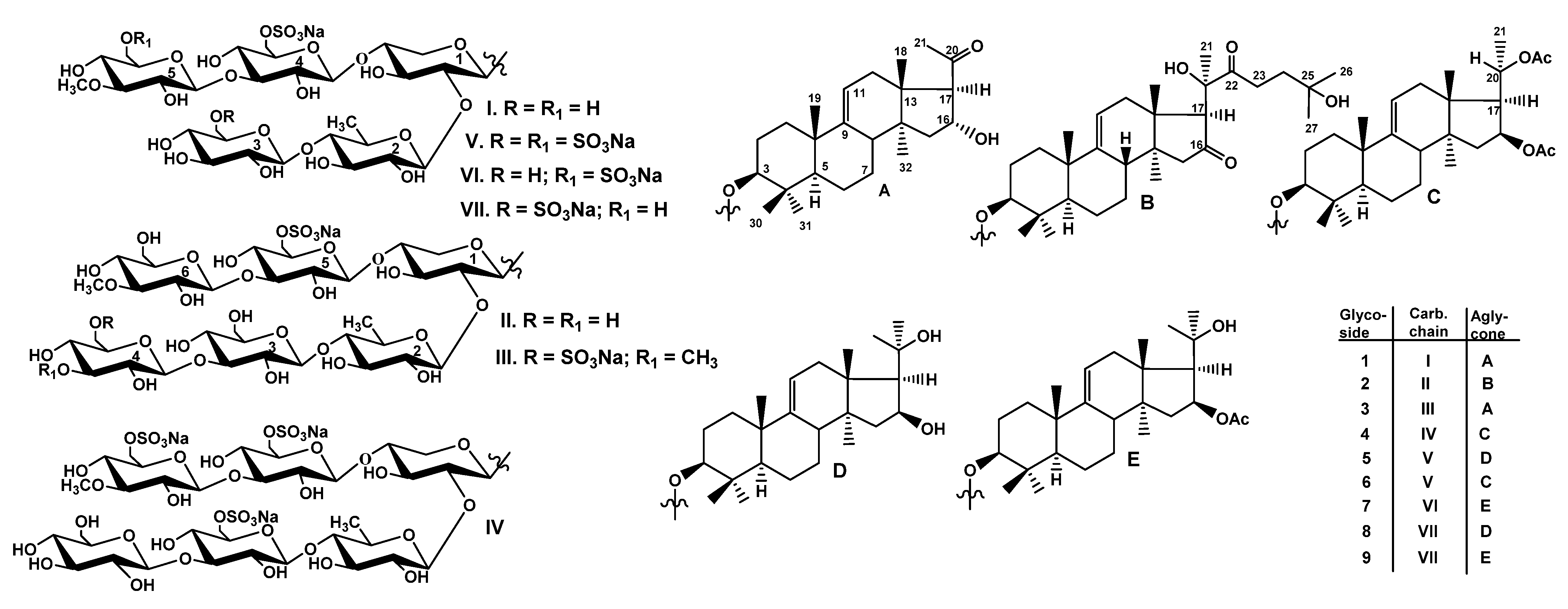
2.1. Structural Elucidation of the Glycosides
The concentrated ethanolic extract of the sea cucumber
Thyonidium (=Duasmodactyla) kurilensis was chromatographed on a Polychrom-1 column (powdered Teflon, Biolar, Latvia). The glycosides were eluted with 50% EtOH and separated by repeated chromatography on Si gel columns using CHCl
3/EtOH/H
2O (100:100:17) and (100:125:25) as mobile phases to give five fractions (I–V). The glycosides
1–
9 (
Figure 1) were isolated as a result of subsequent HPLC of the fractions II–V on a reversed-phase semipreparative column Phenomenex Synergi Fusion RP (10 × 250 mm).
The molecular formula of kuriloside A
3 (
1) was determined to be C
54H
87O
29SNa from the [M
Na − Na]
− ion peak at
m/z 1231.5063 (calc. 1231.5059) in the (−)HR-ESI-MS. Kuriloside A
3 (
1) as well as the reported earlier kurilosides A, A
1, and A
2 [
19] belong to the same group of glycosides, so these compounds have the identical monosulfated pentasaccharide chains that were confirmed by the coincidence of their
1H and
13C NMR spectra corresponding to the carbohydrate chains (
Table S1). The presence of five characteristic doublets at δ
H = 4.64–5.18 (
J = 7.1–7.6 Hz), and corresponding signals of anomeric carbons at δ
C = 102.3–104.7 in the
1H and
13C NMR spectra of the carbohydrate part of
1 indicate the presence of a pentasaccharide chain and
β-configurations of the glycosidic bonds. Monosaccharide composition of
1, established by the analysis of the
1H,
1H-COSY, HSQC, and 1D TOCSY spectra, includes one xylose (Xyl1), one quinovose (Qui2), two glucoses (Glc3 and Glc4), and one 3-O-methylglucose (MeGlc5) residue. The signal of C-6 Glc4 was observed at δ
C = 67.1 due to α-shifting effect of a sulfate group at this position. The positions of interglycosidic linkages were established by the ROESY and HMBC spectra (
Table S1). The analysis of NMR spectra of the aglycone part of
1 (
Table S2) indicated the presence of 22,23,24,25,26,27-hexa-
nor-lanostane aglycone with a 16α-hydroxy,20-oxo-fragment and 9(11)-double bond due to the characteristic signals: (δ
C 149.0 (C-9) and 114.2 (C-11), δ
C = 71.1 (C-16) and δ
H = 5.40 (brt,
J = 7.5 Hz, H-16), δ
C = 208.8 (C-20)). The ROE correlations H-16/H-15β and H-16/H-18 indicated a 16α-OH orientation in the aglycone of kuriloside A
3 (
1). 17αH-orientation, common for the sea cucumber glycosides, was deduced from the ROE-correlation H-17/H-32. The same aglycone was found earlier in kuriloside F [
19].
