
Not Drug-like, but Like Drugs: Cnidaria Natural Products

 ,
,  , , , , and
, , , , and
Abstract
:1. Introduction
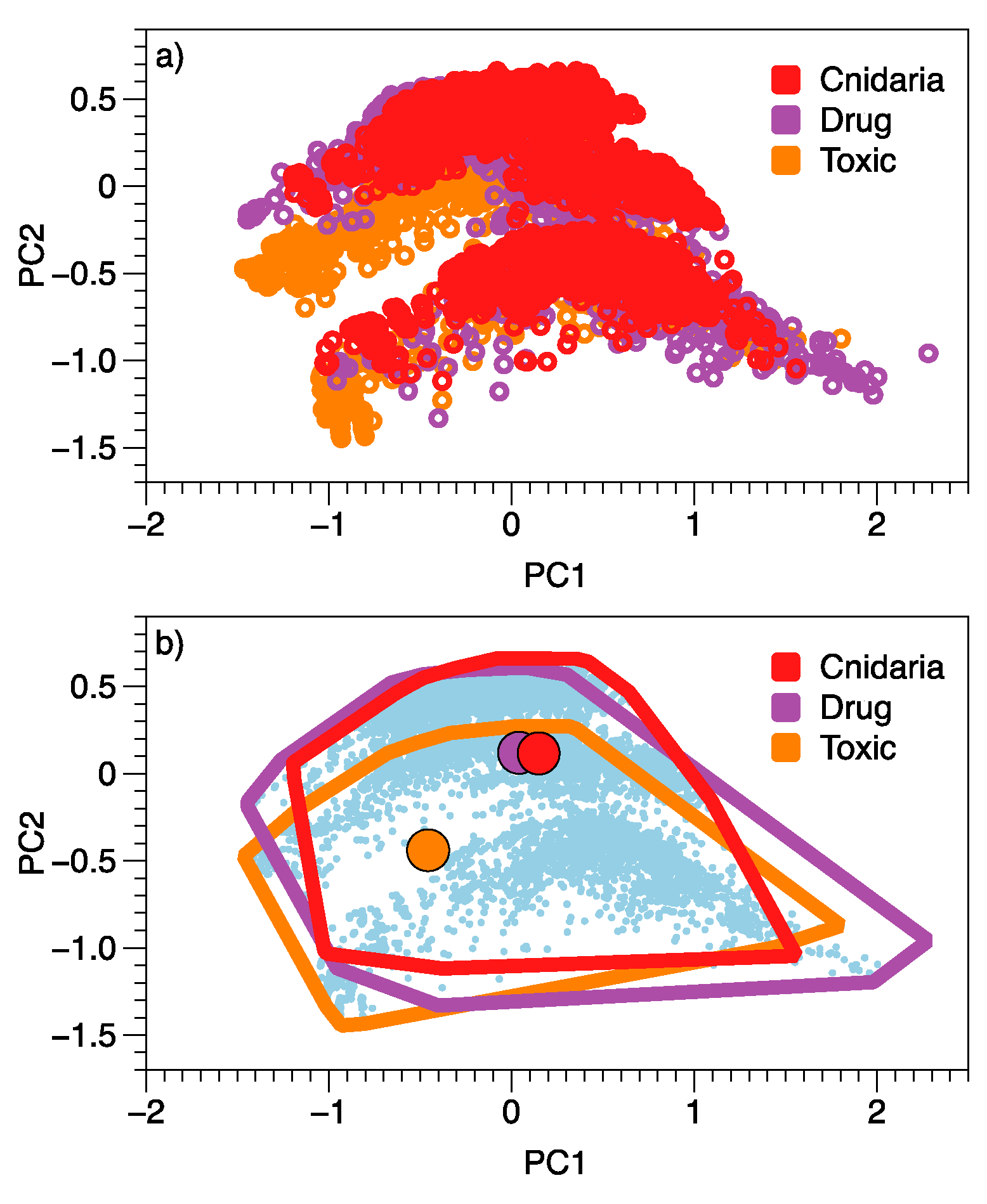
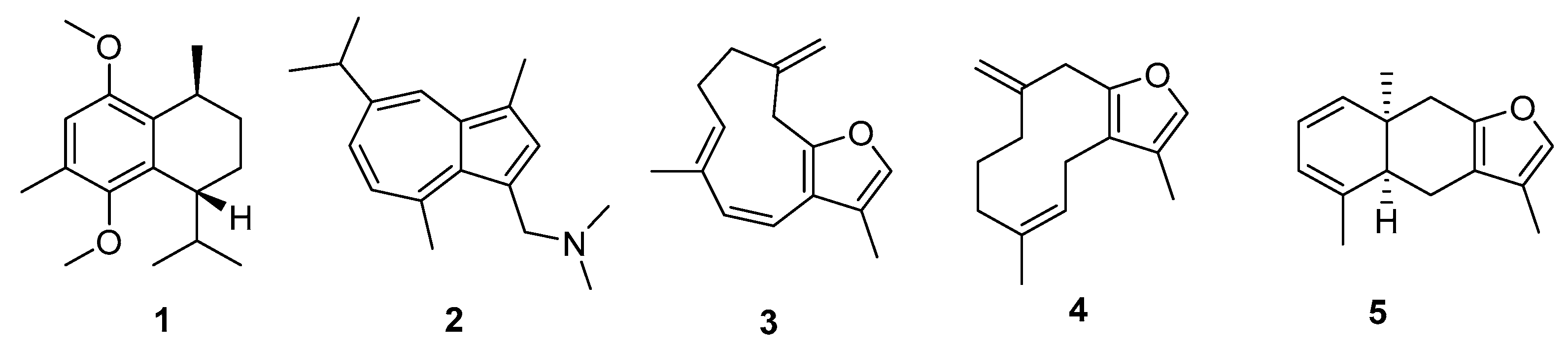
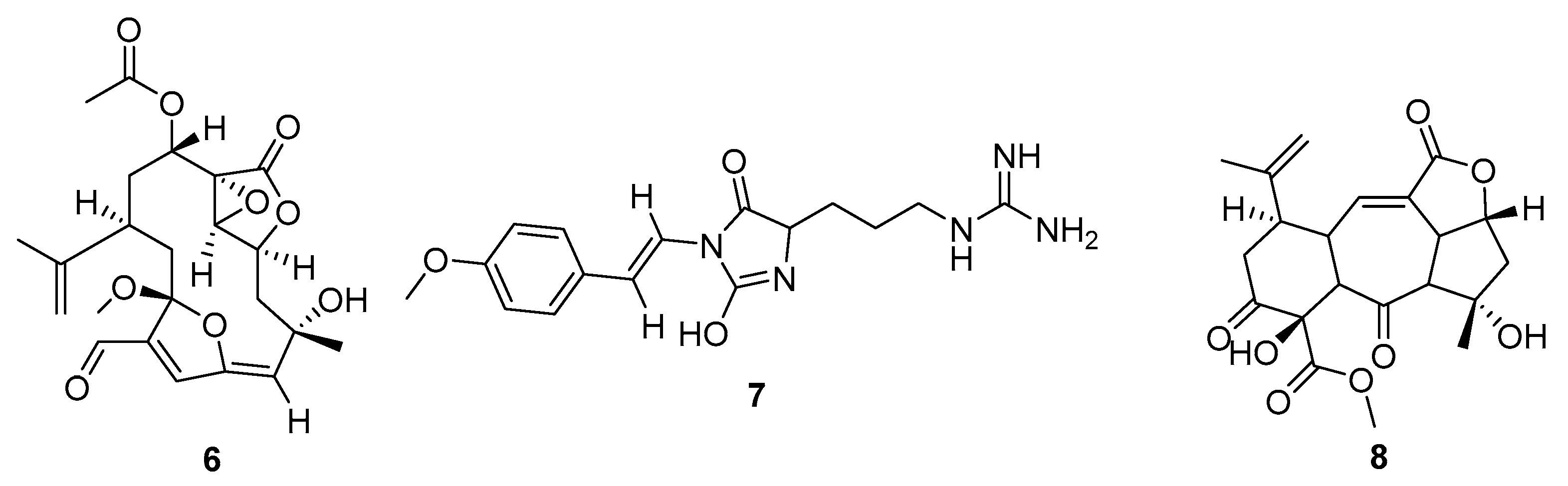
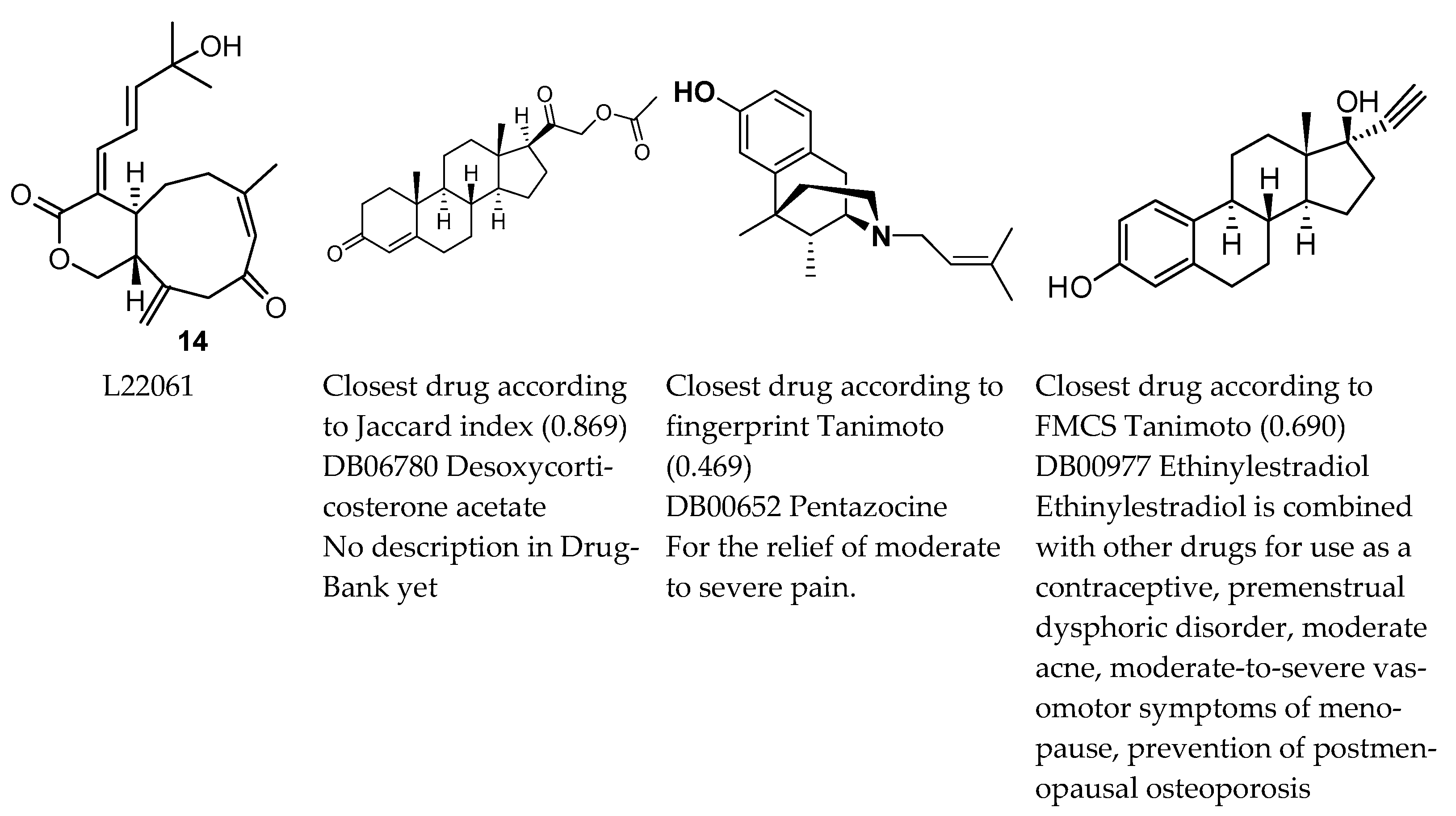
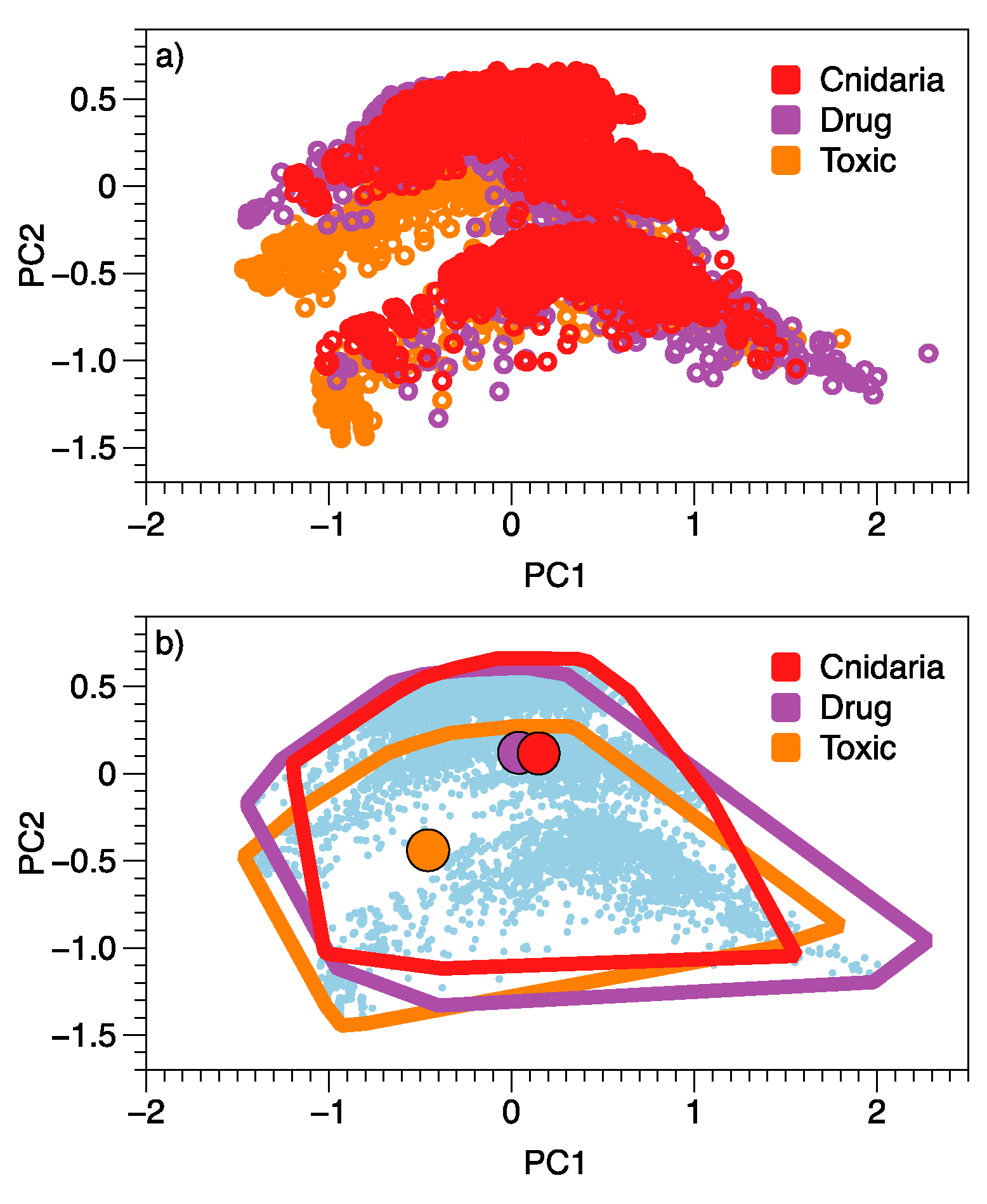
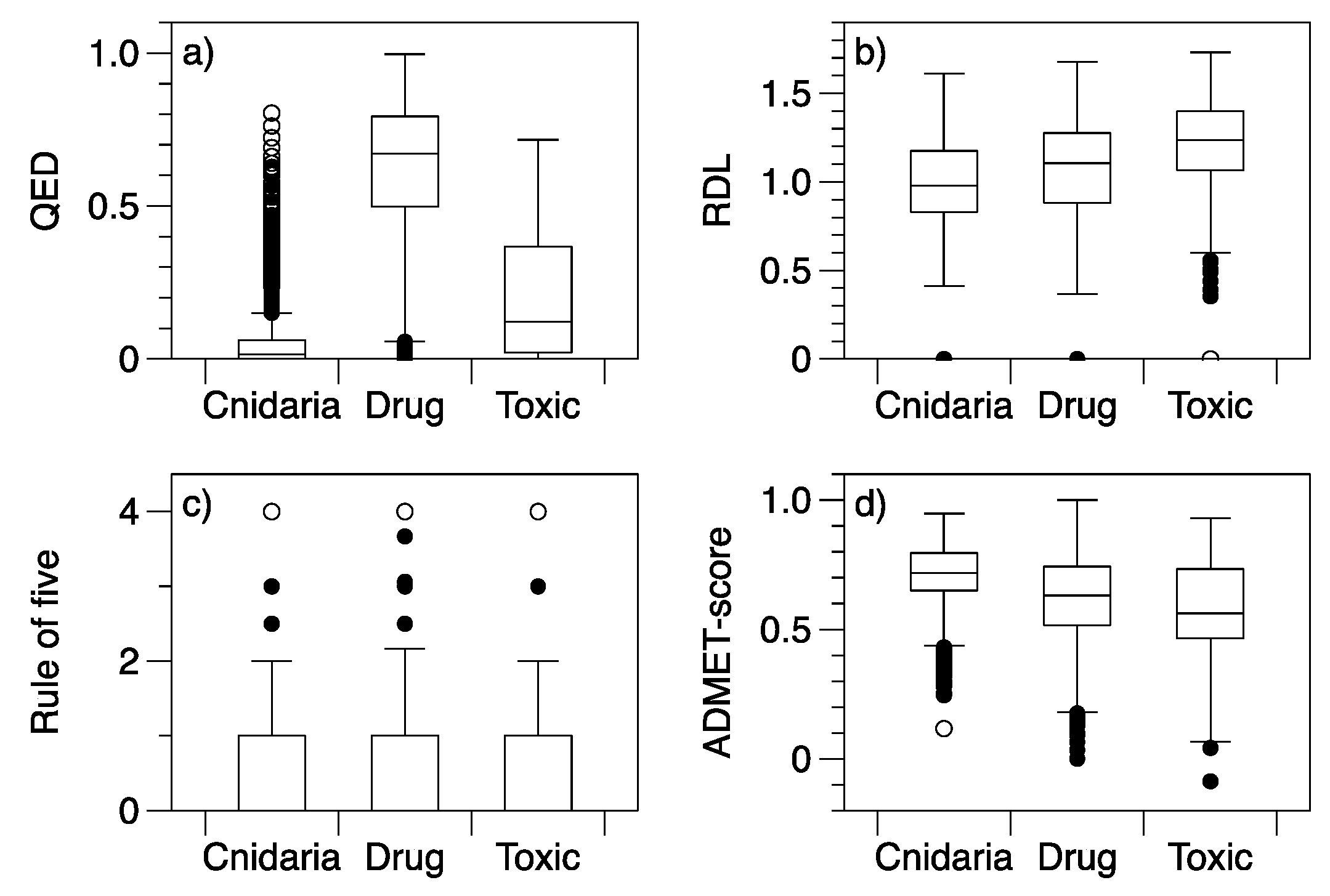
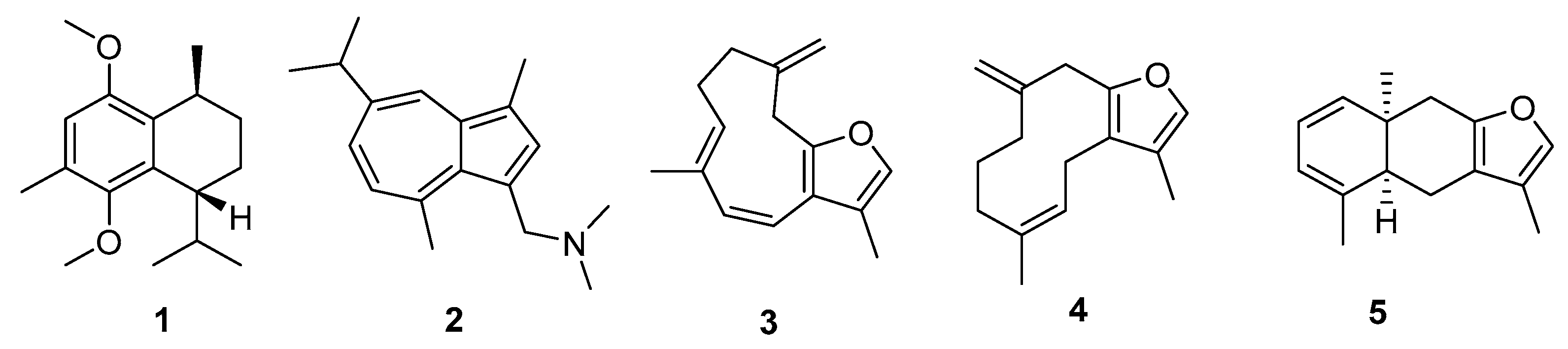
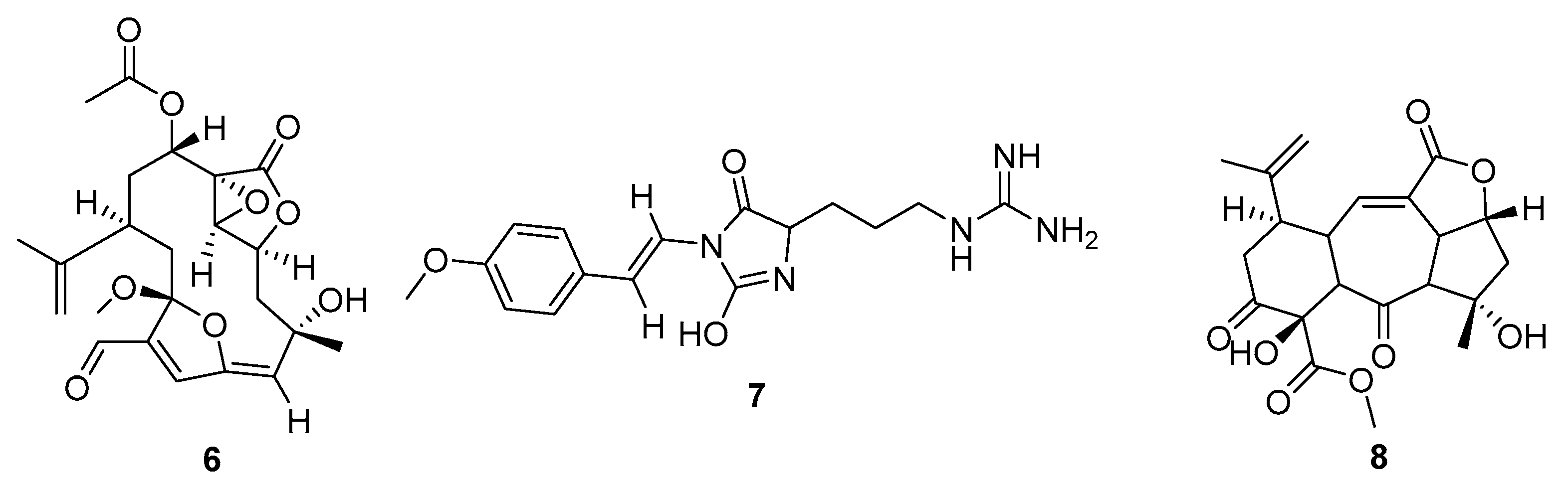
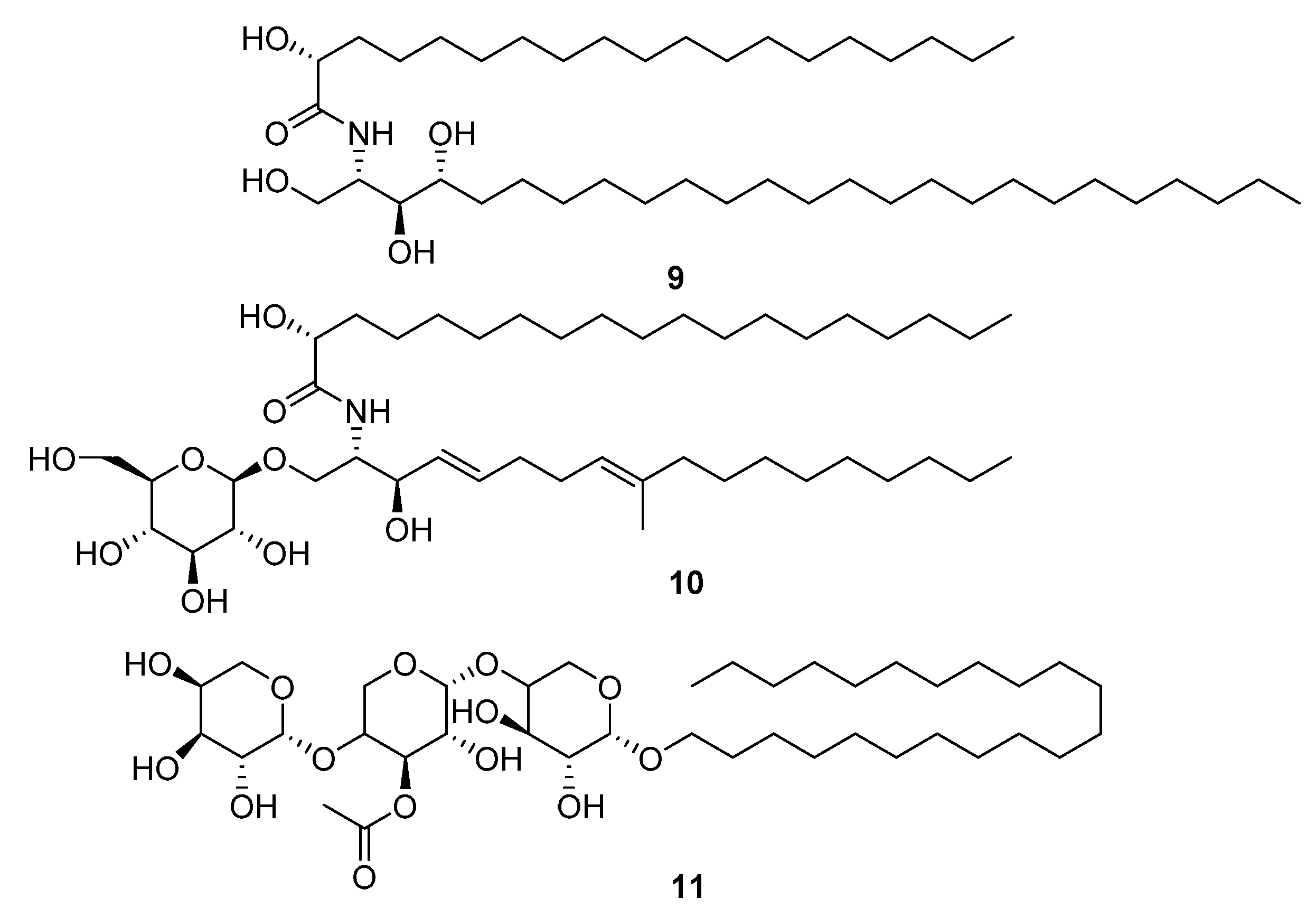
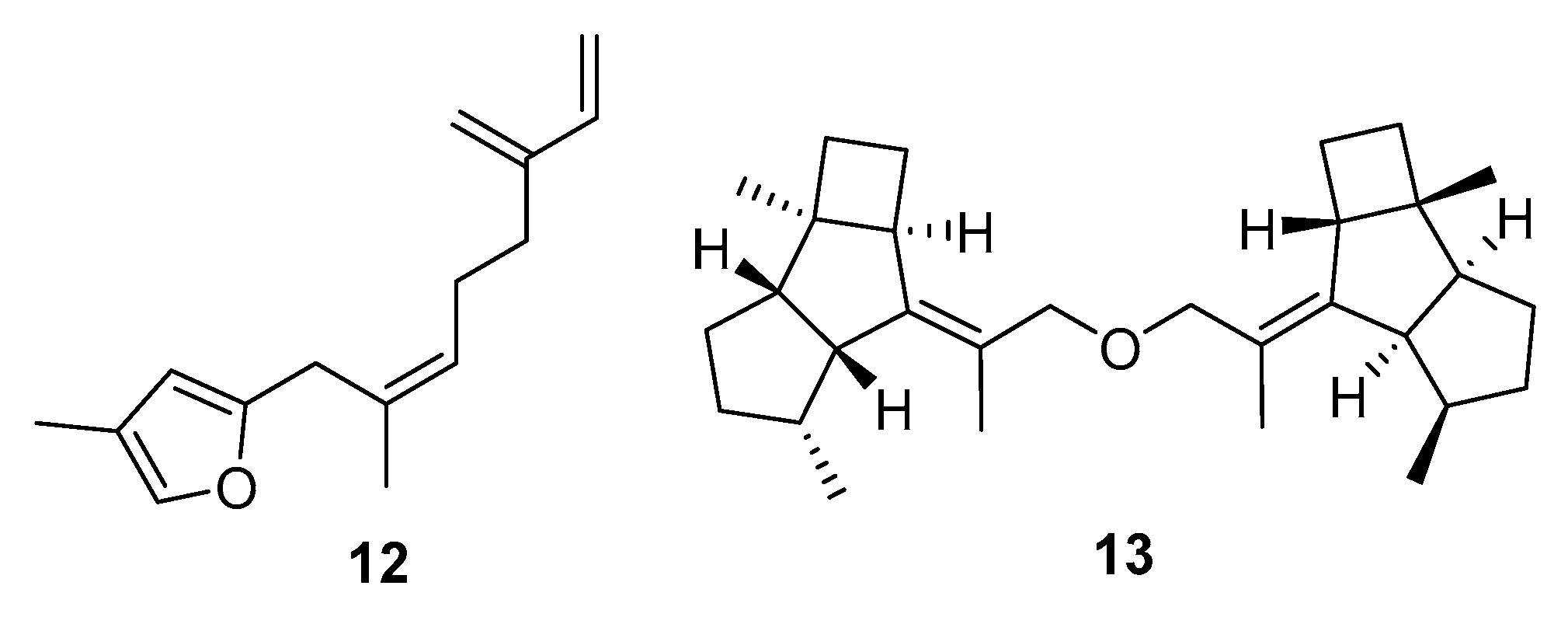
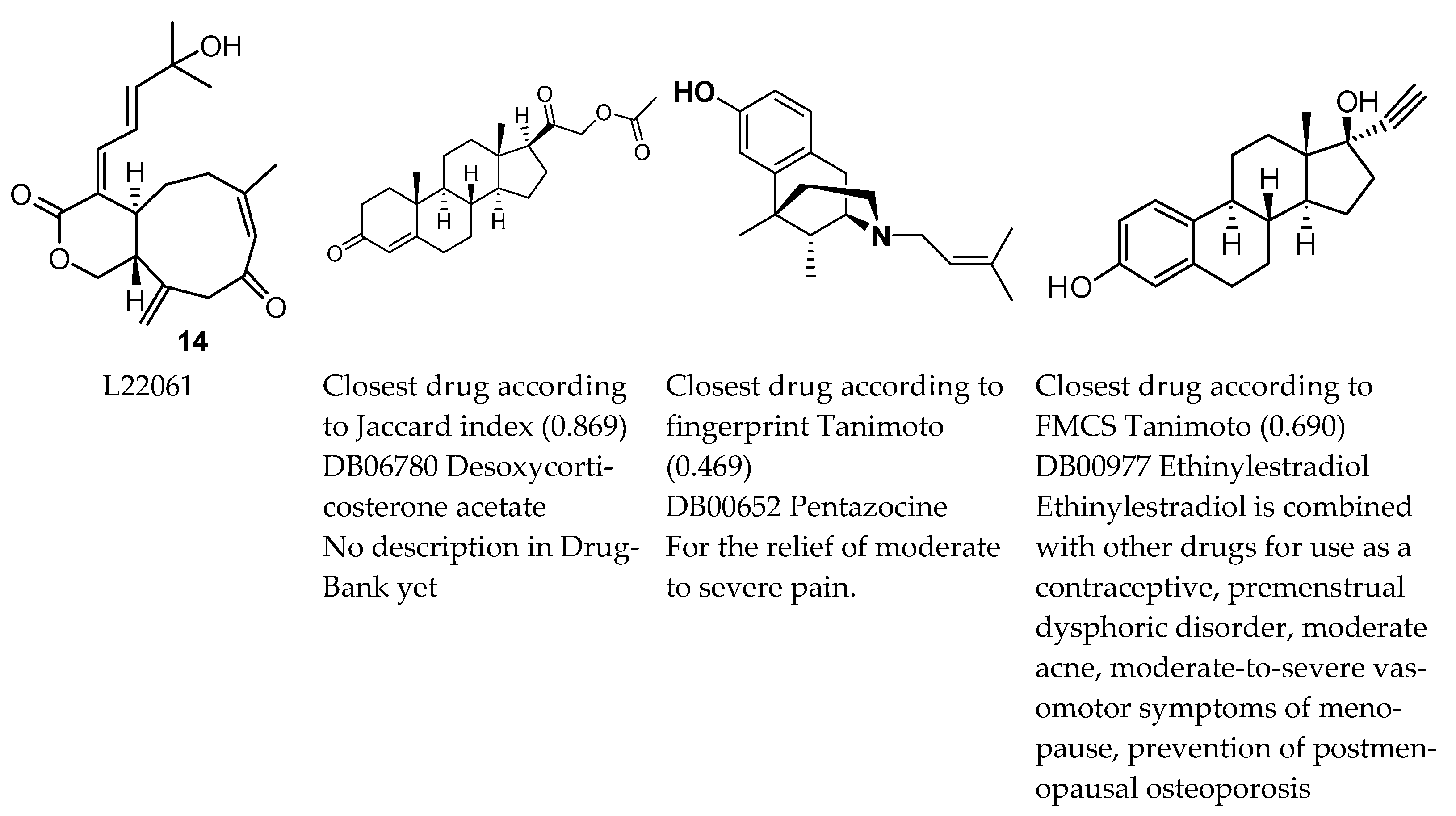
2. Results
3. Discussion
4. Materials and Methods
4.1. Data Collection and Variable Generation
4.2. Drug Likeness Approaches
4.3. Drug Similarity Approaches
4.4. Ligand Similarity and Docking
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine Natural Products. Nat. Prod. Rep. 2019, 36, 122–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobson, A.D.; Jackson, S.A.; Kennedy, J.; Margassery, L.M.; Flemer, B.; O’Leary, N.; Morrissey, J.P.; O’Gara, F. Marine sponges–molecular biology and biotechnology. In Springer Handbook of Marine Biotechnology; Se-Kwon, K., Ed.; Springer: Berlin/Heidelberg, Germany, 2015; pp. 219–254. [Google Scholar] [CrossRef]
- Karuppiah, V.; Li, Z. Marine sponge metagenomics. In Springer Handbook of Marine Biotechnology; Se-Kwon, K., Ed.; Springer: Berlin/Heidelberg, Germany, 2015; pp. 457–473. [Google Scholar] [CrossRef]
