Lakhai, J.M.; Rosing, H.; Hillebrand, M.J.; Bosch, T.M.; Lopez-Lazaro, L.; Schellens, J.H.; Beijnen, J.H. Metabolism of trabectedin (ET-743, Yondelis) in patients with advanced cancer. Cancer Chemother. Pharmacol. 2007, 59, 825–837. [Google Scholar] [CrossRef] [PubMed]
- Gago, F.; Hurley, L.H. Devising a structural basis for the potent cytotoxic effects of Ecteinascidin 743. In Small Molecule DNA and RNA Binders: From Synthesis to Nulceic Acid; The Royal Society of Chemistry: London, UK, 2002; pp. 643–675. [Google Scholar]
- Jimeno, J.; Maki, R.G.; Casali, P.; Faircloth, G.; Martinez, N.; Nieto, A.; Cañigueral, S.; Rinehart, K. Therapeutic impact of ET-743 (yondelis; trabectidin), a new marine-derived compound, in sarcoma. Curr. Opin. Orthop. 2003, 14, 419–428. [Google Scholar] [CrossRef]
- Pommier, Y.; Kohlhagen, G.; Bailly, C.; Waring, M.; Mazumder, A.; Kohn, K.W. DNA sequence-and structure-selective alkylation of guanine N2 in the DNA minor groove by ecteinascidin 743, a potent antitumor compound from the Caribbean tunicate Ecteinascidia turbinata. Biochemistry 1996, 35, 13303–13309. [Google Scholar] [CrossRef]
- Erba, E.; Bergamaschi, D.; Bassano, L.; Damia, G.; Ronzoni, S.; Faircloth, G.; d’Incalci, M. Ecteinascidin-743 (ET-743), a natural marine compound, with a unique mechanism of action. Eur. J. Cancer 2001, 37, 97–105. [Google Scholar] [CrossRef]
- Takebayashi, Y.; Pourquier, P.; Zimonjic, D.B.; Nakayama, K.; Emmert, S.; Ueda, T.; Urasaki, Y.; Kanzaki, A.; Akiyama, S.-I.; Popescu, N.; et al. Antiproliferative activity of ecteinascidin 743 is dependent upon transcription-coupled nucleotide-excision repair. Nat. Med. 2001, 7, 961–966. [Google Scholar] [CrossRef]
- Soares, D.G.; Escargueil, A.E.; Poindessous, V.; Sarasin, A.; de Gramont, A.; Bonatto, D.; Henriques, J.A.P.; Larsen, A.K. Replication and homologous recombination repair regulate DNA double-strand break formation by the antitumor alkylator ecteinascidin 743. Proc. Natl. Acad. Sci. USA 2007, 104, 13062–13067. [Google Scholar] [CrossRef]
- Liguori, M.; Solinas, G.; Germano, G.; Mantovani, A.; Allavena, P. Tumor-associated macrophages as incessant builders and destroyers of the cancer stroma. Cancers 2011, 3, 3740–3761. [Google Scholar] [CrossRef]
- Germano, G.; Frapolli, R.; Belgiovine, C.; Anselmo, A.; Pesce, S.; Liguori, M.; Erba, E.; Uboldi, S.; Zucchetti, M.; Pasqualini, F. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell. 2013, 23, 249–262. [Google Scholar] [CrossRef]
- D’incalci, M.; Badri, N.; Galmarini, C.; Allavena, P. Trabectedin, a drug acting on both cancer cells and the tumour microenvironment. Br. J. Cancer 2014, 111, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Leal, J.; Martínez-Díez, M.; García-Hernández, V.; Moneo, V.; Domingo, A.; Bueren-Calabuig, J.; Negri, A.; Gago, F.; Guillén-Navarro, M.; Avilés, P. PM01183, a new DNA minor groove covalent binder with potent in vitro and in vivo anti-tumour activity. Br. J. Pharmacol. 2010, 161, 1099–1110. [Google Scholar] [CrossRef] [PubMed]
- Baena, J.; Modrego, A.; Zeaiter, A.; Kahatt, C.; Alfaro, V.; Jimenez-Aguilar, E.; Mazarico, J.M.; Paz-Ares, L. Lurbinectedin in the treatment of relapsed small cell lung cancer. Future Oncol. 2021, 17, 2279–2289. [Google Scholar] [CrossRef] [PubMed]
- USFDA. Highlights of Prescribing Information: ZEPZELCA™. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/213702s000lbl.pdf (accessed on 30 June 2022).
- Trigo, J.; Subbiah, V.; Besse, B.; Moreno, V.; López, R.; Sala, M.A.; Peters, S.; Ponce, S.; Fernández, C.; Alfaro, V.; et al. Lurbinectedin as second-line treatment for patients with small-cell lung cancer: A single-arm, open-label, phase 2 basket trial. Lancet Oncol. 2020, 21, 645–654. [Google Scholar] [CrossRef]
- Fernandez-Teruel, C.; Gonzalez, I.; Trocóniz, I.F.; Lubomirov, R.; Soto, A.; Fudio, S. Population-pharmacokinetic and covariate analysis of lurbinectedin (PM01183), a new RNA polymerase II Inhibitor, in pooled phase I/II trials in patients with cancer. Clin. Pharmacokinet. 2019, 58, 363–374. [Google Scholar] [CrossRef]
- Elez, M.E.; Tabernero, J.; Geary, D.; Macarulla, T.; Kang, S.P.; Kahatt, C.; Pita, A.S.-M.; Teruel, C.F.; Siguero, M.; Cullell-Young, M. First-in-human phase I study of Lurbinectedin (PM01183) in patients with advanced solid tumors. Clin. Cancer Res. 2014, 20, 2205–2214. [Google Scholar] [CrossRef]
- Moneo, V.; Martínez, P.; de Castro, B.; Cascajares, S.; Avila, S.; Garcia-Fernandez, L.F.; Galmarini, C.M. Abstract A174: Comparison of the antitumor activity of trabectedin, lurbinectedin, zalypsis and PM00128 in a panel of human cells deficient in transcription/NER repair factors. Am. Assoc. Cancer Res. 2017, 5, 628–638. [Google Scholar] [CrossRef]
- Nuñez, G.S.; Robles CM, G.; Giraudon, C.; Martínez-Leal, J.F.; Compe, E.; Coin, F.; Aviles, P.; Galmarini, C.M.; Egly, J.-M. Lurbinectedin specifically triggers the degradation of phosphorylated RNA polymerase II and the formation of DNA breaks in cancer cells. Mol. Cancer Ther. 2016, 15, 2399–2412. [Google Scholar] [CrossRef]
- Pernice, T.; Bishop, A.G.; Guillen, M.J.; Cuevas, C.; Aviles, P. Development of a liquid chromatography/tandem mass spectrometry assay for the quantification of PM01183 (lurbinectedin), a novel antineoplastic agent, in mouse, rat, dog, Cynomolgus monkey and mini-pig plasma. J. Pharm. Biomed. Anal. 2016, 123, 37–41. [Google Scholar] [CrossRef]
- Romano, M.; Frapolli, R.; Zangarini, M.; Bello, E.; Porcu, L.; Galmarini, C.M.; García-Fernández, L.F.; Cuevas, C.; Allavena, P.; Erba, E. Comparison of in vitro and in vivo biological effects of trabectedin, lurbinectedin (PM01183) and zalypsis®(PM00104). Int. J. Cancer 2013, 133, 2024–2033. [Google Scholar] [CrossRef]
