Abstract
Intracellular Ca2+ plays a pivotal role in the control of a large series of cell functions in all types of cells, from neurotransmitter release and muscle contraction to gene expression, cell proliferation and cell death. Ca2+ is transported through specific channels and transporters in the plasma membrane and subcellular organelles such as the endoplasmic reticulum and mitochondria. Therefore, dysregulation of intracellular Ca2+ homeostasis may lead to cell dysfunction and disease. Accordingly, chemical compounds from natural origin and/or synthesis targeting directly or indirectly these channels and proteins may be of interest for the treatment of cell dysfunction and disease. In this review, we show an overview of a group of marine drugs that, from the structural point of view, contain one or various heterocyclic units in their core structure, and from the biological side, they have a direct influence on the transport of calcium in the cell. The marine compounds covered in this review are divided into three groups, which correspond with their direct biological activity, such as compounds with a direct influence in the calcium channel, compounds with a direct effect on the cytoskeleton and drugs with an effect on cancer cell proliferation. For each target, we describe its bioactive properties and synthetic approaches. The wide variety of chemical structures compiled in this review and their significant medical properties may attract the attention of many different researchers.
1. Introduction
A variety of heterocyclic compounds modulate different biological functions. Here we will focus on heterocyclic compounds that affect intracellular calcium signal through their effect on ion channels, cytoskeleton and impact on tumor cell proliferation.
The divalent cation Ca2+ is a fundamental ion in cellular metabolism as well as participating in the construction and maintenance of bones and teeth. Furthermore, ionic intracellular Ca2+ is also critically involved in cell signaling, as this ion is considered one of the most important second messengers. Thus, intracellular Ca2+ ions modulate a large number of cell functions from exocytosis and muscle contraction to regulation of gene expression, cell proliferation and cell death [1,2]. Intracellular Ca2+ is also a particular second messenger because it is neither created nor destroyed inside the cell; instead, it is transported throughout the cell and intracellular membranes by means of channels and other specific transport systems that play an essential role by regulating its intracellular concentration. Ca2+ binds and modifies the activity of hundreds of Ca2+-binding proteins like, for instance, calmodulin that, in turn, modify the activity of another even larger series of critical enzymes and other proteins [3]. This is how it is involved in regulating muscle contraction, hormone secretion and neurotransmitter exocytosis, gene expression, cell mobility, apoptosis, fertilization or synaptic plasticity, among other functions [4,5]. It is also involved in the regulation of some kinases, such as protein kinase C, as the action of cofactor or other enzymes of the Krebs cycle, which entails regulation of cellular energy metabolism [6]. All of these processes occur as a result of variations in the cytosolic Ca2+ concentration that are characterized by its amplitude, duration, frequency and subcellular location [7]. The transport of Ca2+ in the cell is possible by an enormous electrochemical gradient. At rest, cytosolic Ca2+ is 50–100 nM, while outside of the cell, the concentration is between 1 and 2 mM. Within the cell, the main storage for Ca2+ is the endoplasmic reticulum with concentrations in the 1–2 mM level at rest. However, in other subcellular organelles such as mitochondria and the nucleus, the resting [Ca2+] is similar to the cytosol and can only be increased temporarily after stimulation [8]. Electrical gradients such as plasma membrane potential, negative inside the inner part of the plasma membrane, and the huge mitochondrial potential, also negative inside the mitochondrial matrix, play a pivotal role. They favor Ca2+ entry from outside to the cytosol as well as mitochondrial Ca2+ uptake from the cytosol, in this latter case, even in the absence of Ca2+ chemical gradients. However, in contrast to the endoplasmic reticulum (ER), mitochondria are not Ca2+ stores, they may take Ca2+ quickly, particularly from high Ca2+ concentration hotspots, but this Ca2+ returns slowly back to the cytosol [9].
Ion channels are transmembrane proteins involved in the passive transport of ions, both cations and anions, across biological membranes as the plasma membrane of eukaryotic cells or endomembranes that limit subcellular organelles such as the endoplasmic reticulum, the nuclear envelope or mitochondria, to cite the most important ones. Ion channels are important to cells because they modulate electric gradients required for life. In addition, ion channels that transport the divalent cation Ca2+ are critically involved in cell signaling. Therefore, many drugs from natural origin and/or chemical synthesis, including several marine heterocyclic compounds that target calcium signaling, are potentially useful for preventing and/or treating multiple diseases, including some of the most prevalent ones from hypertension to cancer and Alzheimer’s disease [10].
The cytoskeleton is a subcellular structure made of different proteins and other biological compounds that subserve important cell functions across cell lifespan from cell division to cell differentiation, cell migration and cell death. Specifically, cytoskeleton actin filaments play an important role in the modulation of ion channel function, including Ca2+ channels that are a key for exocytosis; as a consequence, actin filaments are essential in exocytosis [11]. Accordingly, chemical compounds targeting any of the proteins involved in the cytoskeleton may have a deep impact on the pathophysiology of the different cell types.
One of the most critical threats to human life is cancer. Cancer is, in most cases, the result of cell transformation secondary to mutations either inherited, spontaneous, /or induced by physical or chemical factors in genes involved in critical pathways of cell signaling related to cell proliferation or DNA repair. The number of signaling pathways and critical regulatory proteins is large but still limited, and changes in the activity of any of these proteins may have an impact on cell proliferation and cancer development. Ca2+ signaling contributes direct or indirectly to many of these tumor characteristics. Multiple studies suggest changes in the intracellular Ca2+ homeostasis across carcinogenesis; thus, Ca2+ entry is necessary for all tumor cell proliferation [12,13]. Hundreds of chemical compounds, including some heterocycles, have an impact on calcium-dependent cell proliferation and therefore have been considered as a drug for cancer treatment.
In this review, we have tried to cover marine drugs containing heterocycle subunits, which have a direct influence on intracellular calcium signal (Figure 1). Our aim is to provide a general overview of their biological properties, effects on the calcium channels and synthetic approaches to these compounds. The review is organized into three main sections, which are related to their principal biological properties: effects over calcium channels, effects over cytoskeleton and effects on tumor cell proliferation.
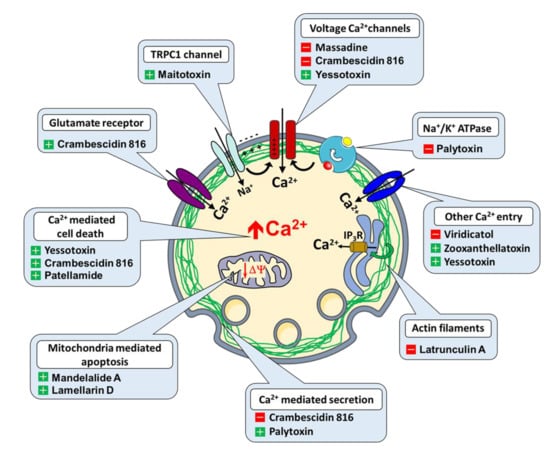
Figure 1.
Ion channels, transport proteins and Ca2+ regulated functions targeted by heterocycles (+ activator, − inhibitor).
2. Heterocyclic Marine Drugs with Effect over Calcium Channels
2.1. Viridicatol
3-hydroxy-4-(3-hydroxyphenyl)-1H-quinolin-2-one, also named viridicatol, is a quinoline alkaloid obtained from a deep-sea derived fungus of the genus Penicillium griseofulvum (Figure 2).
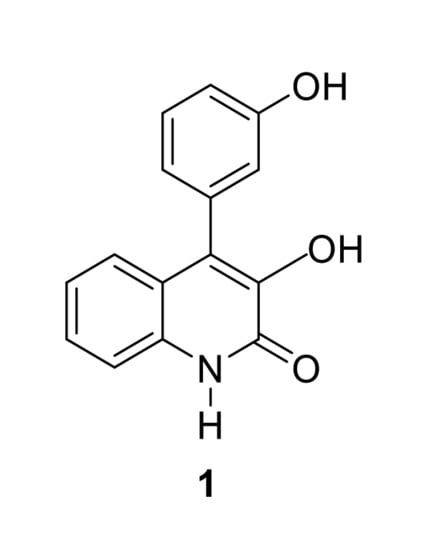
Figure 2.
Viridicatol.
Recent data suggest that, in mast cells, viridicatol may inhibit Ca2+ signaling induced by ionophores. As a consequence, viridicatol may prevent mast cell degranulation, which could block the activation of mast cells that are involved in the inflammatory response [14]. Evidence showed that viridicatol also abolishes intracellular Ca2+ increases in rat basophilic leukemia cells and mast cells isolated from mice treated with ovalbumin to induce food allergy, thus alleviating allergic symptoms. Accordingly, this compound may be considered to treat and/or prevent allergic reactions mediated by mast cell activation and degranulation.
Regarding its total synthesis, in 1964, Luckner and Mohammed [15] described an early approach to viridicatol 1 based on an unexpected regioselective ring expansion process in the reaction of isatin with α-aryldiazomethane (prepared by reaction of an aryl aldehyde with hydrazine and subsequent oxidation of the arylhydrazone intermediate with mercuric oxide). Later on, Kamal, Babu and coworkers reported an improved one-pot methodology for the synthesis of viridicatol [16]. In this protocol, N-tosylhydrazine adds to aldehyde 3 in the presence of a base (K2CO3) to generate in situ the α-aryldiazomethane, which then reacts with isatin 2 to obtain quinolinone viridicatol 1 (Scheme 1).
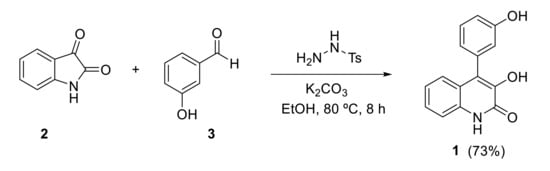
Scheme 1.
Synthesis of viridicatol by Kamal and Babu.
The discovery of the promising biological properties of this marine drug, reported in the late nineties, inspired the development of new synthetic approaches.