- Rocha, J.; Peixe, L.; Gomes, N.; Calado, R. Cnidarians as a source of new marine bioactive compounds—An overview of the last decade and future steps for bioprospecting. Mar. Drugs 2011, 9, 1860–1886. [Google Scholar] [CrossRef]
- Khalesi, M.K. Corals. In Springer Handbook of Marine Biotechnology; Se-Kwon, K., Ed.; Springer: Berlin/Heidelberg, Germany, 2015; pp. 179–217. [Google Scholar] [CrossRef]
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2015, 32, 116–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Pilkington, L.I. A chemometric analysis of deep-Sea natural products. Molecules 2019, 24, 3942. [Google Scholar] [CrossRef] [Green Version]
- Feher, M.; Schmidt, J.M. Property distributions: Differences between drugs, natural products, and molecules from combinatorial chemistry. J. Chem. Inf. Comput. Sci. 2003, 43, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Gui, Y.; Chen, L.; Yuan, G.; Lu, H.-Z.; Xu, X. Use of natural products as chemical library for drug discovery and network pharmacology. PLoS ONE 2013, 8, e62839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mol, V.P.L.; Raveendran, T.V.; Naik, B.G.; Kunnath, R.J.; Parameswaran, P.S. Calamenenes-aromatic bicyclic sesquiterpenes-from the Indian gorgonian Subergorgia reticulata (Ellis and Solander, 1786). Nat. Prod. Res. 2011, 25, 169–174. [Google Scholar] [CrossRef]