- Belgiovine, C.; Bello, E.; Liguori, M.; Craparotta, I.; Mannarino, L.; Paracchini, L.; Beltrame, L.; Marchini, S.; Galmarini, C.M.; Mantovani, A.; et al. Lurbinectedin reduces tumour-associated macrophages and the inflammatory tumour microenvironment in preclinical models. Br. J. Cancer 2017, 117, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, H.; Sen, T.; Rudin, C.M. Targeted therapies and biomarkers in small cell lung cancer. Front. Oncol. 2020, 10, 741. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.M.; Wolchok, J.D.; Old, L.J. Antibody therapy of cancer. Nat. Rev. Cancer 2012, 12, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, I.; Asgher, M.; Sher, F.; Hussain, S.M.; Nazish, N.; Joshi, N.; Sharma, A.; Parra-Saldívar, R.; Bilal, M.; Iqbal, H.M.N. Exploring marine as a rich source of bioactive peptides: Challenges and opportunities from marine pharmacology. Mar. Drugs 2022, 20, 208. [Google Scholar] [CrossRef]
- Sun, X.; Ponte, J.F.; Yoder, N.C.; Laleau, R.; Coccia, J.; Lanieri, L.; Qiu, Q.; Wu, R.; Hong, E.; Bogalhas, M.; et al. Effects of drug–antibody ratio on pharmacokinetics, biodistribution, efficacy, and tolerability of antibody–maytansinoid conjugates. Bioconjugate Chem. 2017, 28, 1371–1381. [Google Scholar] [CrossRef]
- Van de Donk, N.W.; Dhimolea, E. Brentuximab Vedotin; MAbs; Taylor & Francis: Abingdon, UK, 2012; pp. 458–465. [Google Scholar]
- Chang, H.P.; Cheung, Y.; Shah, D.K. Whole-body pharmacokinetics and physiologically based pharmacokinetic model for monomethyl auristatin E (MMAE). J. Clin. Med. 2021, 10, 1332. [Google Scholar] [CrossRef]
- Horie, R.; Watanabe, T. Seminars in Immunology. In CD30: Expression and Function in Health and Disease; Elsevier: Amsterdam, The Netherlands, 1998; pp. 457–470. [Google Scholar]
- Van der Weyden, C.; Pileri, S.; Feldman, A.; Whisstock, J.; Prince, H. Understanding CD30 biology and therapeutic targeting: A historical perspective providing insight into future directions. Blood Cancer J. 2017, 7, e603. [Google Scholar] [CrossRef]
- Moskowitz, C.H.; Nademanee, A.; Masszi, T.; Agura, E.; Holowiecki, J.; Abidi, M.H.; Chen, A.I.; Stiff, P.; Gianni, A.M.; Carella, A. Brentuximab vedotin as consolidation therapy after autologous stem-cell transplantation in patients with Hodgkin’s lymphoma at risk of relapse or progression (AETHERA): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2015, 385, 1853–1862. [Google Scholar] [CrossRef]
- Hansen, H.P.; Trad, A.; Dams, M.; Zigrino, P.; Moss, M.; Tator, M.; Schön, G.; Grenzi, P.C.; Bachurski, D.; Aquino, B. CD30 on extracellular vesicles from malignant Hodgkin cells supports damaging of CD30 ligand-expressing bystander cells with brentuximab-vedotin, in vitro. Oncotarget 2016, 7, 30523. [Google Scholar] [CrossRef]
- Malecek, M.-K.; Watkins, M.P.; Bartlett, N.L. Polatuzumab vedotin for the treatment of adults with relapsed or refractory diffuse large B-cell lymphoma. Expert Opin. Biol. Ther. 2021, 21, 831–839. [Google Scholar] [CrossRef]
- Pfeifer, M.; Zheng, B.; Erdmann, T.; Koeppen, H.; McCord, R.; Grau, M.; Staiger, A.; Chai, A.; Sandmann, T.; Madle, H. Anti-CD22 and anti-CD79B antibody drug conjugates are active in different molecular diffuse large B-cell lymphoma subtypes. Leukemia 2015, 29, 1578–1586. [Google Scholar] [CrossRef] [PubMed]
- International Non-Hodgkin’s Lymphoma Prognostic Factors Project, A predictive model for aggressive non-Hodgkin’s lymphoma. N. Engl. J. Med. 1993, 329, 987–994. [CrossRef] [PubMed]
- USFDA. Clinical Review: Polatuzumab Vedotin™. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/761121Orig1s000MedR.pdf (accessed on 30 June 2022).
- USFDA. Highlights of Prescribing Information: POLIVY™. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/761121s000lbl.pdf (accessed on 30 June 2022).
- FDA Approved Drug Products: Padcev (Enfortumab Vedotin-Ejfv) for IV Injection. Available online: http://www.accessdata.fda.gov/drugsatfda_docs/label/2019/761137s000lbl.pdf (accessed on 30 June 2022).
- Hanna, K.S. Clinical overview of enfortumab vedotin in the management of locally advanced or metastatic urothelial carcinoma. Drugs 2020, 80, 1–7. [Google Scholar] [CrossRef] [PubMed]
- McGregor, B.A.; Sonpavde, G. Enfortumab Vedotin, a fully human monoclonal antibody against Nectin 4 conjugated to monomethyl auristatin E for metastatic urothelial carcinoma. Expert Opin. Investig. Drugs 2019, 28, 821–826. [Google Scholar] [CrossRef] [PubMed]
- FDA Approved Drug Products: Blenrep Belantaman Mafodotin-Blmf Intravenous Injection. Available online: http://www.accessdata.fda.gov/drugsatfda_docs/label/2022/761158s006lbl.pdf (accessed on 30 June 2022).
- McMillan, A.; Warcel, D.; Popat, R. Antibody-drug conjugates for multiple myeloma. Expert Opin. Biol. Ther. 2021, 21, 889–901. [Google Scholar] [CrossRef]
- Trudel, S.; Lendvai, N.; Popat, R.; Voorhees, P.M.; Reeves, B.; Libby, E.N.; Richardson, P.G.; Hoos, A.; Gupta, I.; Bragulat, V.; et al. Antibody-drug conjugate, GSK2857916, in relapsed/refractory multiple myeloma: An update on safety and efficacy from dose expansion phase I study. Blood Cancer J. 2019, 9, 37. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Marine natural products and related compounds in clinical and advanced preclinical trials. J. Nat. Prod. 2004, 67, 1216–1238. [Google Scholar] [CrossRef]
- Rinehart, K.L., Jr.; Gloer, J.B.; Cook, J.C., Jr.; Mizsak, S.A.; Scahill, T.A. Structures of the didemnins, antiviral and cytotoxic depsipeptides from a caribbean tunicate. J. Am. Chem. Soc. 1981, 103, 1857–1859. [Google Scholar] [CrossRef]
- Rinehart, K.L.; Kishore, V.; Bible, K.C.; Sakai, R.; Sullins, D.W.; Li, K.-M. Didemnins and tunichlorin: Novel natural products from the marine tunicate trididemnum solidum. J. Nat. Pproducts 1988, 51, 1–21. [Google Scholar] [CrossRef]
- BioWorld. EU Court Sides with Pharmamar in Aplidin Approval Dispute. Available online: https://www.bioworld.com/articles/499498-eu-court-sides-with-pharmamar-in-aplidin-approval-dispute (accessed on 30 June 2022).