The (−)ESI-MS/MS of 1 demonstrated the fragmentation of [MNa − Na]− ion at m/z 1231.5. The peaks of fragment ions were observed at m/z 1069.5 [MNa – Na − C6H10O5 (Glc)]−, 1055.4 [MNa – Na − C7H12O5(MeGlc)]−, 923.4 [MNa − Na − C6H10O5(Glc) − C6H10O4(Qui)]−, 747.3 [MNa − Na − C6H10O5(Glc) − C6H10O4(Qui) − C7H12O5(MeGlc)]−, 695.1 [MNa −Na − C24H37O3(Agl) − C6H10O5 (Glc) − H]−, 565.1 [MNa − Na − C24H37O2(Agl) − C6H10O5(Glc) − C6H10O4 (Qui) − H]−, 549.1 [MNa − Na − C24H37O3(Agl) − C6H10O5(Glc) − C6H10O4(Qui) − H]−, 417.1 [MNa – Na − C24H37O3(Agl) − C6H10O5(Glc) − C6H10O4(Qui) − C5H8O4(Xyl) − H]−, 241.0 [MNa − Na − C24H37O3(Agl) − C6H10O5(Glc) − C6H10O4(Qui) − C5H8O4(Xyl) − C7H12O5(MeGlc) − H]−, corroborating the structure of kuriloside A3 (1).
All these data indicate that kuriloside A3 (1) is 3β-O-{β-d-glucopyranosyl-(1→4)-β-d-quinovopyranosyl-(1→2)-[3-O-methyl-β-d-glucopyranosyl-(1→3)-6-O-sodium sulfate-β-d-glucopyranosyl-(1→4)]-β-d-xylopyranosyl}-22,23,24,25,26,27-hexa-nor-16α-hydroxy,20-oxo-lanost-9(11)-ene.
The molecular formula of kuriloside D
1 (
2) was determined to be C
66H
107O
36SNa from the [M
Na − Na]
− ion peak at
m/z 1507.6291 (calc. 1507.6268) in the (−)HR-ESI-MS. The hexasaccharide monosulfated carbohydrate chain of
2 was identical to that of previously reported kuriloside D [
19] since their
1H and
13C NMR spectra corresponding to the carbohydrate moieties were coincident (
Table S3). Actually, six signals of anomeric doublets at δ
H = 4.70–5.28 (d,
J = 7.5–8.2 Hz) and corresponding signals of anomeric carbons at δ
C = 103.7–105.7 indicated the presence of a hexasaccharide chain in kuriloside D
1 (
2). The presence of xylose (Xyl1), quinovose (Qui2), three glucose (Glc3, Glc4, Glc5), and 3-O-methylglucose (MeGlc6) residues were deduced from the analysis of the
1H,
1H-COSY, HSQC, and 1D TOCSY spectra of
2. The positions of the interglycosidic linkages were elucidated based on the ROESY and HMBC correlations (
Table S3). The presence in the
13C NMR spectrum of kuriloside D
1 (
2) of the only signal of the
O-methyl group at δ
C 60.5 and the upfield shift of the signal of C-3 Glc4 to δ
C 71.5 indicated the presence of a non-methylated terminal Glc4 residue. Analysis of the
1H and
13C NMR spectra of the aglycone part of
2 indicated the presence of a lanostane aglycone (the signals of lactone ring are absent and the signals of methyl group C-18 are observed at δ
C 16.9 and δ
H 1.30 (s, H-18) with normal side chain (30 carbons) and 9(11)-double bond (the signals at δ
C 149.0 (C-9), 114.9 (C-11), and δ
H 5.35 (brd,
J = 6.2 Hz; H-11) (
Table 1). The comparison of the
13C NMR spectra of
2 and kuriloside D showed their great similarity, except for the signals of the side chain from C-23 to C-27. Two strongly deshielded signals at δ
C 216.3 (C-16) and 217.6 (C-22) corresponded to carbonyl groups, whose positions were established on the base of the HMBC correlations H-15/C-16, H-21/C-22, H-23/C-22, and H-24/C-22. The signals of protons assigned to the methylene group adjacent to 22-oxo group were deshielded to δ
H 3.67 (dd,
J = 10.6; 18.2 Hz; H-23a) and 3.43 (dt,
J = 7.8; 18.2 Hz; H-23b) and correlated in the
1H,
1H-COSY spectrum of
2 with one signal only at δ
H 2.27 (t,
J = 7.8 Hz; H-24). These data, along with the deshielded signal of quaternary carbon at δ
C 69.0 (C-25) and the almost coinciding signals of methyl groups C-26 and C-27 (δ
C 30.0 and 29.5, δ
H 1.42 and 1.41, correspondingly), indicated the attachment of the hydroxy-group to C-25. Therefore, the side chain of kuriloside D
1 (
2) is characterized by the 22-oxo-25-hydroxy-fragment (
Table 1).
The (−)ESI-MS/MS of 2 demonstrated the fragmentation of [MNa − Na]− ion at m/z 1507.6. The peaks of fragment ions were observed at m/z 1349.5 [MNa − Na − C8H15O3 + H]−, corresponding to the loss of the aglycone fragment from C(20) to C(27), 1187.5 [MNa − Na − C8H15O3 − C6H10O5(Glc) + H]−, 1025.4 [MNa − Na − C8H15O3 − C6H10O5(Glc) − C6H10O5 (Glc) + H]−, 879.4 [MNa − Na − C8H15O3 − C6H10O5(Glc) − C6H10O5(Glc) − C6H10O4(Qui) + H]−, 565.1 [MNa − Na − C30H47O4(Agl) − C6H10O5(Glc) − C6H10O5(Glc) − C6H10O4(Qui) − H]−, 417.1 [MNa − Na − C30H47O5(Agl) − C6H10O5(Glc) − C6H10O5(Glc) − C6H10O4(Qui) − C5H8O4(Xyl) − H]−, 241.0 [MNa − Na − C30H47O5(Agl) − C6H10O5(Glc) − C6H10O5(Glc) − C6H10O4(Qui) − C5H8O4(Xyl) − C7H12O5(MeGlc) − H]−, corroborating the structure of kuriloside D1 (2).