Kobayashi and Harayama reported a synthetic approach for viridicatol based on a Knoevenagel condensation of cyanoacetanilides with aldehydes, followed by an oxidative cyclization, in which the cyano substituent works as a leaving group [17]. Cyanoacetanilide 4 reacts with 3-hydroxybenzaldehyde in a one-pot Knoevenagel/epoxidation. Mild conditions using piperidine as catalyst were enough to promote the Knoevenagel condensation, and treatment with tBuOOK and KF allowed the epoxidation in the same pot. Intermediate epoxide 5 undergoes an epoxide-arene cyclization by treatment with sulfuric acid resulting in 3-hydroxy-4-arylquinolin-2(1H)-one 6. N-PMB derivative 6 is then transformed to the desired N-H deprotected quinolinone 1 when treated with trifluoroacetic acid (Scheme 2).
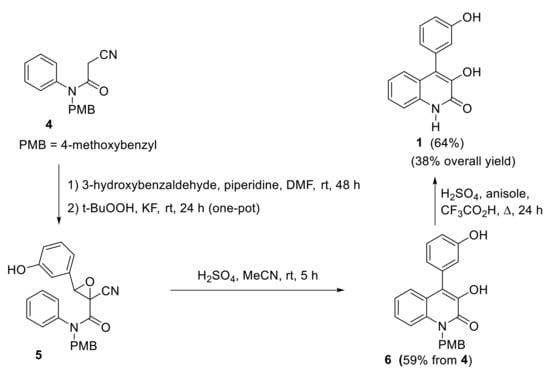
Scheme 2.
Kobayashi and Harayama’s synthesis of viridicatol.
The synthesis of viridicatol also has recently been reported by other groups. As an example, Mamedov and coworkers developed a two-step process to 3-hydroxy-4-arylquinolin-2(1H)-ones starting with a Darzens condensation followed by an intramolecular Friedel–Crafts alkylation [18]. Dichloroacetanilide 7 reacts with 3-nitrobenzaldehyde in a Darzens condensation to produce epoxide 8, which in an intramolecular Friedel–Crafts alkylation with acetic acid and sulfuric acid produces quinolinone 9. Three further steps are required to obtain the natural product. Viridicatol 1 was synthesized in five steps from dichloroacetanilide 7 with 56% overall yield (Scheme 3).
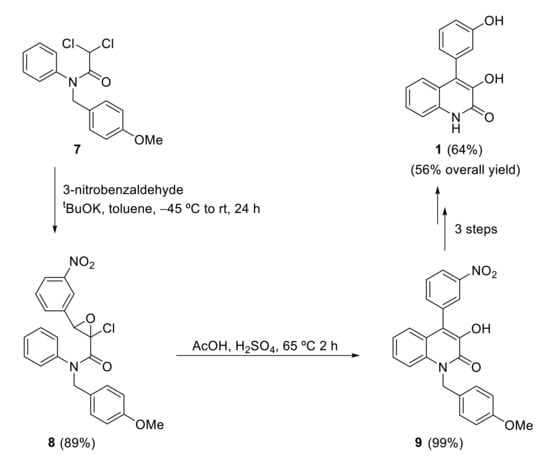
Scheme 3.
Synthesis of viridicatol by Mamedov’s group.
2.2. Zooxanthellatoxin-A
Zooxanthellatoxin-A (ZT-A) is a novel polyhydroxypropylene isolated from a symbiotic marine alga Simbiodinium sp. Evidence indicates that this compound may work as a novel calcium agonist, inducing Ca2+-dependent aggregation in platelets and Ca2+-dependent muscle contraction in vascular smooth muscle cells. The final effect of calcium rinse induced by ZT-A seems mediated by activation of protein tyrosine kinase followed by phospholipase C-gamma 2 and protein kinase C, thus leading to MAP kinase activation and the ensuing release of arachidonic acid finally converted to TXA2 eliciting platelet aggregation in rabbits [19]. This compound also induced aorta contraction, and this effect was entirely dependent on the presence of extracellular Ca2+, suggesting that ZT-A may promote Ca2+ entry into vascular smooth muscle cells, but the exact calcium entry channel activated by ZT-A remains unknown [20].
Its structure and absolute stereochemistry were determined by Nakamura and coworkers by synthesizing the fragments obtained during degradation experiments (Figure 3).
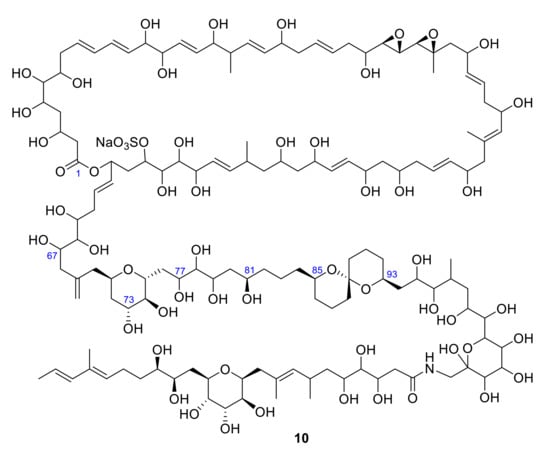
Figure 3.
Structure of zooxanthellatoxin A 10 (ZT-A).
Treatment of ZT-A 10 with an excess amount of NaIO4, followed by reduction with NaBH4, afforded fragments 11 and 12, which possess a 1,3-diepoxide and a sulfate ester as characteristic functionalities (Scheme 4). DQF-COSY, 1D and 2D TOCSY (called HOHAHA at the time), HMBC, and NOE NMR spectra provided enough information to resolve the structure (carbon segments, olefinic proton signals and all connectivities) [21].
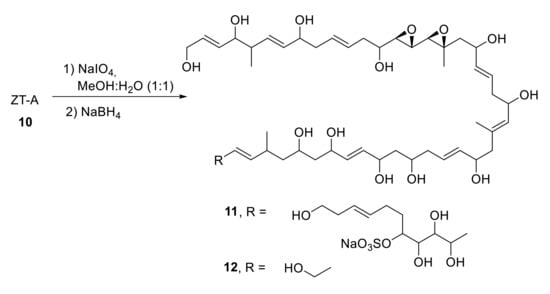
Scheme 4.
Degradation of ZT-A.
In 1996, spiroacetal alcohol peracetates 13 and 14 were obtained from ZT-A by oxidation with excess amounts of NaIO4, followed by reduction with NaBH4 and acetylation (Scheme 5) [22]. Their relative configuration was determined on the basis of NOE data and spin coupling constants.
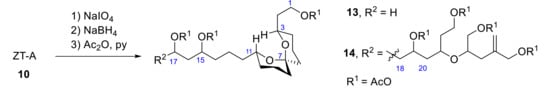
Scheme 5.
Obtention of spiroacetal alcohol peracetates 13 and 14 from ZT-A (10).
The synthesis of fragment 13 was achieved from advanced intermediate 15. Two deprotection steps, spiroacetalization in the presence of HCl-MeOH and final acetylation, provided a stable spiroacetal 16 (R = AcO) in 10% yield (Scheme 6). The 1H NMR spectra of 16 and its triol (17) was similar to 13 and its triol, so it was assumed that the relative configuration of the spiroacetal unit was identical. Thus, the absolute stereochemistry of the degradation product 13 was determined to be 3S, 11R and 15R. These carbons correspond to C93, C85, and C81 of zooxanthellatoxin A.
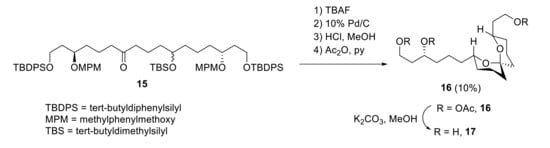
Scheme 6.
Synthesis of spiroacetal fragments 16 and 17 from intermediate 15.
In 1998, oxidation of ZT-A with limited amounts of NaIO4, followed by reduction with NaBH4 and acetylation, provided the tetrahydropyran 18 [23], which was further deacetylated and subjected to NaIO4 oxidation to give 19 (Figure 4). Once acyclic tetraol 19 was synthesized, the stereochemistry of this synthetic product was established by transformation into its corresponding (R)- and (S)-α-methoxy-α-trifluoromethylphenylacetic (MTPA) esters (20 and 21). The spectra of the corresponding (R)-MTPA synthetic ester 19 and (R)-MTPA ester of natural degradation product were identical. Thus, the absolute configuration was confirmed to be 71R,73R,74S,75R.
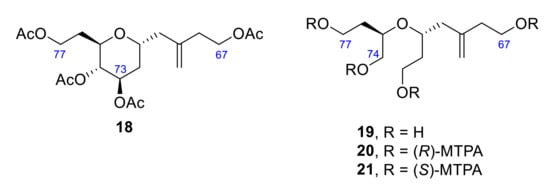
Figure 4.
Structure of tetrahydropyran 18 and its acyclic derivatives 19, 20 and 21.
To complete the structure assignment, just two fragments remained to be synthesized. The acid portion 22 is obtained by alkaline hydrolysis from the natural ZT-A. It was divided into two portions, 23 and 24, that had to be synthesized in order to confirm the relative stereochemistry of this last part of the molecule (Scheme 7).
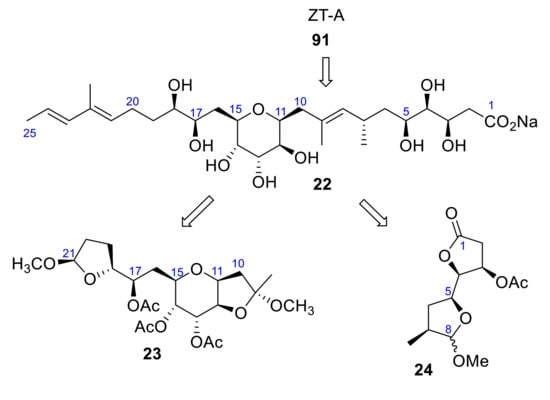
Scheme 7.