- Mol, V.P.L.; Abdulaziz, A.; Sneha, K.G.; Praveen, P.J.; Raveendran, T.V.; Parameswaran, P.S. Inhibition of pathogenic Vibrio harveyi using calamenene, derived from the Indian gorgonian Subergorgia reticulata, and its synthetic analog. 3 Biotech 2020, 10, 248. [Google Scholar] [CrossRef]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012, 4, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitale, R.M.; Thellung, S.; Tinto, F.; Solari, A.; Gatti, M.; Nuzzo, G.; Ioannou, E.; Roussis, V.; Ciavatta, M.L.; Manzo, E. Identification of the hydantoin alkaloids parazoanthines as novel CXCR4 antagonists by computational and in vitro functional characterization. Bioorg. Chem. 2020, 105, 104337. [Google Scholar] [CrossRef]
- Ahmed, S.; Ibrahim, A.; Arafa, A.S. Anti-H5N1 virus metabolites from the Red Sea soft coral, Sinularia candidula. Tetrahedron Lett. 2013, 54, 2377–2381. [Google Scholar] [CrossRef]
- Guan, L.; Yang, H.; Cai, Y.; Sun, L.; Di, P.; Li, W.; Liu, G.; Tang, Y. ADMET-score—A comprehensive scoring function for evaluation of chemical drug-likeness. MedChemComm 2019, 10, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Mignani, S.; Huber, S.; Tomas, H.; Rodrigues, J.; Majoral, J.P. Compound high-quality criteria: A new vision to guide the development of drugs, current situation. Drug Discov. Today 2016, 21, 573–584. [Google Scholar] [CrossRef]