All these data indicate that kuriloside D1 (2) is 3β-O-{β-d-glucopyranosyl-(1→3)-β-d-glucopyranosyl-(1→4)-β-d-quinovopyranosyl-(1→2)-[3-O-methyl-β-d-glucopyranosyl-(1→3)-6-O-sodium sulfate-β-d-glucopyranosyl-(1→4)]-β-d-xylopyranosyl}-16,22-dioxo-25-hydroxylanost-9(11)-ene.
The molecular formula of kuriloside G (
3) was determined to be C
61H
98O
37S
2Na
2 from the [M
2Na − Na]
− ion peak at
m/z 1509.5102 (calc. 1509.5132) and the [M
2Na − 2Na]
2− ion-peak at
m/z 743.2624 (calc. 743.2626) in the (−)HR-ESI-MS. In the
1H and
13C NMR spectra of the carbohydrate part of kuriloside G (
3), six characteristic doublets at δ
H 4.65–5.19 (
J = 7.0–8.1 Hz) and signals of anomeric carbons at δ
C 102.1–104.8, correlated with each anomeric proton by the HSQC spectrum, were indicative of a hexasaccharide chain and
β-configurations of glycosidic bonds (
Table 2). The signals of each monosaccharide unit were found as an isolated spin system based on the
1H,
1H-COSY, and 1D TOCSY spectra of
3. Further analysis of the HSQC and ROESY spectra resulted in the assigning of the monosaccharide residues as one xylose (Xyl1), one quinovose (Qui2), two glucoses (Glc3 and Glc5), and two 3-
O-methylglucose (MeGlc4 and MeGlc6) residues.
The positions of interglycosidic linkages were established by the ROESY and HMBC spectra of
3 (
Table 2) where the cross-peaks between H-1 Xyl1 and H-3 (C-3) of an aglycone, H-1 Qui2 and H-2 (C-2) Xyl1; H-1 Glc3 and H-4 (C-4) Qui2; H-1 MeGlc4 and H-3 Glc3; H-1 Glc5 and H-4 Xyl1; H-1 MeGlc6 and H-3 (C-3) Glc5 were observed.
The signals of C-6 MeGlc4 and C-6 Glc5 in the
13C NMR spectrum of
3 were observed at δ
C 67.0 and δ
C 67.1, correspondingly, due to α-shifting effects of the sulfate groups at these positions. Thus, the hexasaccharide disulfated chain of kuriloside G (
3) was first found in the sea cucumber glycosides. The NMR spectra of the aglycone part of
3 coincided with that of kuriloside A
3 (
1), indicating the identity of these aglycones (
Table S2).
The (−)ESI-MS/MS of 3 demonstrated the fragmentation of [M2Na − Na]− ion at m/z 1509.5. The peaks of fragment ions were observed at m/z 1389.6 [M2Na – Na − NaHSO4]−, 1333.5 [M2Na – Na − C7H12O5(MeGlc)]−, 1231.5 [M2Na – Na − C7H11O8SNa(MeGlcSO3Na)]−, 1069.4 [M2Na – Na − C7H11O8SNa(MeGlcSO3Na) − C6H10O5(Glc)]−, 923.4 [M2Na – Na − C7H11O8SNa(MeGlcSO3Na) − C6H10O5(Glc)] − C6H10O4(Qui)]−.
All these data indicate that kuriloside G (3) is 3β-O-{6-O-sodium sulfate-3-O-methyl-β-d-glucopyranosyl-(1→3)-β-d-glucopyranosyl-(1→4)-β-d-quinovopyranosyl-(1→2)-[3-O-methyl-β-d-glucopyranosyl-(1→3)-6-O-sodium sulfate-β-d-glucopyranosyl-(1→4)]-β-d-xylopyranosyl}-22,23,24,25,26,27-hexa-nor-16α-hydroxy,20-oxo-lanost-9(11)-ene.
The molecular formula of kuriloside H (4) was determined to be C64H101O42S3Na3 from the [M3Na − Na]− ion peak at m/z 1683.4701 (calc. 1683.4730), [M3Na − 2Na]2− ion peak at m/z 830.2425 (calc. 830.2419), and [M3Na − 3Na]3− ion peak at m/z 545.8332 (calc. 545.8315) in the (−)HR-ESI-MS. The presence of three-charged ions in the (−)HR-ESI-MS of kuriloside H (4) was indicative for the trisulfated glycoside.
The
1H and
13C NMR spectra corresponding to the carbohydrate chain of kuriloside H (
4) (
Table 3) demonstrated six signals of anomeric protons at δ
H 4.63–5.21 (d,
J = 7.1–8.6 Hz) and the signals of anomeric carbons at δ
C 102.8–104.7 deduced by the HSQC spectrum, indicative of hexasaccharide moiety with
β-glycosidic bonds. The signals of each sugar residue were assigned by the analysis of the
1H,
1H-COSY, 1D TOCSY, ROESY, and HSQC spectra, enabling the identification of monosaccharide units in the chain of
4 as one xylose (Xyl1), one quinovose (Qui2), three glucoses (Glc3, Glc4 and Glc5), and one 3-
O-methylglucose (MeGlc6). Therefore, the monosaccharide composition of
4 was the same as in kuriloside D
1 (
2).