- White, K.M.; Rosales, R.; Yildiz, S.; Kehrer, T.; Miorin, L.; Moreno, E.; Jangra, S.; Uccellini, M.B.; Rathnasinghe, R.; Coughlan, L.; et al. Plitidepsin has potent preclinical efficacy against SARS-CoV-2 by targeting the host protein eEF1A. Science 2021, 371, 926–931. [Google Scholar] [CrossRef]
- Gomez, J.; Extremera, S.; Nieto, A. Overall survival (OS) results of randomized phase III study (ADMYRE trial) of plitidepsin and dexamethasone (DXM) vs. DXM alone in patients with relapsed/refractory multiple myeloma (RRMM): Evaluation of the crossover impact. Am. Soc. Clin. Oncol. J. 2018, 36, 8018. [Google Scholar] [CrossRef]
- Spicka, I.; Ocio, E.M.; Oakervee, H.E.; Greil, R.; Banh, R.H.; Huang, S.-Y.; D’Rozario, J.M.; Dimopoulos, M.A.; Martínez, S.; Extremera, S.; et al. Randomized phase III study (ADMYRE) of plitidepsin in combination with dexamethasone vs. dexamethasone alone in patients with relapsed/refractory multiple myeloma. Ann. Hematol. 2019, 98, 2139–2150. [Google Scholar] [CrossRef] [PubMed]
- Ribrag, V.; Caballero, D.; Fermé, C.; Zucca, E.; Arranz, R.; Briones, J.; Gisselbrecht, C.; Salles, G.; Gianni, A.M.; Gomez, H. Multicenter phase II study of plitidepsin in patients with relapsed/refractory non-Hodgkin’s lymphoma. Haematologica 2013, 98, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Alonso, M.J.; González-Santiago, L.; Martínez, T.; Losada, A.; Galmarini, C.M.; Muñoz, A. The mechanism of action of plitidepsin. Curr. Opin. Investig. Drugs 2009, 10, 536–542. [Google Scholar] [PubMed]
- Alonso-Álvarez, S.; Pardal, E.; Sánchez-Nieto, D.; Navarro, M.; Caballero, M.D.; Mateos, M.V.; Martín, A. Plitidepsin: Design, development, and potential place in therapy. Drug Des. Dev. Ther. 2017, 11, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Terlau, H.; Olivera, B.M. Conus venoms: A rich source of novel ion channel-targeted peptides. Physiol. Rev. 2004, 84, 41–68. [Google Scholar] [CrossRef]
- Jin, A.-H.; Muttenthaler, M.; Dutertre, S.; Himaya, S.; Kaas, Q.; Craik, D.J.; Lewis, R.J.; Alewood, P.F. Conotoxins: Chemistry and biology. Chem. Rev. 2019, 119, 11510–11549. [Google Scholar] [CrossRef]
- Kaas, Q.; Westermann, J.-C.; Craik, D.J. Conopeptide characterization and classifications: An analysis using conoserver. Toxicon 2010, 55, 1491–1509. [Google Scholar] [CrossRef]
- Kumar, P.S.; Kumar, D.S.; Umamaheswari, S. A perspective on toxicology of conus venom peptides. Asian Pac. J. Trop. Med. 2015, 8, 337–351. [Google Scholar] [CrossRef]
- Bozorgi, H.; Motaghi, E.; Zamani, M.; Ghavimi, R. Neuronal calcium channels blocker, ziconotide (ɷ-conotoxin MVIIA), reverses morphine withdrawal-induced memory impairments via alteration in hippocampal NMDA receptor expression in rats. Toxin Rev. 2018, 39, 1–10. [Google Scholar]
- Wermeling, D.; Drass, M.; Ellis, D.; Mayo, M.; McGuire, D.; O’Connell, D.; Hale, V.; Chao, S. Pharmacokinetics and pharmacodynamics of intrathecal ziconotide in chronic pain patients. J. Clin. Pharmacol. 2003, 43, 624–636. [Google Scholar] [CrossRef] [PubMed]
- Schmidtko, A.; Lötsch, J.; Freynhagen, R.; Geisslinger, G. Ziconotide for treatment of severe chronic pain. Lancet 2010, 375, 1569–1577. [Google Scholar] [CrossRef]
- Smith, H.S.; Deer, T.R. Safety and efficacy of intrathecal ziconotide in the management of severe chronic pain. Ther. Clin. Risk Manag. 2009, 5, 521–534. [Google Scholar] [CrossRef]
- Nair, A.; Poornachand, A.; Kodisharapu, P. Ziconotide: Indications, adverse effects, and limitations in managing refractory chronic pain. Indian J. Palliat. Care 2018, 24, 118–119. [Google Scholar] [PubMed]
- Deer, T.R.; Prager, J.; Levy, R.; Rathmell, J.; Buchser, E.; Burton, A.; Caraway, D.; Cousins, M.; de Andrés, J.; Diwan, S.; et al. Polyanalgesic consensus conference 2012: Recommendations for the management of pain by intrathecal (intraspinal) drug delivery: Report of an interdisciplinary expert panel. Neuromodulation 2012, 15, 436–466. [Google Scholar] [CrossRef]
- Deer, T.; Hagedorn, J.M. How has ziconotide impacted non-cancer pain management? Expert Opin. Pharmacother. 2020, 21, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Matis, G.; de Negri, P.; Dupoiron, D.; Likar, R.; Zuidema, X.; Rasche, D. Intrathecal pain management with ziconotide: Time for consensus? Brain Behav. 2021, 11, e02055. [Google Scholar] [CrossRef] [PubMed]
- Drugs. Protamine Sulfate. Available online: https://www.drugs.com/international/protamine-sulfate.html (accessed on 30 June 2022).