- Lipinski, C.A. Rule of five in 2015 and beyond: Target and ligand structural limitations, ligand chemistry structure and drug discovery project decisions. Adv. Drug Deliv. Rev. 2016, 101, 34–41. [Google Scholar] [CrossRef]
- Stone, S.; Newman, D.J.; Colletti, S.L.; Tan, D.S. Cheminformatic analysis of natural product-based drugs and chemical probes. Nat. Prod. Rep. 2022. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Bender, A.; Glen, R.C. Molecular similarity: A key technique in molecular informatics. Org. Biomol. Chem. 2004, 2, 3204–3218. [Google Scholar] [CrossRef]
- Martinez-Mayorga, K.; Madariaga-Mazon, A.; Medina-Franco, J.L.; Maggiora, G. The impact of chemoinformatics on drug discovery in the pharmaceutical industry. Expert Opin. Drug Discov. 2020, 15, 293–306. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.A.; Maggiora, G.M. Concepts and Applications of Molecular Similarity; Wiley: New York, NY, USA, 1991; ISBN 978-0-471-62175-1. [Google Scholar]
- O’Boyle, N.M.; Sayle, R.A. Comparing structural fingerprints using a literature-based similarity benchmark. J. Cheminformatics 2016, 8, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maggiora, G.M. On outliers and activity cliffs-why QSAR often disappoints. J. Chem. Inf. Model. 2006, 46, 1535. [Google Scholar] [CrossRef] [PubMed]