However, in the
13C NMR spectrum of
4 three signals at δ
C 67.6 (C-6 Glc3), 67.4 (C-6 Glc5), and 67.0 (C-6 MeGlc6), characteristic for sulfated by C-6 hexose units, were observed instead of one signal at δ
C 67.0 (C-6 Glc5) in the spectrum of
2. The signal of the OMe-group observed at δ
C 60.4 indicated one terminal monosaccharide residue was methylated. Actually, the protons of the OMe-group (δ
H 3.75, s) correlated in the HMBC spectrum with C-3 MeGlc6 (δ
C 86.1), which was, in turn, attached to C-3 Glc5 (ROE-correlation H-1 MeGlc6 (δ
H 5.13 (d,
J = 7.4 Hz)/H-3 Glc5 (δ
H 4.13 (t,
J = 8.6 Hz)). At the same time, the fourth (another terminal) monosaccharide unit was glucose (the signal of C-3 Glc4 was shielded to δ
C 77.7 due to the absence of
O-methylation). The positions of all interglycosidic linkages were elucidated based on the ROESY and HMBC correlations (
Table 3).
Hence, kuriloside H (
4) has a hexasaccharide chain with a non-methylated terminal Glc4 residue and three sulfate groups. This carbohydrate chain is first found in the glycosides of the sea cucumbers and kuriloside H (
4) is the most polar glycoside discovered so far as well as two tetrasulfated pentaosides isolated from
Psolus fabricii [
20].
The analysis of the
13C NMR spectrum of the aglycone part of
4 demonstrated its identity to the aglycone of kurilosides A
1 and C
1, isolated earlier [
19]. Therefore, kuriloside H (
4) contains a 22,23,24,25,26,27-hexa-
nor-lanostane aglycone with 9(11)-double bond and acetoxy-groups at C-16 and C-20.
β-orientation of the acetoxy group at C-16 and (20
S)-configuration were established on the base of coincidence of the coupling constants (
J16/17 = 7.7 Hz and
J17/20 = 10.6 Hz), observed in the
1H NMR spectra of
4 and kuriloside A
1, and confirmed by the ROE-correlation H-16/H-32 in the spectrum of
4 (
Table S4).
The (−)ESI-MS/MS of kuriloside H (4) demonstrated the fragmentation of the [M3Na − Na]− ion at m/z 1683.5. The peaks of fragment ions were observed at m/z 1503.5 [M3Na – Na − CH3COOH − NaHSO4]−, 1443.5 [M3Na – Na − 2CH3COOH − NaHSO4]−, 1281.4 [M3Na – Na − 2CH3COOH − NaHSO4 − C6H10O5(Glc)]−, 1165.4 [M3Na – Na − 2CH3COOH − NaHSO4 − C7H11O8SNa(MeGlcOSO3)]−, and 1003.4 [M3Na – Na − 2CH3COOH − NaHSO4 − C7H11O8SNa(MeGlcOSO3) − C6H10O5(Glc)]−, corroborating its carbohydrate chain structure.
All these data indicate that kuriloside H (4) is 3β-O-{β-d-glucopyranosyl-(1→3)-6-O-sodium sulfate-β-d-glucopyranosyl-(1→4)-β-d-quinovopyranosyl-(1→2)-[6-O-sodium sulfate-3-O-methyl-β-d-glucopyranosyl-(1→3)-6-O-sodium sulfate-β-d-glucopyranosyl-(1→4)]-β-d-xylopyranosyl}-22,23,24,25,26,27-hexa-nor-16β,(20S)-diacetoxy-lanost-9(11)-ene.
The molecular formula of kuriloside I (
5) was determined to be C
54H
87O
35S
3Na
3 from the [M
3Na − Na]
− ion peak at
m/z 1437.3952 (calc. 1437.3991), [M
3Na − 2Na]
2− ion peak at
m/z 707.2049 (calc. 707.2049), and [M
3Na − 3Na]
3− ion peak at
m/z 463.8076 (calc. 463.8069) in the (−)HR-ESI-MS, indicating the presence of three sulfate groups. The
1H and
13C NMR spectra corresponding to the carbohydrate part of kuriloside I (
5) (
Table 4) demonstrated five characteristic doublets at δ
H 4.63–5.13 (d,
J = 6.6–7.8 Hz) and corresponding signals of anomeric carbons at δ
C 102.4–104.7 deduced by the HSQC spectrum, which indicated the presence of five monosaccharide residues in the carbohydrate chain of
5. The signals at δ
C 67.0, 67.6, and 67.7 indicated the presence of three sulfate groups as in the carbohydrate chain of kuriloside H (
4). Indeed, the comparison of the
13C NMR spectra of kurilosides I (
5) and H (
4) showed that they differed by the absence in the spectrum of
5 of the signals corresponding to non-sulfated terminal glucose residue attached to C-3 Glc3 in the carbohydrate chain of
4. The signal of C-3 Glc3 in the
13C NMR spectrum of
5 was observed at δ
C 76.9 (instead of δ
C 86.3 in the spectrum of
4), demonstrating the absence of a glycosylation effect. The presence of xylose (Xyl1), quinovose (Qui2), two glucose (Glc3, Glc4), and one 3-
O-methylglucose (MeGlc5) residue was deduced from the analysis of the
1H,
1H-COSY, HSQC and 1D TOCSY spectra of
5. The positions of interglycosidic linkages were elucidated based on the ROESY and HMBC correlations (
Table 4) and indicated the presence of the branched at the C-4 Xyl1 pentasaccharide chain in
5, with the same architecture as in the other pentaosides of
T. kurilensis. Thus, kuriloside I (
5) contains a new pentasaccharide branched trisulfated chain.