- Lindblad, B. Protamine sulphate: A review of its effects: Hypersensitivity and toxicity. Eur. J. Vasc. Surg. 1989, 3, 195–201. [Google Scholar] [CrossRef]
- Sorgi, F.; Bhattacharya, S.; Huang, L. Protamine sulfate enhances lipid-mediated gene transfer. Gene Ther. 1997, 4, 961–968. [Google Scholar] [CrossRef]
- DeLucia, A., 3rd; Wakefield, T.W.; Kadell, A.; Wrobleski, S.K.; VanDort, M.; Stanley, J.C. Tissue distribution, circulating half-life, and excretion of intravenously administered protamine sulfate. J. Am. Soc. Artif. Intern. Organs 1993, 39, M715–M718. [Google Scholar]
- Blajchman, M. An overview of the mechanism of action of antithrombin and its inherited deficiency states. Blood Coagul. Fibrinolysis 1994, 5, S5–S11. [Google Scholar] [CrossRef] [PubMed]
- Hirsh, J.; Warkentin, T.E.; Dalen, J.E.; Deykin, D.; Poller, L. Heparin: Mechanism of action, pharmacokinetics, dosing considerations, monitoring, efficacy, and safety. Chest 1995, 108, 258S–275S. [Google Scholar] [CrossRef] [PubMed]
- Okajima, Y.; Kanayama, S.; Maeda, Y.; Urano, S.; Kitani, T.; Watada, M.; Nakagawa, M.; Ijichi, H. Studies on the neutralizing mechanism of antithrombin activity of heparin by protamine. Thromb. Res. 1981, 24, 21–29. [Google Scholar] [CrossRef]
- Medpagetoday. FDA Clears Andexanet Alfa for Rapid NOAC Reversal. Available online: https://www.medpagetoday.com/cardiology/prevention/72701 (accessed on 30 June 2022).
- Klhsite. About KLH. Available online: https://www.klhsite.com/about-klh/ (accessed on 30 June 2022).
- DrugBank. Keyhole Limpet Hemocyanin. Available online: https://go.drugbank.com/drugs/DB05299 (accessed on 30 June 2022).
- Weigle, W.O. Immunochemical properties of hemocyanin. Immunochemistry 1964, 1, 295–302. [Google Scholar] [CrossRef]
- Curtis, J.; Hersh, E.M.; Harris, J.; McBride, C.; Freireich, E.J. The human primary immune response to keyhole limpet haemocyanin: Interrelationships of delayed hypersensitivity, antibody response and in vitro blast transformation. Clin. Exp. Immunol. 1970, 6, 473–491. [Google Scholar]
- Curtis, J.E.; Hersh, E.M.; Butler, W.T.; Rossen, R.D. Antigen dose in the human immune response: Dose-response relationships in the human immune response to keyhole limpet hemocyanin. J. Lab. Clin. Med. 1971, 78, 61–69. [Google Scholar]
- Herscowitz, H.; Harold, W.; Stavitsky, A. Immunochemical and immunogenic properties of a purified keyhole limpet haemocyanin. Immunology 1972, 22, 51–61. [Google Scholar]
- Söhngen, S.M.; Stahlmann, A.; Harris, J.R.; Müller, S.A.; Engel, A.; Markl, J. Mass determination, subunit organization and control of oligomerization states of keyhole limpet hemocyanin (KLH). Eur. J. Biochem. 1997, 248, 602–614. [Google Scholar] [CrossRef]
- Harris, J.; Markl, J. Keyhole limpet hemocyanin (KLH): A biomedical review. Micron 1999, 30, 597–623. [Google Scholar] [CrossRef]
- Nseyo, U.O.; Lamm, D.L. Seminars in Surgical Oncology. In Immunotherapy of Bladder Cancer; Wiley Online Library: Hoboken, NJ, USA, 1997; pp. 342–349. [Google Scholar]
- International, B. DigiFab. Available online: https://digifab.health/en-us (accessed on 30 June 2022).
- Swaminathan, A.; Lucas, R.M.; Dear, K.; McMichael, A.J. Keyhole limpet haemocyanin–a model antigen for human immunotoxicological studies. Br. J. Clin. Pharmacol. 2014, 78, 1135–1142. [Google Scholar] [CrossRef]
- Shekelle, R.B.; Missell, L.; Paul, O.; Shryock, A.M.; Stamler, J.; Vollset, S.E.; Heuch, I.; Bjelke, E.; Curb, J.D.; Reed, D.M. Fish consumption and mortality from coronary heart disease. N. Engl. J. Med. 1985, 313, 820–824. [Google Scholar]
- Dolecek, T.; Granditis, G. Dietary polyunsaturated fatty acids and mortality in the multiple risk factor intervention trial (MRFIT). World Rev. Nutr. Diet. 1991, 66, 205–216. [Google Scholar] [PubMed]
- Kromhout, D.; Feskens, E.J.; Bowles, C.H. The protective effect of a small amount of fish on coronary heart disease mortality in an elderly population. Int. J. Epidemiol. 1995, 24, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Sasaki, S.; Amano, K.; Kesteloot, H. Fish consumption and mortality from all causes, ischemic heart disease, and stroke: An ecological study. Prev. Med. 1999, 28, 520–529. [Google Scholar] [CrossRef]
- Kris-Etherton, P.M.; Harris, W.S.; Appel, L.J. Fish consumption, fish oil, omega-3 fatty acids, and cardiovascular disease. Circulation 2002, 106, 2747–2757. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.G.; Contaifer, D.; Madurantakam, P.; Carbone, S.; Price, E.T.; van Tassell, B.; Brophy, D.F.; Wijesinghe, D.S. Dietary bioactive fatty acids as modulators of immune function: Implications on human health. Nutrients 2019, 11, 2974. [Google Scholar] [CrossRef]
- Maki, K.C.; Johns, C.; Harris, W.S.; Puder, M.; Freedman, S.D.; Thorsteinsson, T.; Daak, A.; Rabinowicz, A.L.; Sancilio, F.D. Bioequivalence demonstration for Ω-3 acid ethyl Ester formulations: Rationale for modification of current guidance. Clin. Ther. 2017, 39, 652–658. [Google Scholar] [CrossRef]
- Guthrie, G.; Burrin, D. Impact of parenteral lipid emulsion components on cholestatic liver disease in neonates. Nutrients 2021, 13, 508. [Google Scholar] [CrossRef]
- Saremi, A.; Arora, R. The utility of omega-3 fatty acids in cardiovascular disease. Am. J. Ther. 2009, 16, 421–436. [Google Scholar] [CrossRef]
- Ballantyne, C.M.; Braeckman, R.A.; Soni, P.N. Icosapent ethyl for the treatment of hypertriglyceridemia. Expert Opin. Pharmacother. 2013, 14, 1409–1416. [Google Scholar] [CrossRef]
- Bazarbashi, N.; Miller, M. Icosapent ethyl: Drug profile and evidence of reduced residual cardiovascular risk in patients with statin-managed LDL-C cholesterol. Expert Rev. Cardiovasc. Ther. 2020, 18, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Backes, J.; Anzalone, D.; Hilleman, D.; Catini, J. The clinical relevance of omega-3 fatty acids in the management of hypertriglyceridemia. Lipids Health Dis. 2016, 15, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Touloukian, R.J.; Seashore, J.H. Hepatic secretory obstruction with total parenteral nutrition in the infant. J. Pediatric Surg. 1975, 10, 353–360. [Google Scholar] [CrossRef]