- Costantino, L.; Barlocco, D. Privileged structures as leads in medicinal chemistry. Curr. Med. Chem. 2006, 13, 65–85. [Google Scholar] [CrossRef]
- Goldberg, F.W.; Kettle, J.G.; Kogej, T.; Perry, M.W.; Tomkinson, N.P. Designing novel building blocks is an overlooked strategy to improve compound quality. Drug Discov. Today 2015, 20, 11–17. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Lin, Y.S.; Fazary, A.E.; Chen, C.H.; Kuo, Y.H.; Shen, Y.C. Asterolaurins G–J, new xenicane diterpenoids from the Taiwanese soft coral Asterospicularia laurae. Helv. Chim. Acta 2011, 94, 273–281. [Google Scholar] [CrossRef]
- Shultz, M.D. Two decades under the influence of the rule of five and the changing properties of approved oral drugs. J. Med. Chem. 2018, 62, 1701–1714. [Google Scholar] [CrossRef]
- Doak, B.C.; Kihlberg, J. Drug discovery beyond the rule of 5—Opportunities and challenges. Expert Opin. Drug Discov. 2017, 12, 115–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanif, N.; Murni, A.; Yamauchi, M.; Higashi, M.; Tanaka, J. A new trinor-guaiane sesquiterpene from an Indonesian soft coral Anthelia sp. Nat. Prod. Commun. 2015, 10, 1907–1917. [Google Scholar] [CrossRef] [Green Version]