Structural fragments 23 and 24.
Fragment 23 was obtained from compound 25 in moderate yield after 12 steps (Scheme 8). By measuring the optical rotation and circular dichroism (CD) spectra, it was confirmed that the synthetic and the natural fragment were identical. Application of the Mosher method to dimethyl acetal derivative from 26 established the stereochemistry at C17. NOESY spectrum of 23 gave enough information to determine the configuration of C9 and C21 [24].
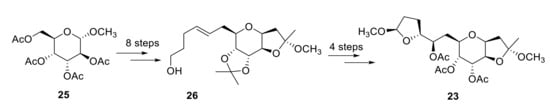
Scheme 8.
Synthesis of fragment 23.
To confirm the stereochemistry of the rest of the carbons and the terminal carboxylic acid portion (C1–C8) of 22, γ-lactones 24 were synthesized (Scheme 7). The similarity between the [α] D values suggested that both fragments, natural and synthetic, had the same configuration, 3R,4R,5S,7S,11S,12S,13R,14S,15R,17R,18R [25].
More recently, a similar class of compounds, the zooxanthellamides (ZADs), were isolated. The zooxanthellamide Cs have improved biological properties with respect to zooxanthellatoxin A, showing a higher vasoconstrictive activity. Structure–activity relationship studies showed that the enormous macrolactone moiety is important for the observed properties. Nakamura’s efforts to the synthesis of zooxanthellatoxin A could be applied to the similar structures of this zooxanthellamides [26].
2.3. Massadine
Massadine is another marine drug reported to modulate Ca2+-signaling. This compound is a highly oxygenated alkaloid obtained from Stylissa aff, a marine sponge that has been described as a blocker of the enzyme geranylgeranyltransferase type I (GGTase I) with an IC50 in the low μM range [27]. Massadine decreases Ca2+ entry mediated by voltage-dependent Ca2+ channels in neuron-like PC12 cells loaded with the calcium probe fura2. This effect, in contrast to those of other compounds, was reversible [28]. Massadine holds two critical structures: a lipophilic, brominated side-chain involved likely in the good membrane solubility, and a hydrophilic, amino-imidazole substructure, possibly involved in the interaction with the channel itself. As intracellular Ca2+ overload is involved in neurotoxicity, the interaction with voltage-dependent Ca2+ channels may account for the neurotoxicity of massadine.
This hexacyclic marine natural product, belonging to the family of pyrrole-imidazole alkaloids, contains a highly congested and stereochemically rich tetracyclic core. Due to its complexity, in the last decades, various researchers have published numerous articles describing the partial or total synthesis of massadine 27 and massadine chloride 28 (Figure 5).
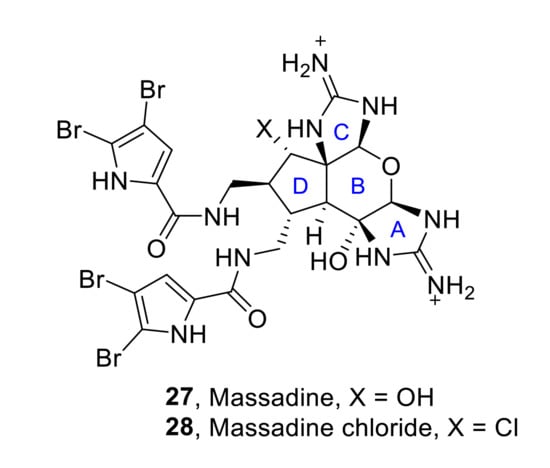
Figure 5.
Structures of massadine 27 and massadine chloride 28.
The first total synthesis of massadine 27 and massadine chloride 28 was reported by P. S. Baran et al. in 2008 [29,30]. Despite the constitutionally isomeric nature between massadine chloride and axinellamines, a rearrangement was not observed under any of the conditions tested. Thus, it was necessary to propose a different synthetic approach, which key intermediate is the spirocycle 29. Thus, starting from intermediate 29, the construction of the different heterocyclic units of massadines was achieved in sequential steps. First of all, the formation of the desired 2-aminoimidazole 30a (32%) (along with its hydroxy analog 30b (24%)) was performed by reaction of 29 with cyanamide. The subsequent formation of the tetrahydropyranyl ring was achieved by oxidation with dimethyldioxirane (DMDO) followed by acid-mediated closure with neat trifluoroacetic acid (TFA). Finally, reduction of the azide moieties, followed by reaction with bromopyrrole, afforded the final desired natural products, massadine 27 (H, OH = α) and massadine chloride 28 (H, OH = α), in 40% and 34% yield, respectively and their epimers (H, OH = β), although these epi-series have not been found in nature (Scheme 9).
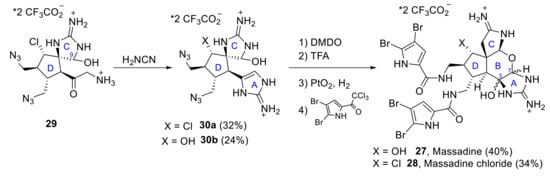
Scheme 9.
Baran’s total synthesis of massadine 27 and massadine chloride 28.
A different approach to synthesize the functionalized core structure 35 of massadine in an enantioselective manner has been developed by E. Carreira et al. Two key steps are involved in the successful construction of the D-ring subunit: an Ugi-4-component reaction affords intermediate 31, followed by 11 steps to reach the appropriately functionalized norbornene derivative 32 [31,32], and end-group-differentiating ozonolysis that yields compound 33. The corresponding synthesis of the spirocyclic CD-ring subunit 35 of massadine was then achieved by reaction of intermediate 34 with 2-iodoxybenzoic acid. Carreira and coworkers carried out the synthesis of this advanced intermediate in a sequence of a total of 37 linear steps and with an overall yield of about 0.9% (Scheme 10) [33].
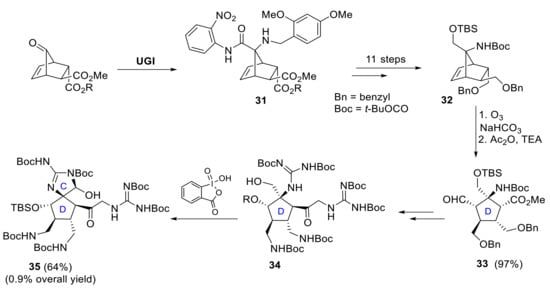
Scheme 10.
Synthesis of norbornene derivative 31 by Ugi-4-component reaction and further elaboration to CD-ring subunit 35.
Lee and coworkers proposed a different strategy for synthesizing the carbocyclic core 40 of massadine [34]. Key steps involved a stereoselective (3 + 2) cycloaddition and a protonolytic N-N bond cleavage in obtaining the tertiary α-amino group and the β-cyano group in a cis arrangement. The preparation of functionalized cyclopentane 40 started with the formation of the cyclopentene carboxaldehyde 37 by the reaction of hexanodial 36 with a catalytic amount of dibenzylammonium trifluoroacetate. The key α-amino-β-cyanation protocol was achieved by the reaction of advanced intermediate 38 with lithiumtrimethylsilyldiazomethane and subsequent acid-mediated N-N bond cleavage (Scheme 11).
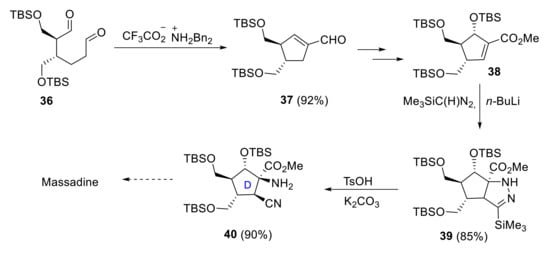
Scheme 11.
Synthesis of the D-ring subunit 36 of massadine proposed by Lee.
In 2018, Cannon reported an enantioselective route to the CD-bicycle of massadine 41 [35]. His strategy consisted of the formation of the cyclopentanone moiety 42 through a thermal nitrone dipolar cycloaddition of intermediate 43. Differentially substituted guanidine 44 was prepared by reduction of the ketone 42 followed by silver-mediated guanylation of the isoxazolidine. Final formation in good yields of CD-bicycles 41 involved a double hydrogenolysis process with different effects: the hydrogenolysis of the N-O bond opened the isoxazolidine ring, while the hydrogenolysis of the CBz protecting group induced the spirocyclic ring closure to form C ring (Scheme 12).
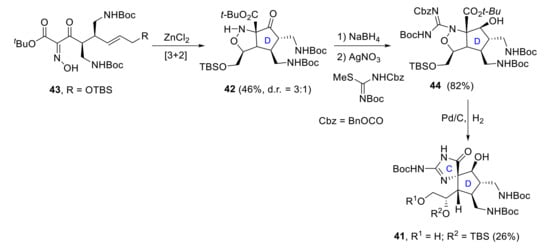
Scheme 12.
Cannon’s synthesis of the CD-ring subunit 41 of massadine.
2.4. Crambescidin 816
Crambescidin 816 (Cramb816) is a member of a family of guanidine alkaloids obtained from Crambe crambe, a Mediterranean marine sponge. This compound with antitumoral activity has several biological effects related to the modulation of Ca2+ signaling. In the first place, electrophysiological evidence obtained in neuroblastoma cells and neurons indicates that Cramb816 inhibits Cav1 or L-type, voltage-dependent Ca2+ channels with higher potency than dihydropyridines such as nifedipine. Cramb816 inhibited partially also voltage-dependent Na+ channels. These effects may account for the inhibition of synaptic transmission in the central nervous system [36]. Consistently with these effects, Cramb816 also prevented the Ca2+-dependent ileum contraction induced by acetylcholine at very low concentrations [37]. Paradoxically, Cramb816 induces a dose-dependent increase in intracellular Ca2+ and reduces viability in neurons. This effect is prevented by glutamate receptor antagonists suggesting that Cramb816 activates glutamate receptors, thus promoting Ca2+ overload and cell death in cortical neurons [38].