- Rangel, S.J.; Calkins, C.M.; Cowles, R.A.; Barnhart, D.C.; Huang, E.Y.; Abdullah, F.; Arca, M.J.; Teitelbaum, D.H. Parenteral nutrition–associated cholestasis: An American pediatric surgical association outcomes and clinical trials committee systematic review. J. Pediatric Surg. 2012, 47, 225–240. [Google Scholar] [CrossRef]
- Kauffmann-Guerrero, D.; Huber, R.M. Orphan drugs in development for the treatment of small-cell lung cancer: Emerging data on lurbinectedin. Lung Cancer 2020, 11, 27–31. [Google Scholar] [CrossRef]
- Zuo, W.; Kwok, H.F. Development of marine-derived compounds for cancer therapy. Mar. Drugs 2021, 19, 342. [Google Scholar] [CrossRef]
- Dyshlovoy, S.A.; Honecker, F. Marine compounds and cancer: 2017 updates. Mar. Drugs 2018, 16, 41. [Google Scholar] [CrossRef] [PubMed]
- Dyshlovoy, S.A.; Honecker, F. Marine compounds and cancer: Updates 2020. Mar. Drugs 2020, 18, 643. [Google Scholar] [CrossRef]
- Saeed, A.; Su, J.; Ouyang, S. Marine-derived drugs: Recent advances in cancer therapy and immune signaling. Biomed. Pharmacother. 2021, 134, 111091. [Google Scholar] [CrossRef]
- Nastrucci, C.; Cesario, A.; Russo, P. Anticancer drug discovery from the marine environment. Recent Pat. Anti-Cancer Drug Discov. 2012, 7, 218–232. [Google Scholar]
- Glaser, K.B.; Mayer, A.M. A renaissance in marine pharmacology: From preclinical curiosity to clinical reality. Biochem. Pharmacol. 2009, 78, 440–448. [Google Scholar] [CrossRef]
- Jimenez, P.C.; Wilke, D.V.; Branco, P.C.; Bauermeister, A.; Rezende-Teixeira, P.; Gaudêncio, S.P.; Costa-Lotufo, L.V. Enriching cancer pharmacology with drugs of marine origin. Br. J. Pharmacol. 2020, 177, 3–27. [Google Scholar] [CrossRef] [PubMed]
- Galasso, C.; Gentile, A.; Orefice, I.; Ianora, A.; Bruno, A.; Noonan, D.M.; Sansone, C.; Albini, A.; Brunet, C. Microalgal derivatives as potential nutraceutical and food supplements for human health: A focus on cancer prevention and Interception. Nutrients 2019, 11, 1226. [Google Scholar] [CrossRef] [PubMed]
- Shikov, A.N.; Pozharitskaya, O.N.; Krishtopina, A.S.; Makarov, V.G. Naphthoquinone pigments from sea urchins: Chemistry and pharmacology. Phytochem. Rev. 2018, 17, 509–534. [Google Scholar] [CrossRef]
- Wei, J.; Liu, R.; Hu, X.; Liang, T.; Zhou, Z.; Huang, Z. MAPK signaling pathway-targeted marine compounds in cancer therapy. J. Cancer Res. Clin. Oncol. 2021, 147, 3–22. [Google Scholar] [CrossRef]
- Wei, J.; Gou, Z.; Wen, Y.; Luo, Q.; Huang, Z. Marine compounds targeting the PI3K/Akt signaling pathway in cancer therapy. Biomed. Pharmacother. 2020, 129, 110484. [Google Scholar] [CrossRef]
- Cappello, E.; Nieri, P. From life in the sea to the clinic: The marine drugs approved and under clinical trial. Life 2021, 11, 1390. [Google Scholar] [CrossRef]
- Lu, W.Y.; Li, H.J.; Li, Q.Y.; Wu, Y.C. Application of marine natural products in drug research. Bioorganic Med. Chem. 2021, 35, 116058. [Google Scholar] [CrossRef]
- Jiménez, C. Marine natural products in medicinal chemistry. ACS Med. Chem. Lett. 2018, 9, 959–961. [Google Scholar] [CrossRef]
- Ghareeb, M.A.; Tammam, M.A.; El-Demerdash, A.; Atanasov, A.G. Insights about clinically approved and preclinically investigated marine natural products. Curr. Res. Biotechnol. 2020, 2, 88–102. [Google Scholar] [CrossRef]
- Prokopov, I.A.; Kovaleva, E.L.; Minaeva, E.D.; Pryakhina, E.A.; Savin, E.V.; Gamayunova, A.V.; Pozharitskaya, O.N.; Makarov, V.G.; Shikov, A.N. Animal-derived medicinal products in Russia: Current nomenclature and specific aspects of quality control. J. Ethnopharmacol. 2019, 240, 111933. [Google Scholar] [CrossRef] [PubMed]
- Altmann, K.-H. Drugs from the oceans: Marine natural products as leads for drug discovery. Chimia 2017, 71, 646–652. [Google Scholar] [CrossRef] [PubMed]
- Trame, M.N.; Biliouris, K.; Lesko, L.J.; Mettetal, J.T. Systems pharmacology to predict drug safety in drug development. Eur. J. Pharm. Sci. 2016, 94, 93–95. [Google Scholar] [CrossRef] [PubMed]
- Saginc, G.; Voellmy, F.; Linding, R. Cancer systems biology: Harnessing off-target effects. Nat. Chem. Biol. 2017, 13, 1204–1205. [Google Scholar] [CrossRef]
- Garon, S.L.; Pavlos, R.K.; White, K.D.; Brown, N.J.; Stone, C.A., Jr.; Phillips, E.J. Pharmacogenomics of off-target adverse drug reactions. Br. J. Clin. Pharmacol. 2017, 83, 1896–1911. [Google Scholar] [CrossRef]
- Huang, W.; Whittaker, K.; Zhang, H.; Wu, J.; Zhu, S.W.; Huang, R.P. Integration of antibody array technology into drug discovery and development. Assay Drug Dev. Technol. 2018, 16, 74–95. [Google Scholar] [CrossRef]
- Han, K.; Wang, M.; Zhang, L.; Wang, C. Application of molecular methods in the identification of ingredients in Chinese herbal medicines. Molecules 2018, 23, 2728. [Google Scholar] [CrossRef]
- Chatterjee-Kishore, M.; Miller, C.P. Exploring the sounds of silence: RNAi-mediated gene silencing for target identification and validation. Drug Discov. Today 2005, 10, 1559–1565. [Google Scholar] [CrossRef]
- Zhou, Y.; Takacs, G.P.; Lamba, J.K.; Vulpe, C.; Cogle, C.R. Functional dependency analysis identifies potential druggable targets in acute myeloid leukemia. Cancers 2020, 12, 3710. [Google Scholar] [CrossRef]
- Kim, H.S.; Lee, K.; Bae, S.; Park, J.; Lee, C.K.; Kim, M.; Kim, E.; Kim, M.; Kim, S.; Kim, C.; et al. CRISPR/Cas9-mediated gene knockout screens and target identification via whole-genome sequencing uncover host genes required for picornavirus infection. J. Biol. Chem. 2017, 292, 10664–10671. [Google Scholar] [CrossRef]