- Tian, S.; Wang, J.; Li, Y.; Li, D.; Xu, L.; Hou, T. The application of in silico drug-likeness predictions in pharmaceutical research. Adv. Drug Deliv. Rev. 2015, 86, 2–10. [Google Scholar] [CrossRef]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, D668–D672. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Cheng, D.; Shrivastava, S.; Tzur, D.; Gautam, B.; Hassanali, M. DrugBank: A knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2007, 36, D901–D906. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2017, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Lim, E.; Pon, A.; Djoumbou, Y.; Knox, C.; Shrivastava, S.; Guo, A.C.; Neveu, V.; Wishart, D.S. T3DB: A comprehensively annotated database of common toxins and their targets. Nucleic Acids Res. 2010, 43, D781–D786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wishart, D.; Arndt, D.; Pon, A.; Sajed, T.; Guo, A.C.; Djoumbou, Y.; Knox, C.; Wilson, M.; Liang, Y.; Grant, J.; et al. T3DB: The toxic exposome database. Nucleic Acids Res. 2015, 43, D928–D934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminformatics 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenk, R.; Schipani, A.; James, D.; Krasowski, A.; Gilbert, I.H.; Frearson, J.; Wyatt, P.G. Lessons learnt from assembling screening libraries for drug discovery for neglected diseases. ChemMedChem 2008, 3, 435–444. [Google Scholar] [CrossRef]