The analysis of the
13C and
1H NMR spectra of the aglycone part of
5 indicated the presence of 22,23,24,25,26,27-hexa-
nor-lanostane aglycone having a 9(11)-double bond (
Table 5). The signals of methine group CH-16 were observed at c δ
C 72.8 (C-16) and at δ
H 4.82 (dd,
J = 7.1; 14.9 Hz, H-16) due to the attachment of the hydroxyl group to this position. The HMBC correlations H-15/C-16 and H-20/C-16 confirmed this. The signals of C-20 and H-20 were shielded to δ
C 66.5 and δ
H 4.38 (dd,
J = 6.0; 9.5 Hz), correspondingly, when compared with the same signals in the spectra of kuriloside H (
4) (δ
C-20 69.4, δ
H-20 5.46 (dd,
J = 6.1; 10.6 Hz)), containing (20S)-acetoxy-group. Hence, it was supposed that the attachment of the hydroxyl group to C-20 was in the aglycone of kuriloside I (
5) instead of the acetoxy group in the aglycone of kuriloside H (
4).
The ROE-correlations H-16/H-17 and H-16/H-32 indicated a 16
β-OH orientation in the aglycone of kuriloside I (
5). (20
S)-configuration in
5 was determined on the base of the closeness of the coupling constant
J20/17 = 9.5 Hz to those in the spectra of kurilosides A
1, C
1 [
19], and H (
4) and corroborated by the observed ROE-correlations H-17/H-21, H-20/H-18 and biogenetic background. Hence, kuriloside I (
5) has an aglycone with a 16
β,(20
S)-dihydroxy-fragment that is unique in marine glycosides.
The (−)ESI-MS/MS of kuriloside I (5) demonstrated the fragmentation of the [M3Na − Na]− ion at m/z 1437.5. The peaks of fragment ions were observed at m/z 1317.4 [M3Na – Na − NaHSO4]−, 1197.4 [M3Na – Na − 2NaHSO4]−, 1173.4 [M3Na − Na − C6H9O8SNa(GlcOSO3)]−, 1039.4 [M3Na – Na − NaHSO4 − C7H11O8SNa(MeGlcOSO3)]−, 1027.3 [M3Na – Na − C6H9O8SNa(GlcOSO3) − C6H10O4(Qui)]−, 907.3 [M3Na – Na − NaHSO4 − C6H9O8SNa(GlcOSO3) − C6H10O4(Qui)]−, 895.4 [M3Na – Na − C6H9O8SNa(GlcOSO3) − C7H11O8SNa(MeGlcOSO3)]−, 667.4 [M3Na – Na − C24H39O2(Agl) − C6H9O8SNa(GlcOSO3) − C6H10O4(Qui) − H]−, 519.0 [M3Na – Na − C24H39O3(Agl) − C6H9O8SNa(GlcOSO3) − C6H10O4(Qui) − C5H8O4(Xyl) − H]−, and 417.1 [M3Na – Na − C24H39O3(Agl) − C6H9O8SNa(GlcOSO3) − C6H10O4(Qui) − C5H8O4(Xyl) − NaHSO3]−, corroborating the structure of the glycoside.
All these data indicate that kuriloside I (5) is 3β-O-{6-O-sodium sulfate-β-d-glucopyranosyl-(1→4)-β-d-quinovopyranosyl-(1→2)-[6-O-sodium sulfate-3-O-methyl-β-d-glucopyranosyl-(1→3)-6-O-sodium sulfate-β-d-glucopyranosyl-(1→4)]-β-d-xylopyranosyl}-22,23,24,25,26,27-hexa-nor-16β,(20S)-dihydroxy-lanost-9(11)-ene.
The molecular formula of kuriloside I
1 (
6) was determined to be C
58H
91O
37S
3Na
3 from the [M
3Na − 2Na]
2− ion peak at
m/z 749.2148 (calc. 747.2155) and [M
3Na − 3Na]
3− ion peak at
m/z 491.8146 (calc. 491.8139) in the (−)HR-ESI-MS. Kuriloside I
1 (
6) as well as kuriloside I (
5) belong to one group because they have identical trisulfated pentasaccharide chains and, therefore, parts of the
1H and
13C NMR spectra corresponding to the carbohydrate chains are coincident (
Table 4). 22,23,24,25,26,27-hexa-
nor-lanostane aglycone of kuriloside I
1 (
6) is identical to that of kurilosides H (
4), A
1 and C
1 [
19] (
Table S4) and characterized by the presence of 16
β,(20
S)-diacetoxy-fragment.
The (−)ESI-MS/MS of 6 demonstrated the fragmentation of the [M3Na − Na]− ion at m/z 1521.4 and [M3Na − 2Na]2− ion at m/z 749.2. The peaks of fragment ions were observed at m/z: 1281.4 [M3Na – Na − 2CH3COOH − NaHSO4]−, 1197.4 [M3Na – Na − CH3COOH − C6H9O8SNa(GlcOSO3)]−, 1137.4 [M3Na – Na − 2CH3COOH − C6H9O8SNa(GlcOSO3)]−, 859.4 [M3Na – Na − 2CH3COOH − C6H9O8SNa(GlcOSO3) − C7H11O8SNa(MeGlcOSO3)]−, 719.2 [M3Na − 2Na − CH3COOH]2−, 629.2 [M3Na − 2Na − NaHSO4]2−, and 557.2 [M3Na − 2Na − 2CH3COOH − C6H9O8SNa(GlcOSO3)]2−, which confirmed its structure, established by the NMR data.