- Huang, H.; Zhang, G.; Zhou, Y.; Lin, C.; Chen, S.; Lin, Y.; Mai, S.; Huang, Z. Reverse screening methods to search for the protein targets of chemopreventive compounds. Front. Chem. 2018, 6, 138. [Google Scholar] [CrossRef] [PubMed]
- Pinzi, L.; Rastelli, G. Molecular docking: Shifting paradigms in drug discovery. Int. J. Mol. Sci. 2019, 20, 4331. [Google Scholar] [CrossRef] [PubMed]
- Kimber, T.B.; Chen, Y.; Volkamer, A. Deep learning in virtual screening: Recent applications and developments. Int. J. Mol. Sci. 2021, 22, 4435. [Google Scholar] [CrossRef] [PubMed]
- Pasala, C.; Katari, S.K.; Nalamolu, R.M.; Aparna, R.B.; Amineni, U. Integration of core hopping, quantum-mechanics, molecular mechanics coupled binding-energy estimations and dynamic simulations for fragment-based novel therapeutic scaffolds against Helicobacter pylori strains. Comput. Biol. Chem. 2019, 83, 107126. [Google Scholar] [CrossRef] [PubMed]
- Nakano, H.; Miyao, T.; Funatsu, K. Exploring topological pharmacophore graphs for scaffold hopping. J. Chem. Inf. Modeling 2020, 60, 2073–2081. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Jarada, T.N.; Rokne, J.G.; Alhajj, R. A review of computational drug repositioning: Strategies, approaches, opportunities, challenges, and directions. J. Cheminformatics 2020, 12, 46. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name (DrugBank ID) | Brand Name (Company) | Source Organism | Type (MW) | Mechanism of Action | Treatment Indications, Approving Agency (Year) |
|---|---|---|---|---|---|
| Spongonucleosides | |||||
| Cytarabine, ara-C (DB00987) | CYTOSAR-U® (Pfizer, New York City, NY, USA) and DEPOCYT® (Pacira Pharma, San Diego, CA, USA; Bedford Lab, Seattle, WA, USA, Enzon Pharmaceuticals, Piscataway, NJ, USA) | Cryptotethia crypta (sponge) | Small molecule (243.22 Da) | Synthetic spongonucleoside analog, arrests cells in S phase by inhibiting DNA synthesis | Remission induction in acute nonlymphocytic leukemia, FDA (1969) [5] Lymphomatous meningitis, FDA (1999) [6] |
| Vidarabine, ara-A (DB00194) | VIRA-A® (King Pharmaceuticals, Bristol, FL, USA) | Cryptotethia crypta (sponge) | Small molecule (267.24 Da) | Synthetic spongonucleoside analog, stops the replication of herpes viral DNA | Acute keratoconjunctivitis and recurrent superficial keratitis caused by HSV-1 and HSV-2, FDA (1976) [7] |
| Fludarabine, F-ara-A (DB01073) | FLUDARA® (Sandoz, Basel, Switzerland) and OFORTA® (Sanofi-Aventis, Paris, France) | Cryptotethia crypta (sponge) | Small molecule (285.23 Da) | Synthetic spongonucleoside analog, inhibits DNA synthesis by inhibiting DNA polymerase alpha, ribonucleotide reductase, and DNA primase | B-cell CLL, FDA (1991) [8] |
| Nelarabine (DB01280) | ARRANON® (GSK, Brentford, UK) ATRIANCE® (Novartis, Basel, Switzerland) | Cryptotethia crypta (sponge) | Small molecule (297.27 Da) | Synthetic spongonucleoside analog, is metabolized into ara-GTP, competes with dGTP, and is incorporated into the DNA, inhibiting DNA elongation | T-cell acute lymphoblastic leukemia and T cell lymphoblastic lymphoma, FDA (2005) [9] |
| Histochrome | Pacific-Ocean Institute of Bioorganic Chemistry, Vladivostok, Russia | Scaphechinus mirabilis (sea urchin) | Small molecule (220 Da) | The drug prevents DNA damage and regulates apoptosis under oxidative stress condition | Used to treat degenerative diseases of the retina and cornea, macular degeneration, etc., myocardial ischemia/reperfusion injury etc., Russia (1999) [10] |
| Microtubule Inhibitors | |||||
| Eribulin mesylate (DB08871) | HALAVEN® (Eisai, Bunkyo, Japan) | Halichondria okadai (sponge) | Small molecule, orphan Ϯ (826.00 Da) | Polyether macrolide, arrests cells in G2/M phase by inhibiting microtubule growth after direct interaction | Metastatic breast cancer, FDA (2010) [11], unresectable or metastatic liposarcoma, FDA (2016) [12] |
| DNA alkylating agent | |||||
| Trabectedin, ET-743 (DB05109) | YONDELIS® (PharmaMar SA, Madrid, Spain) | Ecteinascidia turbinata (tunicate) | Small molecule, orphan Ϯ (761.80 Da) | DNA alkylating agent, forms adducts with DNA guanine residue in the minor groove, bends the DNA helix toward the major groove, and disrupts the association of DNA binding proteins | Soft-tissue sarcoma and relapsed platinum-sensitive ovarian cancer, EMA (2007), [13] unresectable or metastatic liposarcoma or leiomyosarcoma, FDA (2015) [14] |
| Lurbinectedin (DB12674) | ZEPZELCA™ (PharmaMar SA, Madrid, Spain) | Ecteinascidia turbinata (tunicate) | Small molecule, orphan Ϯ (784.90 Da) | DNA alkylating agent, forms adducts with DNA guanine residue in the minor groove, bends the DNA helix toward the major groove, and disrupts the association of DNA binding proteins | Metastatic SCLC, FDA (2020) [15] |
| Antibody-Drug Conjugates | |||||
| Brentuximab vedotin (DB08870) | ADCERTIS® (Seattle Genetics, Bothell, Wahington, WA, USA) | Dolabella auricularia (mollusk) | Biotech (~153 kDa) | The antibody component (IgG1) targets CD30 and MMAE disrupts the microtubules after internalization | Hodgkin lymphoma and systemic anaplastic large-cell lymphoma, FDA (2011) [16] |
| Polatuzumab vedotin (DB12240) | POLIVY™ (Genentech, San Francisco, CA, USA) | Dolabella auricularia (mollusk) | Biotech (~150 kDa) | The antibody component targets CD79b and MMAE disrupts the microtubules after internalization | Relapsed or refractory diffuse large B-cell lymphoma, FDA (2019) [17] |
| Enfortumab vedotin (DB13007) | Padcev® (Astellas Pharma US Inc., Northbrook, IL, USA) | Dolabella auricularia (mollusk) | Biotech (~153 kDa) | The antibody component (IgG1) targets nectin-4 and MMAE disrupts the microtubules after internalization | Treatment of patients with advanced, treatment-resistant urothelial cancer, FDA (2019) [18] |