The crambescidin family contains a unique pentacyclic guanidine unit and a spermidine unit linked by a linear long-chain fatty acid (Figure 6). In 2000, the first derivative compound lacking the spermidine unit was isolated from the marine sponge of the genus Monanchora by Breakman et al. under the name of crambescidin 359 (45) [39].
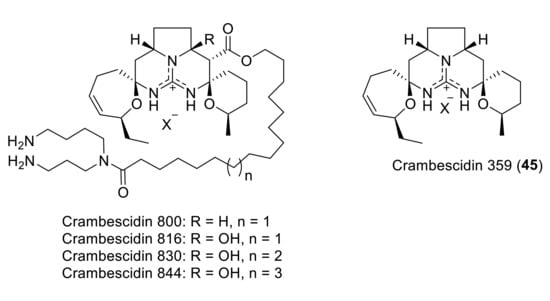
Figure 6.
The crambescidin family.
No total synthesis of crambescidin 816 has been reported so far, although various approaches to the crambescidin core present in crambescidin 359 have been described. Thus, in 1999, shortly before the isolation of crambescidin 359, C.G. Moore achieved its total synthesis during his PhD, although the work was published in 2007 [40]. The key step for the formation of the core tetracycle implies the reaction of bis-enone 46 with guanidine. Subsequent treatment with TBAF and acid-mediated cyclization gave the desired pentacycle 45 in 18% overall yield (Scheme 13).
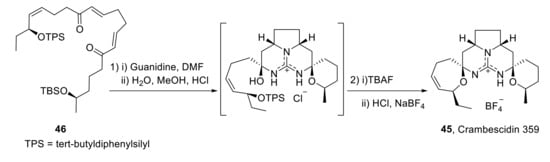
Scheme 13.
Moore’s synthesis of crambrescidin 359 (45).
In 2003, the first total synthesis of crambescidin 359 (45) was published [41], in which the key step consists of the double N,O-acetalization of the guanylated diketone 47 (Scheme 14).
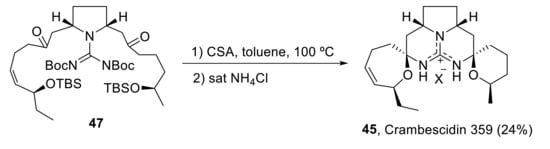
Scheme 14.
Total synthesis of crambescidin 359 (45).
Two years later, Overman et al. reported the total enantioselective synthesis of crambescidin 359 (45) in 29 total steps and 2.8% overall yield. The construction of the pentacyclic core was accomplished in sequential steps such as Biginelli condensation of aminal 48 with β-ketoester 49; acid-mediated spirocyclization of the deprotected pyrrolopyrimidine 50 and transformation of tricyclic urea 51 into the methyl pseudourea derivative, followed by reaction with ammonia, gave crambescidin analogs of type 52, which afford crambescidin 359 (45) after decarboxylation (Scheme 15) [42].
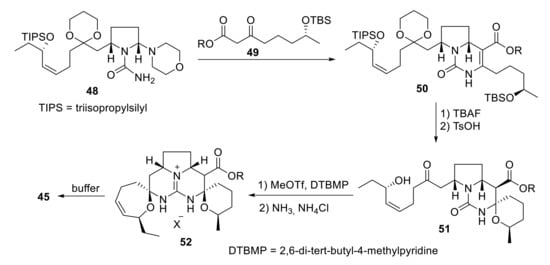
Scheme 15.
Overman’s synthesis of crambescidin 359 (45).
2.5. Maitotoxin
Maitotoxin (MTX) is an extremely potent toxin that is produced by Gambierdiscus toxicus, a dinoflagellate species. This toxin induces ciguatera fish poisoning, a human intoxication syndrome. MTX promotes Ca2+ influx at extremely low concentrations in the pM range. This effect seems mediated by the activation of Ca2+-permeable, non-selective channels belonging to the large superfamily of transient receptor potential channels (TRP channels). Ca2+ and Na+ influx induced by activation of these channels may lead to plasma membrane depolarization, which, in excitable cells, may, in turn, recruit voltage-gated Ca2+ channels leading to massive Ca2+ influx in neurons, muscle or endocrine cells. Recent evidence shows that siRNA-mediated silencing of the TRPC1 channel, the first member of the canonical family of channels, abolishes Ca2+ influx induced by MTX, thus suggesting that MTX targets and activates TRPC1 channels [43].
Maitotoxin is the largest among the secondary metabolites isolated and characterized up to date. This fact makes it an inspiration to many synthetic chemists. On the other hand, small fragments have been synthesized in order to support the absolute stereochemistry assigned in its characterization and as an approach to its total synthesis. In addition, the efforts towards the synthesis of this huge molecule have pushed the limits of organic synthesis, and many new synthetic methods have been developed along the way (Figure 7).
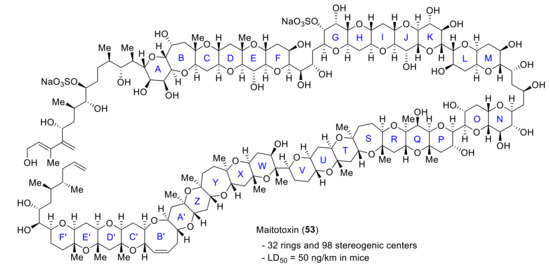
Figure 7.
Structure of maitotoxin (53).
If there is one group that stands out for their efforts towards the synthesis of this challenging molecule is the Nicolaou group. Relying on processes like the Noyori reduction/Achmatowicz rearrangement, hydroxy dithioketal cyclization or SmI2-mediated ring closure, they have synthesized the ABCDEFG, QRSTU [44], WXYZA’ [45] and C’D’E’F’ [46] ring systems. These advances in the synthesis of large maitotoxin domains were reviewed up to 2011 [47]. More recently, in 2014, Nicolaou reported the synthesis of the QRSTUVWXYZA′ system of maitotoxin [48]. Comparison of 13C NMR data supported the original structural assignment in all cases.
Another group that has worked towards the synthesis of maitotoxin is the Oishi group. In 2008, Oishi reported a convergent synthesis of the C’B’A’ZYXW ring 56, relying on acetal or thioacetal formation as the key cyclization step [49]. For instance, the preparation of the ZYXW tetracyclic intermediate 55 relies on the formation of a mixed thioacetal ring by the reaction of hydroxyketone 54 with ethanethiol in the presence of Zn(OTf)2 (Scheme 16).
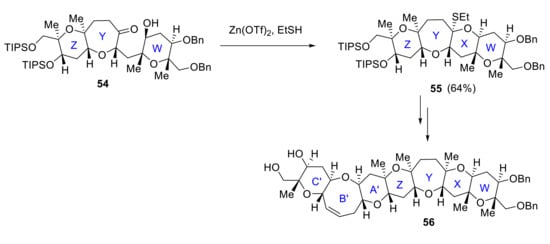
Scheme 16.
Formation of the WXYZA’B’C’ ring by Oishi group.
In 2014, Oishi reported the stereoselective synthesis of the F’E’D’C’ ring system of maitotoxin starting from E’ ring. E’D’ ring system 57 is formed by SmI2-induced cyclization of E’ ring, and E’D’C’ ring 59 formation follows the same procedure, starting from 58 (Scheme 17). The side-chain fragment was installed via Suzuki−Miyaura cross-coupling, and F’ ring formation was achieved by a Pd(II)-catalyzed cyclization yielding the desired compound 60 [50].
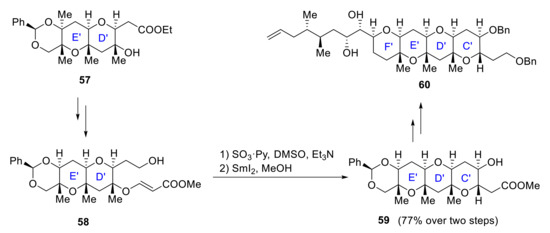
Scheme 17.
Stereoselective formation of the C’D’E’F’ ring by Oishi group.
Finally, in 2014 Oishi also proposed the stereoselective synthesis of the SQR ring of maitotoxin based on the formation of five contiguous stereogenic centers on the Q ring. The synthesis starts from (S) ring 61, which is transformed in several steps to intermediate 62. Removal of NAP group with DDQ, followed by the conversion of the hydroxy ketone to methyl acetal with (MeO)3CH, produces (S)RQ ring 63. After highly diastereoselective dihydroxylation and ring expansion of a six-membered to a seven-membered ring ketone, final SRQ ring 64 is obtained (Scheme 18) [51]. Similar strategies have also been applied to the synthesis of the LMNO ring system [52].
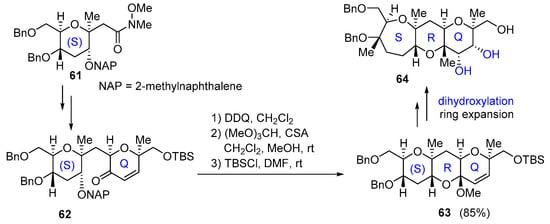
Scheme 18.
Stereoselective formation of the QRS ring by Oishi group.
Other groups have also made great efforts in the synthesis of different parts of this amazing molecule. In 2008, Nakata published a series of papers describing the synthesis of C’D’E’F’ [53], WXYZA’ [54] and BCDE [55] ring systems relying on SmI2 reductive cyclization. More recently, in 2017, Saito and Nakata synthesized the enantiomer of the ZA′B′C′D′-ring system of maitotoxin through a convergent strategy [56].