All these data indicate that kuriloside I1 (6) is 3β-O-{6-O-sodium sulfate-β-d-glucopyranosyl-(1→4)-β-d-quinovopyranosyl-(1→2)-[6-O-sodium sulfate-3-O-methyl-β-d-glucopyranosyl-(1→3)-6-O-sodium sulfate-β-d-glucopyranosyl-(1→4)]-β-d-xylopyranosyl}-22,23,24,25,26,27-hexa-nor-16β,(20S)-diacetoxy-lanost-9(11)-ene.
The molecular formula of kuriloside J (
7) was determined to be C
56H
90O
33S
2Na
2 from the [M
2Na−Na]
− ion peak at
m/z 1377.4687 (calc. 1377.4709) and [M
2Na−2Na]
2− ion peak at
m/z 677.2413 (calc. 677.2408) in the (−)HR-ESI-MS. In the
1H and
13C NMR spectra of the carbohydrate part of kuriloside J (
7) (
Table 6), five signals of anomeric protons at δ
H 4.65–5.12 (d,
J = 7.2–7.9 Hz) and corresponding five signals of anomeric carbons at δ
C 102.0–104.7, deduced by the HSQC spectrum, were observed, which indicated the presence of a pentasaccharide chain similar to compounds
5 and
6. Actually, the comparison of the
13C NMR spectra of sugar parts of kurilosides I (
5) and J (
7) revealed the closeness of the signals of four monosaccharide residues, except the signals of the third unit, attached to C-4 Qui2. The analysis of the signals of this residue in the
1H,
1H-COSY, HSQC, 1D TOCSY, and ROESY spectra of kuriloside J (
7) showed that it is a glucose without a sulfate group (δ
C-6 Glc3 61.8, δ
C-5 Glc3 77.7), while in the carbohydrate chain of
5, this residue is sulfated. The other sulfate groups occupy the same positions at C-6 Glc4 (δ
C-6 Glc4 67.1, δ
C-5 Glc4 75.1) and at C-6 MeGlc5 (δ
C-6 MeGlc5 66.7, δ
C-5 MeGlc5 75.5) as in the sugar chains of kurilosides I (
5) and I
1 (
6). The positions of interglycosidic linkages in the carbohydrate chain of
7, elucidated by the ROESY and HMBC correlations (
Table 6), were the same as in kurilosides of groups A [
19] and I. Thus, kuriloside J (
7) is a branched disulfated pentaoside with the sulfate groups bonding to C-6 Glc4 and C-6 MeGlc5 in the upper semi-chain.
The analysis of the
1H and
13C NMR spectra of the aglycone part of kuriloside J (
7) (
Table 7) revealed the presence of the hexa-
nor-lanostane aglycone having a 9(11)-double bond, similar to the majority of the other glycosides of
T. kurilensis [
19]. The signals at δ
C 171.2 and 21.1 were characteristic for the acetoxy group, bonded to C-16, that was deduced from the characteristic δ
C 75.1 value of C-16 and the ROE-correlation between the signal of
O-acetyl methyl group (δ
H 2.17 (s)) and H-16 (δ
H 5.76 (m). Actually, in the spectrum of
7, the signal of C-16 was deshielded by 2.3 ppm due to the presence of the acetoxy-group when compared with the corresponding signal in the spectrum of kuriloside I (
5), having a 16-hydroxy-group. The presence of hydroxyl group at C-20 was deduced from the characteristic signals at δ
C 64.8 (C-20) and δ
H 4.28 (dd,
J = 6.4; 10.0 Hz, H-20). Hence, the hydroxyl group is attached to C-20 in the aglycones of kuriloside I (
5) and J (
7). The ROE-correlation H-16/H-32 indicated 16
β-O-Ac orientation in the aglycone of kuriloside J (
7), which was confirmed by the coupling constant
J16/17 = 7.9 Hz, indicating both protons, H-16 and H-17, to be α [
21]. (20
S)-configuration in
7 was corroborated by the coupling constant
J17/20 = 10.0 Hz and the ROE-correlations H-17/H-21, H-20/H-18. Hence, kuriloside J (
7) is characterized by the new hexa-
nor-lanostane aglycone with a 16
β-acetoxy,(20
S)-hydroxy-fragment.
The (−)ESI-MS/MS of kuriloside J (7) demonstrated the fragmentation of [M2Na − Na]− ion at m/z 1377.5. The peaks of fragment ions were observed at m/z 1317.4 [M2Na – Na − CH3COOH]−, 1257.4 [M2Na – Na − NaHSO4]−, 1197.5 [M2Na – Na − CH3COOH − NaHSO4]−, 1155.4 [M2Na – Na − CH3COOH − C6H10O5 (Glc)]−, 1039.4 [M2Na – Na − CH3COOH − C7H11O8SNa(MeGlcOSO3)]−, 1009.4 [M2Na – Na − CH3COOH − C6H10O5(Glc) − C6H10O4(Qui)]−, 889.4 [M2Na − Na − NaHSO4 − CH3COOH − C6H10O5(Glc) − C6H10O4(Qui)]−, 667.4 [M2Na – Na − C26H41O3(Agl) − C6H10O5(Glc) − C6H10O4(Qui) − H]−, 519.0 [M2Na – Na − C26H41O3(Agl) − C6H10O5(Glc) − C6H10O4(Qui) − C5H8O4(Xyl) − H]−, 417.1 [M2Na – Na − C26H41O3(Agl) − C6H10O5(Glc) − C6H10O4(Qui) − C5H8O4(Xyl) − NaHSO3]−, corroborating the structure of its aglycone and the carbohydrate chain.
All these data indicate that kuriloside J (7) is 3β-O-{β-d-glucopyranosyl-(1→4)-β-d-quinovopyranosyl-(1→2)-[6-O-sodium sulfate-3-O-methyl-β-d-glucopyranosyl-(1→3)-6-O-sodium sulfate-β-d-glucopyranosyl-(1→4)]-β-d-xylopyranosyl}-22,23,24,25,26,27-hexa-nor-16β-acetoxy,(20S)-hydroxy-lanost-9(11)-ene.