| Belantamab mafodotin (DB15719) | Blenrep® (GlaxoSmithKline, Brentford, UK) | Dolabella auricularia (mollusk) | Biotech (~153 kDa) | The antibody component (IgG1) targets BCMA (B-cell maturation antigen) and MMAF disrupts the microtubules after internalization | Treatment of adult patients with relapsed or refractory multiple myeloma who have received at least 4 prior therapies, including and anti-CD38 monoclonal antibody, a proteasome inhibitor, and an immunomodulatory agent, FDA (2020) [19] |
| Peptides or Proteins Used as Drugs or in Drug Preparations | |||||
| Plitidepsin (DB04977) | APLIDIN® (PharmaMar SA, Madrid, Spain) | Aplidium albicans (sea squirt) | Small molecule, orphan Ϯ (1110.30 Da) | Binds to the gene product of eEF1A2, thus inhibiting cancer cell viability | Tumors in pancreatic, stomach, bladder, and prostate cancers, TGA (2018) [20] |
| Ziconotide (DB06283) | PRIALT® (Azur Pharma, Dublin, Ireland) | Conus magus (marine snail) | Small molecule (2639.20 Da) | Blocks excitatory neurotransmitter release by inhibiting the N-type calcium channels of primary nociceptive afferent nerves and relieves pain | Severe chronic pain, FDA (2004) and EMA (2005) [21] |
| Protamine sulfate (DB09141) | PROSULF® (CP Pharm, Hong Kong, China; Wockhardt Mumbai, India; etc.) PROTAM® (Eipico, Ramadan City, Egypt) | Salmon sperm heads | Biotech (~4.3 kDa) | Reversal of the anticoagulant effect of heparin by forming an inactive complex with heparin | Heparin overdose, FDA (1939) [22,23] |
| Keyhole limpet hemocyanin (DB05299) | IMMUCOTHEL® VACMUNE® (Biosyn Corporation, Carlsbad, CA, USA) | Keyhole limpet (marine mollusk) | Biotech (350 to 390 kDa) | An immunomodulator | IMMUCOTHEL® for bladder cancer, [24], VACMUNE® as protein carrier for vaccine development [25], the Netherlands (1997), Austria and South Korea (2001), Argentina (2004/2005) [26] |
| Fish Oil and its Components Used as Drugs | |||||
| Omega-3-acid ethyl esters (DB09539) | LOVAZA® (GSK, Brentford, UK), OMACOR® (GSK, Brentford, UK), and OMTRYG® (Trygg Pharma, Oslo, Norway) | Fish | Small molecule (330.51 to 356.55 Da) | Reduces triglyceride synthesis by inhibiting 1,2-diacylglycerol acyltransferase | Reduce triglyceride (TG) levels, FDA (2004) [27,28,29] |
| Icosapent ethyl (DB00159) | VASCEPA® (Amarin Pharma, Dublin, Ireland) | Fish | Small molecule (330.51 Da) | Reduces triglyceride synthesis by inhibiting 1,2-diacylglycerol acyltransferase | Reduces the risk of myocardial infarction, stroke, coronary revascularization, and unstable angina, FDA (2012) [30] |
| Omega-3-carboxylic acids (DB09568) | EPANOVA® (AstraZeneca Pharmaceuticals, Landon, UK) | Fish | Small molecule (302.45 to 328.49 Da) | Reduces triglyceride synthesis by inhibiting 1,2-diacylglycerol acyltransferase | Reduce triglyceride (TG) levels, FDA (2014) [31] |
| Fish oil triglycerides (DB13961) | OMEGAVEN® (Fresenius Kabi, Bad Homburg, Germany) | Fish | Small molecule (mixture of fatty acids, 700 to 1000 Da each) | Source of calories and essential fatty acids | PNAC, FDA (2018) [32] |
| Drug Name | Component of Fish Oil and Its Form | Approximate Contents of Major Constituents | Drug Form and Usage |
|---|---|---|---|
| LOVAZA® OMACOR® OMTRYG® | Omega-3-acid ethyl esters | A 1 g capsule contains 465 mg of EPA and 375 mg of DHA | Liquid-filled gel capsule; used orally |
| VASCEPA® | Icosapent ethyl | 0.5 g and 1 g of icosapent ethyl in 0.5 g and 1 g capsules, respectively | Amber colored, liquid-filled soft-gelatin capsule; used orally |
| EPANOVA® | Omega-3-carboxylic acids | 850 mg of polyunsaturated fatty acids among which EPA and DHA are most abundant | Soft-gelatin capsule; used orally |
| OMEGAVEN® | Fish oil triglycerides | 0.1 g of fish oil in 1 mL; EPA 13–26%, DHA 14–27%, and other fatty acids such as palmitic acid, oleic acid, palmitoleic acid, myristic acid, and arachidonic acid are present in low proportions | White-colored, homogenous emulsion; used intravenously |
| Name | Company | Source Organism | Disease [NCT CODE] |
|---|---|---|---|
| Phase III | |||
| Lurbinectedin (alkaloid) | Hoffmann-La Roche, Basel, Switzerland | Ecteinascidia turbinata (tunicate) | 1. Study examining its combination with atezolizumab for higher-stage SCLC. [NCT05091567] 2. Study in combination of topotecan or irinotecan in patients with relapsed small-cell lung cancer. [NCT05153239] |
| Tetrodotoxin (alkaloid) | Wex Pharmaceuticals, Vancouver, Canada | Pseudoalteromonas sp., Pseudomonas sp., Vibrio sp. puffer fish (fugu). | Management of moderate or severe cancer-related pain. [NCT00726011] |
| Plitidepsin (depsipeptide) | PharmaMar, Madrid, Spain | Aplidium albicans (sea squirt) | Management of hospitalized patients with moderate COVID-19. [NCT04784559] |
| Marizomib | European Organization for Research and Treatment of Cancer-EORTC, Brussel, Belgium | Salinispora tropica (bacteria) | Non-small-cell lung cancer, pancreatic cancer, melanoma, lymphoma, multiple myeloma; newly diagnosed glioblastoma [NCT00667082, NCT03345095] |
| Plinabulin | Memorial Sloan Kettering Cancer Center; BeyondSpring Pharmaceuticals, New York City, NY, USA | Halimide (fungus) | Multiple myeloma; non-small-cell lung cancer, brain tumor [NCT05130827] |
| Phase II | |||
| AGS-16C3F (MMAF) | Astellas Pharma Global Development, Tokyo, Japan | Dolabella auricularia (mollusk) | Metastatic renal cell carcinoma [NCT02639182] |
| Tisotumab vedotin (MMAE) | Seagen, Bothell, WA, USA | Dolabella auricularia (mollusk) | Cervical Cancer [NCT03438396] |
| Ladiratuzumab vedotin (MMAE) | Seagen, Bothell, USA | Dolabella auricularia (mollusk) | Small-cell lung cancer, non-small-cell lung cancer, squamous, non-small-cell lung cancer, non-squamous carcinoma, head and neck squamous cell carcinoma, esophageal squamous cell carcinoma, gastric adenocarcinoma, gastroesophageal junction adenocarcinoma, prostate cancer, melanoma [NCT04032704] |
| Telisotuzumab vedotin (MMAE) | AbbVie, North Chicago, IL, USA | Dolabella auricularia (mollusk) | Non-small-cell lung cancer [NCT03539536] |