2.6. Yessotoxin
Yessotoxin (YTX) 65 is a polyether toxin related to ciguatoxins and produced by different dinoflagellates, particularly Lingulodinium polyedrum and Gonyaulax spinifera (Figure 8). This toxin has been reported to induce also Ca2+ influx in different cell types, including lymphocytes, and to promote neurotoxicity in cultured cerebellar neurons [57]. Ca2+ entry induced by yessotoxin is prevented by Nifedipine and SKF96365, a well-known blocker of L-type, voltage-gated Ca2+, and other types of channels. Yessotoxin may modulate L-type Ca2+ channels and also other types of Ca2+ channels [58]. Yessotoxin also induced Ca2+ influx and apoptosis in human hepatocellular carcinoma (Bel7402) cells, and this effect was blocked by external Ca2+ chelation and nifedipine, thus suggesting that YTX-induced apoptosis is mediated by activating a Ca2+ entry pathway and Ca2+ overload [59].
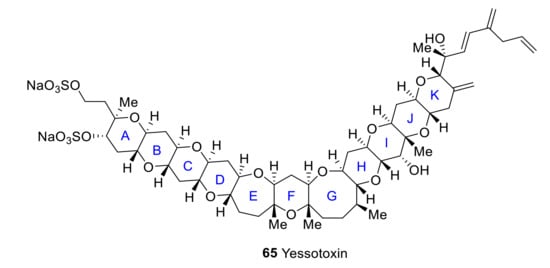
Figure 8.
Structure of yessotoxin.
In 2002, Mori reported a convergent synthesis of the BCDE ring [60]. B and E rings are coupled with an alkyne moiety via alkylation with acetylene. 1,2-diketone 67, obtained by ruthenium oxidation of intermediate alkyne 66, is treated with TsOH to produce tetracyclic dihemiketal 68. O-methylation of the two hemiketals hydroxyl groups 68 and reductive etherification results in the formation of BCDE ring system 69 of yessotoxin (Scheme 19). Later, similar methodologies were applied to synthesize the bigger CDEFGHIJ fragment [61].
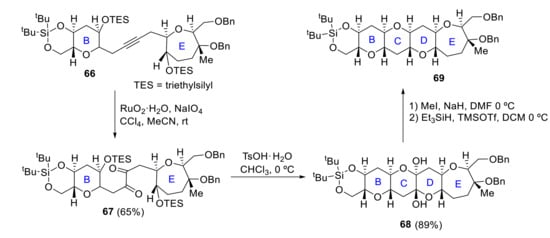
Scheme 19.
Mori’s synthesis of BCDE ring.
Kadota reported in 2006 a convergent synthesis of FGHI ring system 72 in which the key step in the construction of ring G by ring-closing metathesis of diene 70 with second-generation Grubbs catalyst 71 (Scheme 20) [62].
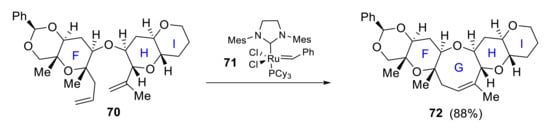
Scheme 20.
Kadota’s synthesis of FGHI ring.
In parallel, Kadota also reported a stereocontrolled synthesis of IJK ring 76, where SmI2- and acid-induced cyclization is the key ring-closure process. Thus, SmI2-mediated cyclization of 73 produces IJ ring 74 while the K ring is later constructed via acid-catalyzed cyclization of epoxy alcohol 75 (Scheme 21) [63].
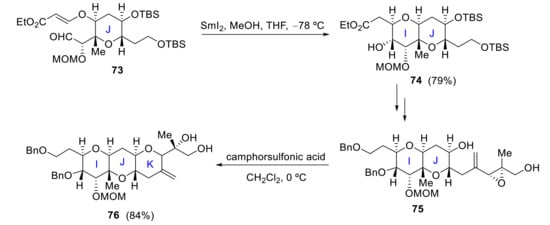
Scheme 21.
Kadota’s approach to the IJK ring system of yessotoxin.
In 2006, Oishi reported the synthesis of ABC and IJ rings. Cyclization of hydroxyenone 77 in the presence of pTsOH produces ABC ring system 78 (Scheme 22), while IJ ring fragment 79 is synthesized starting from the J ring. Acid-catalyzed 6-endo cyclization of the vinyl epoxy alcohol 79 results in IJ ring 80 in 93% yield (Scheme 23) [64].
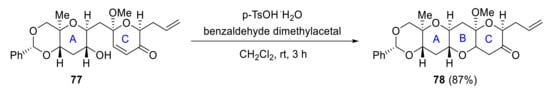
Scheme 22.
Oishi’s synthesis of the ABC ring.
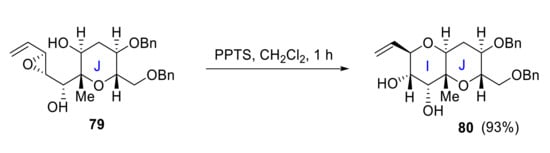
Scheme 23.
Oishi’s synthesis of IJ ring.
In 2005, Oishi reported a route to the FGHIJ fragment 84 [65], and in 2008 he finally achieved a highly convergent synthesis of the A–J ring system [66]. The main features for the construction of the pentacyclic F–J rings are a ring-closing metathesis process (81 to 82) and the formation of a cyclic acetal from a hydroxy ketone (83 to 84) [67] (Scheme 24).
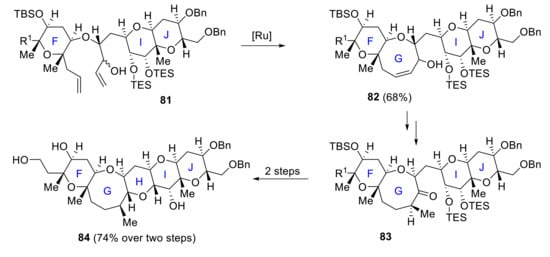
Scheme 24.
Oishi’s synthesis of the FGHIJ ring.
In 2016, Zhang and Rainier proposed a synthesis for ABCDEF and FGHI rings. Fragments AB 85 and EF 86 were easily obtained from known precursors previously synthesized for the total synthesis of brevenal [68]. Ring C is formed by removal of the TBS group of intermediate 87 and intramolecular acetalization, affording hemiketal 88. Final ABCDEF ring 89 was produced from 88 in a few more steps (Scheme 25). Regarding the FGHI fragment, F ring 90 and I ring 91 are coupled by Yamaguchi esterification. After some steps, including olefinic-ester cyclization to obtain the G ring, the final intermediate 92 is obtained. The last H ring is then crafted via reductive cyclization of this TES-protected hydroxy ketone 92 with TMSOTf/Et3SiH, affording the desired FGHI ring system 93 (Scheme 26) [69].
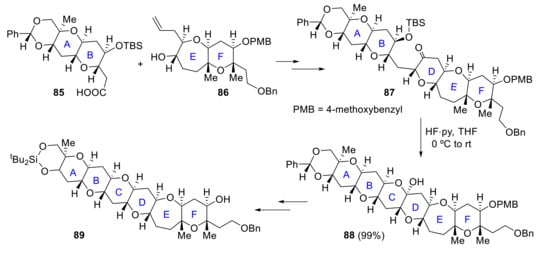
Scheme 25.
Zhang and Rainier’s approach to the ABCDEF ring system of yessotoxin.
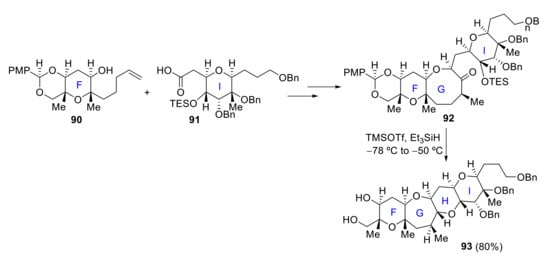
Scheme 26.
Zhang and Rainier’s approach to the FGHI ring system of yessotoxin.
Some groups have also developed different methodologies to synthesize small fragments of this marine drug relying on transition metal catalysis. Regarding palladium catalysis, it can be applied to the construction of the AB ring system as demonstrated by Hirai [70] and Jamison [71] or to the KJ ring system as shown by Yokoyama [72]. Other metals like ruthenium have shown their applicability in this synthesis, like in Trost’s approach to the BCD ring system by a ruthenium-catalyzed cycloisomerization reaction [73].
2.7. Palytoxin
Palytoxin (PTX) is a highly thermostable, polyhydroxylated and partially unsaturated compound with as much as eight double bonds within a long carbon chain. It is produced by Palythoa corals and Ostreopsis dinoflagellates, as well as by bacteria living in these organisms. It can also be isolated from other living organisms close to palytoxin producing organisms like sponges, mussels, starfish and cnidaria. PTX is among the most potent toxins found in nature. PTX produces severe vasoconstriction, disrupts cardiac function, and it is highly lethal to mice. PTX strongly inhibits the Na+/K+ATPase pump leading to a loss of ion gradients and plasma membrane depolarization that triggers activation of voltage-dependent ion channels, including voltage-dependent Ca2+ channels, reverse operation of the Na+/Ca2+ exchanger and induces Ca2+ overload. Most of these effects are prevented by ouabain [74]. As a consequence, PTX induces the release of neurotransmitters from motor nerve terminals [75].
PTX also elicits the release of glutamate from cerebellar granule cells leading to their depletion as well as prostaglandins from rat aorta with intact endothelium [76]. Finally, PTX is also an atypical tumor promoter on mouse skin initiated by irritants, probably because of its proinflammatory action [77].
Initial studies of palytoxin focused on determining its stereochemistry, relying on the synthesis of some of its degradation products [78]. Thus, the structure of palytoxin 94 was elucidated, as depicted in Figure 9. Due to its size and structural and stereochemical complexity, palytoxin is one of the most challenging compounds in the field of total synthesis.
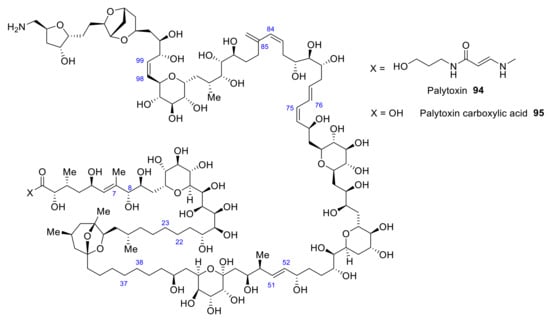
Figure 9.