The molecular formula of kuriloside K (
8) was determined to be C
54H
88O
32S
2Na
2 from the [M
2Na − Na]
− ion peak at
m/z 1335.4573 (calc. 1335.4603) and the [M
2Na − 2Na]
2− ion peak at
m/z 656.2357 (calc. 656.2356) in the (−)HR-ESI-MS. In the
1H and
13C NMR spectra of the carbohydrate part of kuriloside K (
8) (
Table 8), five signals of anomeric protons at δ
H 4.62–5.19 (d,
J = 6.5–8.5 Hz) and five signals of anomeric carbons at δ
C 102.7–104.8, deduced by the HSQC spectrum, were indicative for the pentasaccharide chain with the
β-configuration of glycosidic bonds. The comparison of the
13C NMR spectra of oligosaccharide parts of trisulfated kuriloside I (
5) and kuriloside K (
8) revealed the coincidence of the monosaccharide residues, except for the signals of a terminal, 3-
O-methylglucose (MeGlc5) unit. The analysis of the signals of this residue in the
1H,
1H-COSY, HSQC, 1D TOCSY, and ROESY spectra of kuriloside K (
8) showed the absence of a sulfate group (δ
C-6 MeGlc5 61.6, δ
C-5 MeGlc5 77.5), in contrast with the carbohydrate chain of
5 (δ
C-6 MeGlc5 67.0, δ
C-5 MeGlc5 75.4). The positions of interglycosidic linkages in the carbohydrate chain of
8, deduced by the ROESY and HMBC correlations (
Table 8), showed that kuriloside K (
8) has branching at C-4 Xyl1 in the disulfated pentasaccharide chain with the sulfate groups at C-6 Glc3 and C-6 Glc4.
The NMR spectra as well as the ROE-correlations of the aglycone part of kuriloside K (
8) were coincident to that of kuriloside I (
5), indicating the presence of a 22,23,24,25,26,27-hexa-
nor-lanostane aglycone with 16β,(20
S)-dihydroxy-fragment (
Table 5).
The (−)ESI-MS/MS of 8 demonstrated the fragmentation of the [M2Na − Na]− ion at m/z 1335.4 resulted in the fragment ions observed at m/z: 1215.4 [M2Na – Na − NaHSO4]−, 1159.4 [M2Na – Na − C7H12O5(MeGlc)]−, 1071.4 [M2Na – Na − C6H9O8SNa(GlcOSO3)]−, 925.4 [M2Na – Na − C6H9O8SNa(GlcOSO3) − C6H10O4(Qui)]−, 895.4 [M2Na – Na − C6H9O8SNa(GlcOSO3) − C7H12O5(MeGlc)]−, 713.3 [M2Na – Na − C24H39O2(Agl) − C6H9O8SNa(GlcOSO3) − H]−, 417.1 [M2Na – Na − C24H39O3(Agl) − C6H9O8SNa(GlcOSO3) − C6H10O4(Qui) − C5H8O4(Xyl) − H]−, 241.0 [M2Na – Na − C24H39O3(Agl) − C6H9O8SNa(GlcOSO3) − C6H10O4(Qui) − C5H8O4(Xyl) − C7H12O5(MeGlc) − H]−, which confirmed the chemical structure established by the NMR data.
All these data indicate that kuriloside K (8) is 3β-O-{6-O-sodium sulfate-β-d-glucopyranosyl-(1→4)-β-d-quinovopyranosyl-(1→2)-[3-O-methyl-β-d-glucopyranosyl-(1→3)-6-O-sodium sulfate-β-d-glucopyranosyl-(1→4)]-β-d-xylopyranosyl}-22,23,24,25,26,27-hexa-nor-16β,(20S)-dihydroxy-lanost-9(11)-ene.
The molecular formula of kuriloside K
1 (
9) was determined to be C
56H
90O
33S
2Na
2 from the [M
2Na − Na]
− ion peak at
m/z 1377.4723 (calc. 1377.4709) and the [M
2Na − 2Na]
2− ion peak at
m/z 677.2426 (calc. 677.2408) in the (−)HR-ESI-MS. The comparison of the
1H and
13C NMR spectra of the carbohydrate chains of kuriloside K
1 (
9) and kuriloside K (
8) demonstrated their coincidence (
Table 8) due to the presence of the same pentasaccharide, branched by C-4 Xyl1, sugar parts with the sulfate groups at C-6 Glc3 and C-6 Glc4. The analysis of the NMR spectra of the aglycone part of
9 indicated the presence of 22,23,24,25,26,27-hexa-
nor-lanostane aglycone with 16
β-acetoxy,(20
S)-hydroxy-fragment (
Table 7), identical to that of kuriloside J (
7). Hence, kuriloside K
1 (
9) is an isomer of kuriloside J (
7) by the position of one of the sulfate groups, that was confirmed by the presence of the ion-peaks having coincident
m/z values in their (−)ESI-MS/MS spectra.
The (−)ESI-MS/MS of 9 demonstrated the fragmentation of [M2Na − Na]− ion at m/z 1377.5. The peaks of fragment ions were observed at m/z 1317.4 [M2Na – Na − CH3COOH]−, 1197.5 [M2Na – Na − CH3COOH − NaHSO4]−, 1069.5 [M2Na – Na − C6H10O5(Glc)]−, 1053.4 [M2Na – Na − CH3COOH − C6H9O8SNa(GlcOSO3)]−, 877.4 [M2Na – Na − CH3COOH − C6H9O8SNa(GlcOSO3) − C7H12O5(MeGlc)]−, 731.3 [M2Na – Na − CH3COOH − C6H9O8SNa(GlcOSO3) − C7H12O5(MeGlc) − C6H10O4(Qui)]−, 565.1 [M2Na – Na − C26H41O3(Agl) − C6H9O8SNa(GlcOSO3) − C6H10O4(Qui) − H]−, 417.1 [M2Na – Na − C26H41O4(Agl) − C6H9O8SNa(GlcOSO3) − C6H10O4(Qui) − C5H8O4(Xyl)]−.