| Enapotamab vedotin (MMAE) | Genmab, Copenhagen V, Denmark | Dolabella auricularia (mollusk) | Ovarian cancer, cervical cancer, endometrial cancer, non-small-cell lung cancer, thyroid cancer, melanoma, sarcoma [NCT02988817] |
| Disitamab vedotin (MMAE) | Seagen, Bothell, USA | Dolabella auricularia (mollusk) | Urothelial carcinoma, advanced cancer, gastric cancer, HER2-overexpressing gastric carcinoma, advanced breast cancer, solid tumors [NCT04879329] |
| CX-2029 (MMAE) | CytomX Therapeutics, South San Francisco, CA, USA | Dolabella auricularia (mollusk) | Solid tumors, head and neck cancer, non-small-cell lung cancer, diffuse large B-cell lymphoma, esophageal cancer [NCT03543813] |
| RC88 (MMAE) | RemeGen, Beijing, China | Dolabella auricularia (mollusk) | Solid tumors [NCT04175847] |
| W0101 (auristatin variant) | Pierre Fabre Medicament, Boulogne, France | Dolabella auricularia (mollusk) | Advanced or metastatic solid tumors, insulin-like growth factor type 1 receptor [NCT03316638] |
| ARX788 (auristatin variant) | Ambrx, La Jolla, CA, USA | Dolabella auricularia (mollusk) | HER2-positive metastatic breast cancer [NCT04829604] |
| XMT-1536 | Mersana Therapeutics, Cambridge, MA, USA | Dolabella auricularia (mollusk) | Platinum-resistant ovarian cancer [NCT03319628, NCT04907968] |
| MORAb-202 | Eisai, Bunkyo, Japan | Halichondria okadai (sponge) | Tumors [NCT04300556] |
| GTS-21 | CoMentis, South San Francisco, CA, USA | Nemertines sp. (worm) | Alzheimer’s disease [NCT00414622] |
| Plocabulin (PM060184/PM184) | PharmaMar, Madrid, Spain | Lithoplocamia lithiostoides (sponge) | Solid tumors, advanced colorectal cancer [NCT03427268] |
| Soblidotin (auristatin PE; TZT-1027) | Daiichi Sankyo, Inc., Tokyo, Japan | Synthetic dolastatin 10 derivative | Non-small-cell lung cancer, sarcoma [NCT00061854, NCT00064220] |
| Synthadotin (Tasidotin; ILX-651) | Genzyme (Sanofi), Boston, MA, USA | Synthetic dolastatin 15 derivative | Melanoma, prostate cancer, non-small-cell lung carcinoma [NCT00068211, NCT00082134, NCT00078455] |
| Bryostatin 1 | Neurotrope Bioscience, New York City, NY, USA | Bugula neritina (bryozoan) | Alzheimer’s disease, kidney cancer, acute myelogenous leukemia and myelodysplastic syndrome, colorectal cancer, myelodysplastic syndrome, relapsed multiple myeloma, Hodgkin disease, non-small-cell lung cancer, head and neck cancer, breast cancer, ovarian epithelial cancer, prostate cancer, cervical cancer, esophageal cancer, gastric cancer, relapsed acute myelogenous leukemia, esophageal cancer, gastric cancer. [NCT02221947, NCT00003968, NCT00136461, NCT00003171, NCT00002907, NCT00003936, NCT00005849, NCT00003443, NCT00003205, NCT00004008, NCT00005028, NCT00005965, NCT00005599, NCT00017342, NCT00006081] |
| Phase I | |||
| CAB-ROR2-ADC (MMAE) | BioAtla, San Diego, CA, USA | Dolabella auricularia (mollusk) | Non-small-cell lung cancer, triple negative breast cancer, melanoma, head and neck cancer [NCT03504488] |
| FOR46 (MMAE) | Fortis Therapeutics, San Diego, CA, USA | Dolabella auricularia (mollusk) | Metastatic castration-resistant prostate cancer [NCT05011188] |
| ALT-P7 (MMAE) | Alteogen, Yuseong, S. Korea | Dolabella auricularia (mollusk) | HER2-positive breast cancer [NCT03281824] |
| MRG003 (MMAE) | Shanghai Miracogen, Shanghai, China | Dolabella auricularia (mollusk) | Advanced or metastatic gastric cancer [NCT05188209] |
| SGN-CD228A (MMAE) | Seagen, Bothell, USA | Dolabella auricularia (mollusk) | Cutaneous melanoma, pleural mesothelioma, HER2-negative breast neoplasms, non-small-cell lung cancer, colorectal cancer, pancreatic ductal adenocarcinoma [NCT04042480] |
| SGN-B6A (MMAE) | Seagen, Bothell, USA | Dolabella auricularia (mollusk) | Carcinoma, non-small-cell lung, squamous cell carcinoma of head and neck, HER2-negative breast neoplasms, esophageal squamous cell carcinoma, esophageal, adenocarcinoma, gastroesophageal junction adenocarcinoma, ovarian neoplasms, cutaneous squamous cell cancer, exocrine pancreatic adenocarcinoma, urinary bladder neoplasms, uterine cervical neoplasms, stomach neoplasms [NCT04389632] |
| Cofetuzumab pelidotin (auristatin variant) | AbbVie, North Chicago, USA | Dolabella auricularia (mollusk) | Cancer, non-small-cell lung cancer (NSCLC) [NCT04189614] |
| PF-06804103 (auristatin variant) | Pfizer, New York City, USA | Dolabella auricularia (mollusk) | Breast neoplasms [NCT03284723] |
| ZW-49 (auristatin variant) | Zymeworks, Vancouver, Canada | Dolabella auricularia (mollusk) | HER2-expressing cancers [NCT03821233] |
| A-166 (duostatin 5) | Sichuan Kelun Pharmaceutical Research Institute, Sichuan, China | Dolabella auricularia (mollusk) | Breast cancer [NCT05311397] |
| STI-6129 (duostatin 5) | Sorrento Therapeutics, San Diego, CA, USA | Dolabella auricularia (mollusk) | Light chain (AL) amyloidosis [NCT04316442] |
| Griffithsin | Center for Predictive Medicine, Louisville, KY, USA | Red alga | HIV prevention [NCT02875119] |
| Hemiasterlin (E7974) | Eisai, Bunkyo, Japan | Sponge | Cancer, malignant tumors [NCT00121732, NCT00130169, NCT00165802] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haque, N.; Parveen, S.; Tang, T.; Wei, J.; Huang, Z. Marine Natural Products in Clinical Use. Mar. Drugs 2022, 20, 528. https://doi.org/10.3390/md20080528
Haque N, Parveen S, Tang T, Wei J, Huang Z. Marine Natural Products in Clinical Use. Marine Drugs. 2022; 20(8):528. https://doi.org/10.3390/md20080528
Chicago/Turabian StyleHaque, Neshatul, Sana Parveen, Tingting Tang, Jiaen Wei, and Zunnan Huang. 2022. "Marine Natural Products in Clinical Use" Marine Drugs 20, no. 8: 528. https://doi.org/10.3390/md20080528
APA StyleHaque, N., Parveen, S., Tang, T., Wei, J., & Huang, Z. (2022). Marine Natural Products in Clinical Use. Marine Drugs, 20(8), 528. https://doi.org/10.3390/md20080528