Structure of palytoxin and palytoxin carboxylic acid.
Seven years after its isolation, Kishi reported its first total synthesis [79]. He focused on obtaining eight different segments (Scheme 27) whose coupling produced palytoxin carboxylic acid 95. Palytoxin carboxylic acid retrosynthesis results in a division in eight fragments: C1-C7 (96), C8-C22 (97), C23-C37 (98), C38-C51 (99), C52-C75 (100), C76-C84 (101), C85-C98 (102), C99-C115 (103).
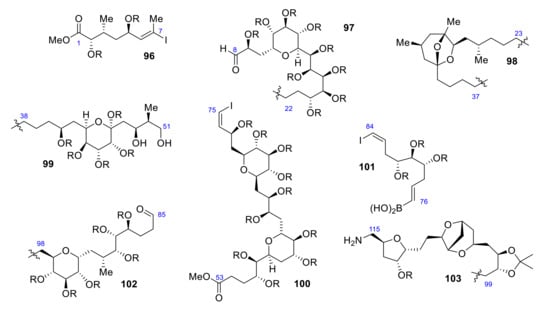
Scheme 27.
Retrosynthesis of palytoxin carboxylic acid.
Before proceeding to the coupling of the fragments, they needed to be synthesized. Each segment requires a different coupling, being some of them relatively straightforward, while others are more challenging.
Coupling between C22-C23 and C37-C38 goes via Wittig reaction followed by hydrogenation, so a C8-C51 chain is synthesized (104).
A Ni(II)/Cr(II)-catalyzed coupling reaction was then used to bond 96 with 104. After acetylation and hydrolysis of the acetonide, 105, left half of 95, is obtained (Scheme 28).
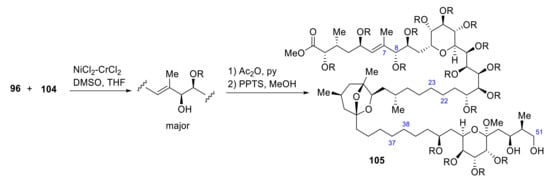
Scheme 28.
Synthesis of fragment 105 of palytoxin carboxylic acid.
Segments 102 and 103 are coupled by a Wittig reaction producing the C98-C99 olefinic bound. 106 reaction with 101, relying on Ni(II)/Cr(II) catalysis, followed by PDC oxidation and Wittig olefination, produces C84-C85 olefinic bond (Scheme 29).
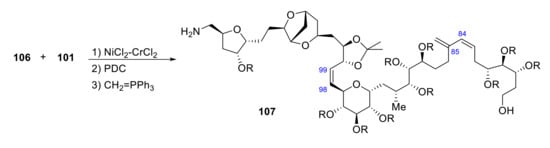
Scheme 29.
Synthesis of fragment 107 of palytoxin carboxylic acid.
107 reacts with 100 producing C75-C76 coupling by a Suzuki reaction (Scheme 30).
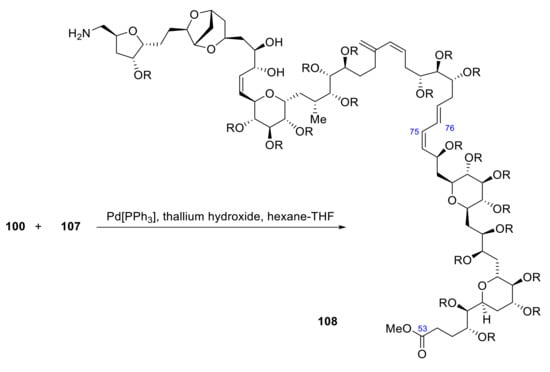
Scheme 30.
Synthesis of fragment 108 of palytoxin carboxylic acid.
Once the initial eight fragments have been coupled to form 105 and 108, the C51-C52 olefinic bond produces the coupling of both fragments to form 95. 105 is oxidized with RuCl2[PPh3] 3 to form an aldehyde, which is coupled with the anion generated from the ketophosphonate derived from methyl ester 108 to form an α,β-unsaturated ketone in 80–90% yield. After, C53 ketone is reduced to an alcohol by lithium borohydride in the presence of a lanthanide salt (EuCl3). As numerous protection groups have been used, a deprotection step is required to obtain the final product. All deprotection steps produce a 30% overall yield of the carboxylic acid 95 (Scheme 31). Finally, a functional group transformation of the carboxylic acid of palytoxin carboxylic acid 95 to the required N-acyl vinylogous urea (via acid-catalyzed lactonization, followed by addition with the corresponding amine bearing a phenylselenide group and final oxidative elimination of the selenide) provides palytoxin 94 [80].
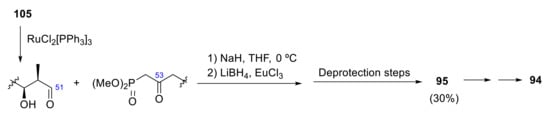
Scheme 31.
Synthesis of palytoxin.
After this first total synthesis, some other approaches have been developed, being the main objective of most of them the quantitative stereospecific synthesis of a fragment. For instance, Still’s approach pursues the synthesis of C30-C43 fragments by a stereospecific macrocyclically controlled remote asymmetric induction process [81]. On the other hand, Nelson’s objective was to synthesize the C58-C71 segment of palytoxin [82]. Since this fragment is close to having a C2-symmetry, being the features that break symmetry the absence of a hydroxyl group in C59, and different (2,6) relative configurations of the two tetrahydropyran rings, this method proposed a two-directional synthesis for a C2-symmetrical fragment, which will be later modified in order to obtain functionalized tetrahydropyrans in a stereoselective manner.
3. Heterocyclic Marine Drugs with a Direct Effect over Cytoskeleton
Latrunculin A
Latrunculin A is a member of the family of natural products and toxins termed latrunculins. At low μM concentrations, it binds actin monomers preventing their polymerization, thus resulting in the disruption of actin filaments of the cytoskeleton [83]. This effect may also result in the modulation of Ca2+ channels, specifically intracellular Ca2+ channels, as the inositol trisphosphate (IP3) receptor, a highly relevant, ligand-gated Ca2+ channel located in the membrane of the ER and responsible for agonist-induced Ca2+ release from intracellular stores. Accordingly, latruculin A may induce Ca2+ release from the ER, then the depletion of intracellular Ca2+ stores may activate store-operated Ca2+ channels and secondary Ca2+-dependent Na+ entry, which may promote membrane depolarization. These effects are similar to those induced by cytochalasin D, a well-known actin disruptor. Latruculin A-elicited depolarization consisted of a positive shift in Vm, which reached the threshold of activation of voltage-dependent Ca2+ channels, thus triggering an action potential. In Ca2+-free medium latruculin A lacked the ability to induce action potentials, while it was abolished upon removal of external Na+. Moreover, membrane depolarization was prevented by pre-injection of BAPTA and heparin, but not ryanodine [84]. Actin polymerization is involved in many different cell functions that are disrupted by latrunculin A. For instance, actin polymerization is required for clathrin-coated vesicle maturation of lamprey synapses. It is also very important for myogenic constriction in cerebral arteries [85]. Another example is egg fertilization that requires Ca2+ waves that depend on an intact cytoskeleton [86].
Latrunculins A and B (Figure 10) were first isolated and characterized by Kashman from the Red Sea sponge Latrunculia magnifica (Keller) [87].
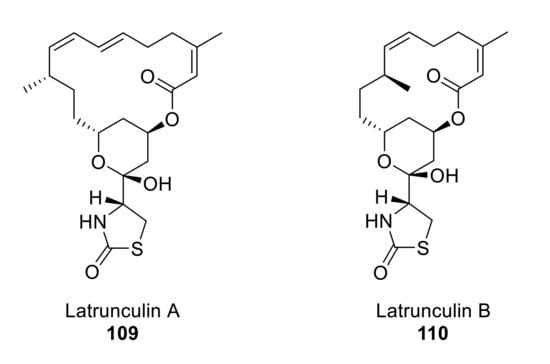
Figure 10.
Structures of latrunculin A and B.
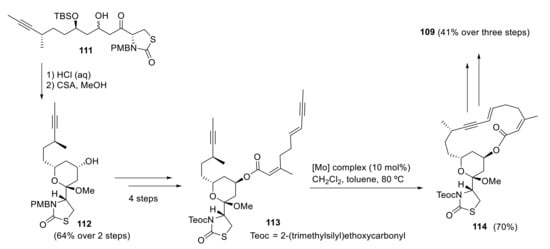
Fürstner and coworkers designed a versatile synthetic route to different members of the latrunculin family through ring-closing enyne-yne metathesis [88,89,90]. Treatment of intermediate 111 with aqueous HCl deprotected the TBS group and also affected the cyclization into hemiacetal, which was methylated with catalytic CSA and methanol to give 112. Ring-closing alkyne metathesis (RCAM) of intermediate 113 (obtained from 112 in four steps) produces the highly strained 16-membered cycle 114 in an enyne-yne metathesis catalyzed by a molybdenum complex. Compound 114, the smallest ring size that has been synthesized by an enyne-yne metathesis, forms 109 in three further steps in 41% overall yield [90] (Scheme 32). The total synthesis of latrunculin A was achieved in 15 linear steps over 1.7% overall yield.
Scheme 32.
Fürstner’s synthesis of latrunculin A.