- Yusof, I.; Segall, M.D. Considering the impact drug-like properties have on the chance of success. Drug Discov. Today 2013, 18, 659–666. [Google Scholar] [CrossRef]
- Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y. admetSAR: A comprehensive source and free tool for assessment of chemical ADMET properties. J. Chem. Inf. Model. 2012, 52, 3099–3105. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Sun, L.; Li, W.; Liu, G.; Tang, Y. In silico prediction of chemical toxicity for drug design using machine learning methods and structural alerts. Front. Chem. 2018, 6, 30. [Google Scholar] [CrossRef]
- Larsson, J.; Gottfries, J.; Muresan, S.; Backlund, A. ChemGPS-NP: Tuned for navigation in biologically relevant chemical space. J. Nat. Prod. 2007, 70, 789–794. [Google Scholar] [CrossRef]
- Zuegg, J.; Cooper, M.A. Drug-Likeness and increased hydrophobicity of commercially available compound libraries for drug screening. Curr. Top. Med. Chem. 2012, 12, 1500–1513. [Google Scholar] [CrossRef] [PubMed]
- Zuur, A.; Ieno, E.N.; Smith, G.M. Analysing Ecological Data; Springer: New York, NY, USA, 2007; ISBN 978-0-387-45972-1. [Google Scholar]
- Anderson, M.J. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001, 26, 32–46. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package. R Package Version 2.5-7. 2020. Available online: https://CRAN.R-project.org/package=vegan (accessed on 19 April 2021).