All these data indicate that kuriloside K1 (9) is 3β-O-{6-O-sodium sulfate-β-d-glucopyranosyl-(1→4)-β-d-quinovopyranosyl-(1→2)-[3-O-methyl-β-d-glucopyranosyl-(1→3)-6-O-sodium sulfate-β-d-glucopyranosyl-(1→4)]-β-d-xylopyranosyl}-22,23,24,25,26,27-hexa-nor-16β-acetoxy,(20S)-hydroxy-lanost-9(11)-ene.
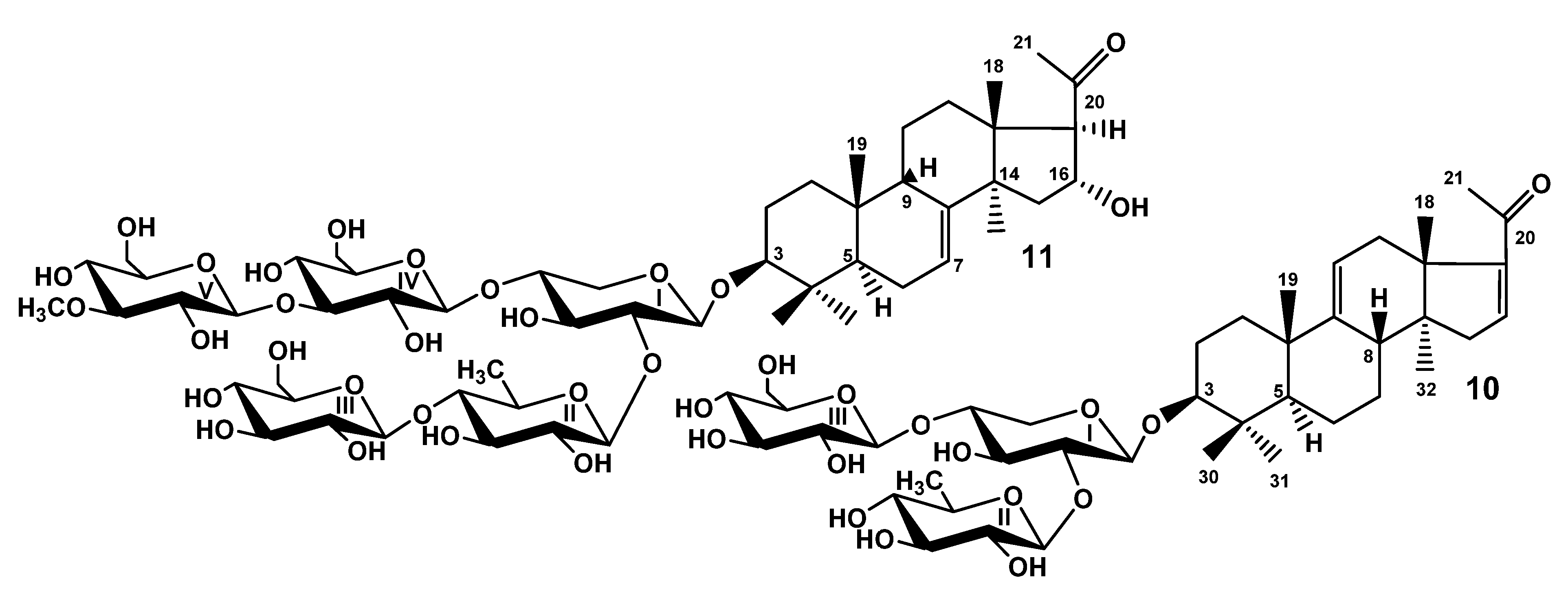
When the studies on the glycosides of
T. kurilensis were started [
22], the complexity of glycosidic mixture became obvious. Therefore, the part of the glycosidic sum was subjected to solvolytic desulfation to facilitate the chromatographic separation and isolation of the glycosides. However, the obtained fraction of desulfated glycosides was separated only recently as part of the effort to discover some minor glycosides possessing interesting structural peculiarities. As a result, the compounds
10 and
11 were isolated (
Figure 2). Their structures were elucidated by thorough analysis of 1D and 2D NMR spectra, similar to the natural compounds
1–
9 and confirmed by the HR-ESI-MS.
The molecular formula of DS-kuriloside L (
10) was determined to be C
41H
64O
15 from the [M − H]
− ion peak at
m/z 795.4169 (calc. 795.4172) in the (−)HR-ESI-MS. Compound
10 has a trisaccharide sugar chain (for NMR data see
Tables S5 and S6, for original spectra see
Figures S69–S76) and a hexa-
nor-lanostane-type aglycone identical to that of kuriloside A
2 [
19].
The molecular formula of DS-kuriloside M (
11) was determined to be C
54H
88O
26 from the [M − H]
− ion peak at
m/z 1151.5469 (calc. 1151.5491) in the (−)HR-ESI-MS. DS-kuriloside M (
11), characterized by the 7(8)-double bond in the hexa-
nor-lanostane nucleus and pentasaccharide chain, differed from the chains of kurilosides of the groups A, I, J, and K by the absence of sulfate groups (see
Tables S7 and S8 for the NMR data,
Figures S77–S85 for the original spectra). Noticeably, all of the isolated kurilosides, with the exception of
11, contained a 9(11)-double bond in the polycyclic systems.



 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}