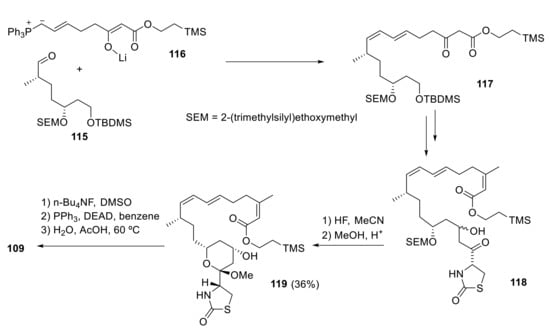
As latrunculin A differs from other members of its family in the presence of a conjugated diene, White’s convergent synthetic approach proposes the formation of an acyclic E,Z-diene formed by Wittig reaction of an E-ylide with an aldehyde [91].
Thus, the reaction of aldehyde 115 with E-ylide 116 gives E,Z-diene 117, which is further elaborated to produce intermediate aldol 118 in five steps. The final steps to 109 from 118 are quite straightforward. Treatment with HF and methanol allows selective deprotection of SEM group and subsequent cyclization, thus closing the desired tetrahydropyran ring through the formation of a methyl ketal. Finally, 119 is transformed to 109 in three additional steps (Scheme 33).
Scheme 33.
White’s synthesis of latrunculin A.
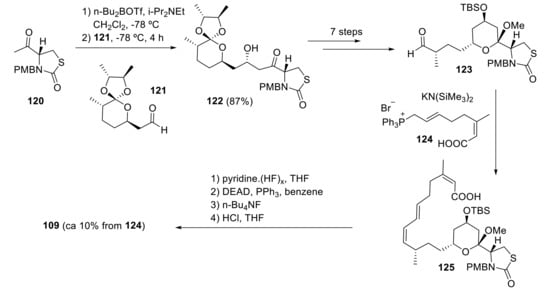
Smith and coworkers designed a short and highly convergent total synthesis of latrunculin A (109), where the longest linear sequence is 17 steps [92]. Treatment of 120 with n-Bu2BOTf and i-Pr2NEt followed by reaction with aldehyde 121 produces 122 (major isomer), which is then transformed to 123 in 22% yield over seven steps. Wittig coupling of aldehyde 123 with ylide 124 furnishes a cis-trans diene 125 that in four steps produces 109 (Scheme 34).
Scheme 34.
Smith highly convergent synthesis of latrunculin A.
Building block 123, as well as an intermediate for the synthesis of latrunculin A 109, is useful for the synthesis of other latrunculin family members [93], as they share a common structure.
Whites’ synthetic approach for latrunculin A 109 goes via the advanced intermediate 126 [94]. After selective deprotection of the SEM ether with concentrated HF, the tetrahydropyran ring 127 is constructed by exposing the resulting diol to acidic (CSA) methanol. Then, ester cleavage to 128, followed by Mitsunobu reaction, gives macrolactone 129. Simple hydrolysis with acetic acid affords the desired latrunculin A 109 (Scheme 35).
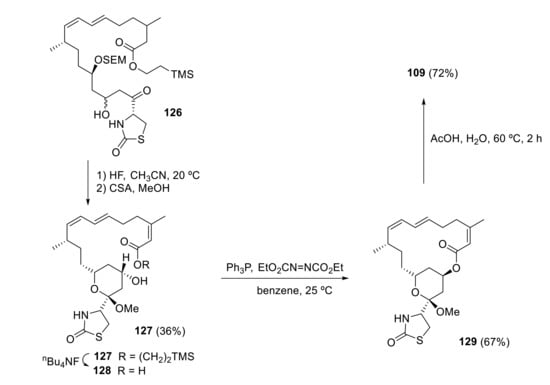
Scheme 35.
White’s synthesis of latrunculin A.
4. Heterocyclic Marine Drugs with Effect on Tumor Cell Proliferation
4.1. Mandelalides A–D
Mandelalides A–D are several glycosylated macrolides isolated from a new species of Lissoclinum ascidian that can be found in South Africa, Algoa Bay, close to Port Elizabeth and the surrounding Nelson Mandela Metropole [95]. These compounds show strong cytotoxicity to both human and mouse cancer cells, including neuroblastoma and lung cancer cells. The action mechanism is related to the inhibition of mitochondria complex V in the nM range. Specifically, treatment with mandelalide A decreases the aerobic respiratory capacity of living cancer cells dramatically [96]. By disrupting the mitochondrial membrane potential, mandelalide A destroys the main driving force for the mitochondrial Ca2+ uptake indirectly. If calcium cannot enter mitochondria, the store-operated calcium entry is inhibited, which is the main Ca2+ entry pathway involved in many tumor cell proliferation. Thus, it may lead to tumor cell cytotoxicity [97].
Two bicyclic domains are part of the overall macrocyclic macrolides, a (glycosylated) 2,6-cis-substituted tetrahydropyran and 2,5-cis-substituted tetrahydrofuran (Figure 11). Altmann has described a highly efficient convergent total synthesis of madelalide A, in which different methodologies have been used for the construction of the two main heterocycles of this macrolide.
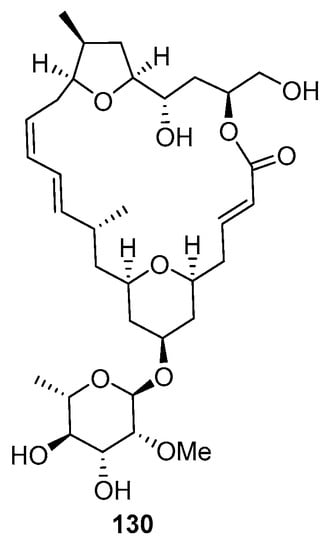
Figure 11.
Structure of mandelalide A (130).
Thus, the tetrahydropyran unit of building block 133 was prepared by a Prins cyclization reaction of the homoallylic alcohol 131 and the acetal 132 (Scheme 36). On the other hand, the synthesis of tetrahydrofuran moiety 137 included more steps and departed from the commercially available acetonide 134. The five-membered oxacycle 136 was obtained by reaction of the intermediate 135 with FeCl3·6H2O, in a tandem reaction involving acetonide cleavage and the subsequent attack of the liberated secondary hydroxy group to the proximal epoxide carbon (Scheme 37).
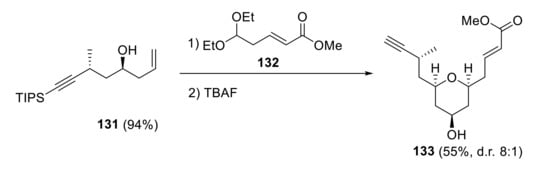
Scheme 36.
Synthesis of the tetrahydropyran unit.
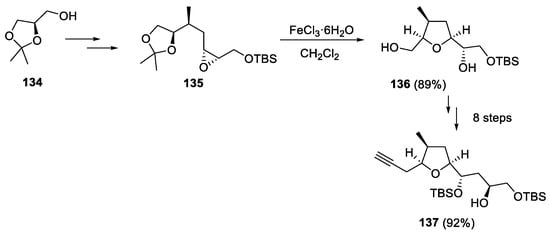
Scheme 37.
Synthesis of the tetrahydrofuran building block 137.
4.2. Patellamide
Patellamide is a cycle peptide and natural product isolated from Prochloron didemni, a cyanobacterial symbiont of Lissoclinum patella. This compound also shows strong cytotoxic and growth inhibitory activity in cancer cells [98]. The mechanism by which patellamide inhibits tumor cell proliferation is unknown, but its effects have been compared to verapamil, a well-known Ca2+ channel blocker also cytotoxic in cancer cells. Calcium entry is mandatory for cell proliferation; any compound that inhibited calcium entry in a sustained way could inhibit cell proliferation [99].
Patellamides A–C contains two oxazoline and two thiazole subunits in their structure (Figure 12).
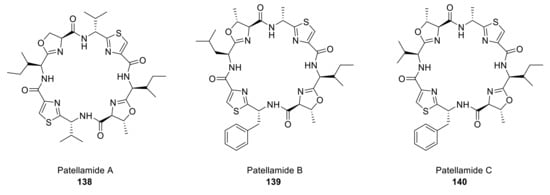
Figure 12.
Structures of the patellamide family.
The original synthetic methodology for patellamide B and C proposed by Hamada and Shioiri is based on the construction of the cyclic peptide structure from peptide building blocks and late formation of the oxazoline ring [98]. All peptides used in this route are previously synthesized from thiazole amino acids following a developed procedure [100]. Patellamides B and C are analogously synthesized but with different initial amino acids. In the last step, intermediates 141 and 142 are treated with SOCl2, thus forming the oxazoline rings and yielding the desired compounds (Scheme 38). A similar methodology was used to prepare patellamide B [101].
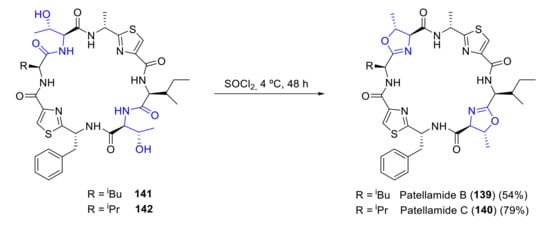
Scheme 38.
Synthesis of patellamides B and C by Hamada and Shioiri.
Schmidt proposed a different approach to patellamide B [102]. In this route, the oxazoline rings are formed early in the synthesis by reacting protected aminoalcohols 143 and 144 with boc-isoleucine imidic ester. Coupling of the two fragments 145 and 146 with diphenyl phosphorazidate produced a linear peptide 147, which cyclization then affords patellamide B 139 (Scheme 39).
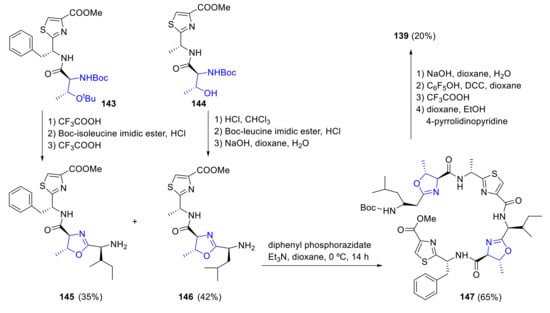
Scheme 39.
Schmidt’s synthesis of patellamide B.