- Cao, Y.; Charisi, A.; Cheng, L.-C.; Jiang, T.; Girke, T. ChemmineR: A Compound Mining Framework for R. Bioinformatics 2008, 24, 1733–1734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Backman, T.W.H.; Horan, K.; Girke, T. fmcsR: Mismatch Tolerant Maximum Common Substructure Searching in R. Bioinformatics 2013, 29, 2792–2794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilson, M.K.; Liu, T.; Baitaluk, M.; Nicola, G.; Hwang, L.; Chong, J. BindingDB in 2015: A public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 2016, 44, D1045–D1063. [Google Scholar] [CrossRef] [PubMed]
- Kratz, F.; Beyer, U. Serum Proteins as Drug Carriers of Anticancer Agents: A Review. Drug Deliv. 1998, 5, 281–299. [Google Scholar] [CrossRef]
- Larsen, M.T.; Kuhlmann, M.; Hvam, M.L.; Howard, K.A. Albumin-based drug delivery: Harnessing nature to cure disease. Mol. Cell Ther. 2016, 4, 3. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PC1 | PC2 | ||||
|---|---|---|---|---|---|
| Variable | Cor. | Contrib. | Variable | Cor. | Contrib. |
| RuleOfFive | 0.100 | 3.17 | HumanOralAbsorption | 0.386 | 54.73 |
| X.nonHatm | 0.095 | 2.90 | glob | 0.113 | 4.67 |
| QPpolrz | 0.090 | 2.60 | WPSA | −0.092 | 3.11 |
| volume | 0.088 | 2.48 | X.stars | −0.106 | 4.14 |
| X.rtvFG | 0.085 | 2.32 | RuleOfFive | −0.116 | 4.92 |
| RuleOfThree | 0.085 | 2.29 | RuleOfThree | −0.129 | 6.15 |
| FOSA | 0.084 | 2.23 | QPPCaco | −0.156 | 8.92 |
| X.noncon | 0.080 | 2.03 | QPPMDCK | −0.163 | 9.80 |
| HumanOralAbsorption | −0.131 | 5.44 | |||
| %HumanOralAbsorption | −0.136 | 5.90 | |||
| QPPMDCK | −0.204 | 13.22 | |||
| QPPCaco | −0.222 | 15.71 | |||
| CNS | −0.259 | 21.35 | |||
| Drug-Likeness Index | KW Chi-Squared | df | p | Significantly Different Pairs |
|---|---|---|---|---|
| QED (high values imply drug like) | 4648.5 | 2 | <0.001 | Drug > Cnidaria |
| Drug > Toxic | ||||
| Toxic > Cnidaria | ||||
| RDL (high values imply drug like) | 1035.6 | 2 | <0.001 | Drug > Cnidaria |
| Toxic > Drug | ||||
| Toxic > Cnidaria | ||||
| Ro5 (number of exceptions) | 57.7 | 2 | <0.001 | Cnidaria > Drug |
| Toxic > Drug | ||||
| ADMET-score (high values imply low predicted toxicity) | 1193.7 | 2 | <0.001 | Cnidaria > Drug |
| Drug > Toxic | ||||
| Cnidaria > Toxic |
| Tanimoto | QED | RDL | Admet Score | Rule-of-five | PC1 | PC2 | ||
|---|---|---|---|---|---|---|---|---|
| Jaccard | R | 0.016 | −0.134 | 0.483 | 0.291 | −0.740 | −0.507 | 0.778 |
| p | 0.239 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | |
| Tanimoto | R | 1.000 | 0.025 | −0.038 | −0.077 | −0.040 | −0.019 | −0.159 |
| p | x | 0.064 | 0.005 | <0.001 | 0.003 | 0.153 | <0.001 |
| Entry | Target Name | UniProID | PDB ID |
|---|---|---|---|
| 1 | Mitogen-activated protein kinase kinase kinase MLT | Q9NYL2 | 6JUU |
| 2 | Mast/stem cell growth factor receptor Kit | P10721 | 6HH1 |
| 3 | Amidophosphoribosyltransferase | Q06203 | 6CZF |
| 4 | Tyrosine-protein kinase BTK | Q06187 | 6AUB |
| 5 | Receptor-type tyrosine-protein kinase FLT3 | P36888 | 4XUF |
| 6 | LIM domain kinase 1 | P53667 | 5NXC |
| 7 | Platelet-derived growth factor receptor alpha | P16234 | 5GRN |
| 8 | Tyrosine-protein kinase ABL1 | P00519 | 4WA9 |
| 9 | Thyroid hormone receptor alpha | P10827 | 4LNW |
| 10 | Histone deacetylase 3 | O15379 | 4A69 |
| 11 | Mitogen-activated protein kinase 14 | Q16539 | 3UVR |
| 12 | Alpha-1-acid glycoprotein 1 | P02763 | 3KQ0 |
| 13 | Vascular endothelial growth factor receptor 1 | P17948 | 3HNG |
| 14 | G-protein coupled estrogen receptor 1 | Q99527 | 2R6Y |
| 15 | Histone deacetylase 8 | Q9BY41 | 1T69 |
| 16 | Matrix metalloproteinase-16 | P51512 | 1RM8 |
| 17 | Thyroid hormone receptor beta | P10828 | 1Q4X |
| 18 | Serum albumin | P02768 | 4LA0 |
| 19 | Sex hormone-binding globulin | P04278 | 1D2S |
| 20 | Glucocorticoid receptor | P04150 | 6DXK |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laguionie-Marchais, C.; Allcock, A.L.; Baker, B.J.; Conneely, E.-A.; Dietrick, S.G.; Kearns, F.; McKeever, K.; Young, R.M.; Sierra, C.A.; Soldatou, S.; et al. Not Drug-like, but Like Drugs: Cnidaria Natural Products. Mar. Drugs 2022, 20, 42. https://doi.org/10.3390/md20010042
Laguionie-Marchais C, Allcock AL, Baker BJ, Conneely E-A, Dietrick SG, Kearns F, McKeever K, Young RM, Sierra CA, Soldatou S, et al. Not Drug-like, but Like Drugs: Cnidaria Natural Products. Marine Drugs. 2022; 20(1):42. https://doi.org/10.3390/md20010042
Chicago/Turabian StyleLaguionie-Marchais, Claire, A. Louise Allcock, Bill J. Baker, Ellie-Ann Conneely, Sarah G. Dietrick, Fiona Kearns, Kate McKeever, Ryan M. Young, Connor A. Sierra, Sylvia Soldatou, and et al. 2022. "Not Drug-like, but Like Drugs: Cnidaria Natural Products" Marine Drugs 20, no. 1: 42. https://doi.org/10.3390/md20010042
APA StyleLaguionie-Marchais, C., Allcock, A. L., Baker, B. J., Conneely, E.-A., Dietrick, S. G., Kearns, F., McKeever, K., Young, R. M., Sierra, C. A., Soldatou, S., Woodcock, H. L., & Johnson, M. P. (2022). Not Drug-like, but Like Drugs: Cnidaria Natural Products. Marine Drugs, 20(1), 42. https://doi.org/10.3390/md20010042