More recently, a new approach relying on contemporary methods for the construction of the oxazoline rings was proposed by VanNieuwenhze [103]. In this strategy, all thiazole and oxazoline rings are installed in a linear intermediate, leaving the macrocyclization for the last step. Starting from thiazole 148, intermediate 149 was synthesized following a convergent condensation of peptide fragments. By treatment with Burgess reagent, both oxazoline subunits are formed through a cyclodehydration process yielding compound 150. Then, cleavage of both the allyl ester (PdPPh3)4 and the Fmoc group (piperidine/DMF) and macrocyclization (PyBOP, DMAP, DIEA) yielded the desired patellamide A 138 (Scheme 40).
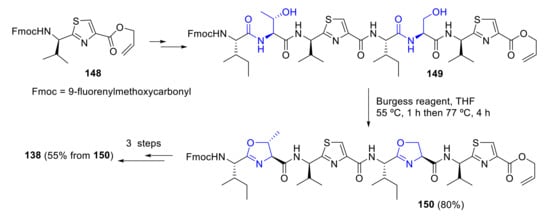
Scheme 40.
Synthesis of patellamide A by VanNieuwenhze.
4.3. Lamellarins
Lamellarins are natural alkaloids originally isolated from a small sea snail that show potential antiviral and anticancer properties. More than 50 lamellarins are available at present, being lamellarin D the leading compound of most of them. Lamellarins are frequently used as starting points for the design of anticancer drugs [104]. Lamellarin D shows strong inhibitory capacity of both nuclear and mitochondrial topoisomerase I, a protein involved in DNA reparation. It also interacts directly with mitochondria to elicit the death of cancer cells. In fact, the antitumoral effect of lamellarin D is seemingly mediated by apoptosis activation even in multidrug-resistant cancer cell lines. Lamellarin D acts as a direct inductor of mitochondrial permeability transition pore. Treatment with lamellarin D significantly reduces the threshold level of calcium required for opening the mitochondrial permeability transition pore induction. This leads, accordingly, to caspase-independent mitochondrial depolarization, mitochondrial swelling and release of cytochrome c. All these data indicate that lamellarin D facilitates calcium-dependent mitochondrial permeability transition pore opening leading to apoptosis in cancer cells [105].
Lamellarins A–D were isolated in 1985 by Faulkner and coworkers from the marine mollusk Lamellaria sp. [106]. Since then, more than 70 lamellarins and related compounds have been isolated from mollusks, sponges, and tunicates. The structures of some members of the family are depicted in Figure 13, although we will only focus on lamellarin D. Two approaches are possible to build the central functionalized pyrrole ring. The first one includes the functionalization of the pyrrole moiety, and the second one consists of employing the appropriate acyclic precursor to synthesize the pyrrole. Due to its biological activity, many groups have devoted their time to develop methods for its synthesis [107,108,109,110] as well as the synthesis of other members of the family [111,112,113,114].
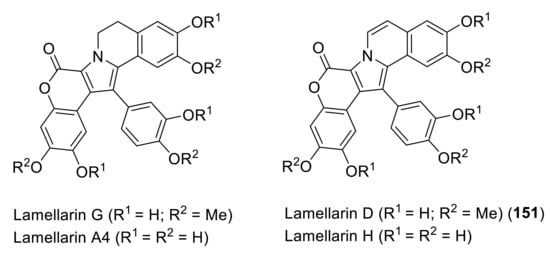
Figure 13.
Structures of some members of the lamellarin family.
In 2017, Chandrasekhar et al. proposed a total synthesis of lamellarin D in which the central pyrrole ring was synthesized through Ru(II)-catalyzed (3 + 2) annulation strategy [115]. The total synthesis of lamellarin D 151 was carried out in seven steps and in a 29% overall yield. The key intermediate 154 was obtained via Ru-catalyzed oxidative annulation of enamide 152 and diarylalkyne 153 in 87% yield. Bromination of this product, followed by Suzuki–cross-coupling with 155 and final MOM deprotection/lactonization, provided tetrasubstituted pyrrole 156 in 64% yield over three steps. N-alkylation and intramolecular Iwao cyclization generated an intermediate, which was treated with BCl3 to furnish lamellarin D 151 (Scheme 41).
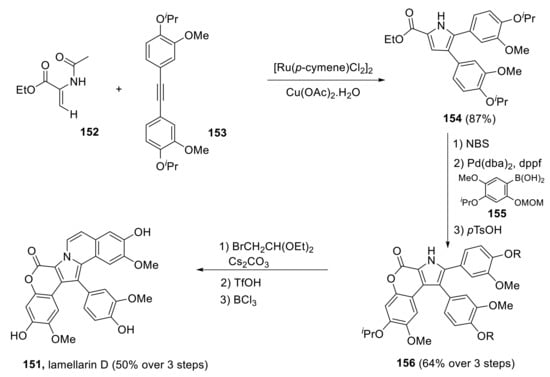
Scheme 41.
Total synthesis of lamellarin D by Chandrasekhar.
Khan et al. carried out a short and scalable total synthesis of lamellarin D trimethyl ether 162 and other analogs in 2019 [116]. The construction of the pyrrole ring was performed by annulation of aziridine ester 158 with β-bromo-β-nitrostyrene 157, giving the central 1,2,4-trisubstituted pyrrole core in a highly regioselective manner. Coupling of pyrrole-2-carboxylic acid 159 with phenol 160, followed by Pd-catalyzed intramolecular oxidative C-H arylation, afforded the pentacyclic 161, lamellarin G trimethyl ether, in 44% yield. Lamellarin D trimethyl ether 162 was obtained by dehydrogenation with DDQ in excellent yield (Scheme 42).
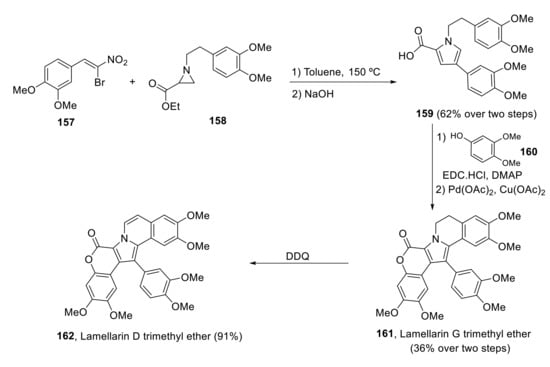
Scheme 42.
Khan’s synthesis of lamellarin D trimethyl ether.
More recently, Michael and coworkers have published the synthesis of lamellarin D trimethyl ether 162 by using enaminones as intermediates to alkaloids and other nitrogen heterocycles [117]. Eschenmoser sulfide contraction between bromoketone 164 and thiolactam 163, in the presence of triphenylphosphine and trimethylamine, afforded the desired heptamethoxylated enaminone 165 in high yields (96–100%). Conventional heating of enaminone 165 in ethyl bromoacetate provided a tricyclic product, which was oxidated by DDQ to give the unsaturated ring 166. A final saponification/lactonization process furnished lamellarin D trimethyl ether 162 in yields consistently above 96% (Scheme 43). Overall, yield over seven steps was 86%, the highest reported to date.
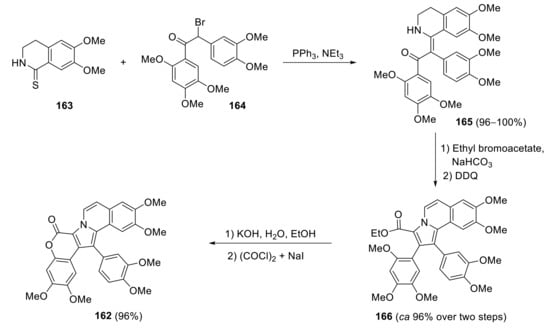
Scheme 43.
Michael’s synthesis of lamellarin D trimethyl ether.
5. Conclusions
In this review, we have tried to show an overview of a group of marine drugs that, from the structural point of view, contain one or various heterocyclic units in their core structure and, from the biological side, have a direct influence on the transport of calcium in the cell. The marine compounds covered in this review are divided into three groups, which correspond to their direct biological activity, such as compounds with a direct influence in the calcium channel, compounds with a direct effect on the cytoskeleton and drugs with an effect on cancer cell proliferation. For each target, we have described its bioactive properties and synthetic approaches. The wide variety of chemical structures compiled in this review and their significant medical properties will attract the attention of many researchers.
Author Contributions
A.B. designed the study and performed the literature search. P.G.-A., L.F.-P., C.D.-P., C.V. and L.N. wrote the first draft of the manuscript and A.B. revised it and prepared the final draft. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Spanish Ministry of Science and Innovation, grant number RTI2018-099298-B-100 to CV and LN, and Junta de Castilla y León, grant number VA294-P18 to LN.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
C.D.-P. and L.F.-P. acknowledge predoctoral Grants funded by the European Social Fund and the “Junta de Castilla y León”.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Humeau, J.; Bravo-San Pedro, J.M.; Vitale, I.; Nuñez, L.; Villalobos, C.; Kroemer, G.; Senovilla, L. Calcium signaling and cell cycle: Progression or death. Cell Calcium 2018, 70, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Villalobos, C.; Faught, W.J.; Frawley, L.S. Dynamic changes in spontaneous intracellular free calcium oscillations and their relationship to prolactin gene expression in single, primary mammotropes. Mol. Endocrinol. 1998, 12, 87–95. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. Elementary and global aspects of calcium signalling. J. Physiol. 1997, 499, 291–306. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- González, D.R.; Treuer, A.V.; Castellanos, J.; Dulce, R.A.; Hare, J.M. Impaired S-nitrosylation of the ryanodine receptor caused by xanthine oxidase activity contributes to calcium leak in heart failure. J. Biol. Chem. 2010, 285, 28938–28945. [Google Scholar] [CrossRef]
- Petersen, O.H.; Michalak, M.; Verkhratsky, A. Calcium signalling: Past, present and future. Cell Calcium 2005, 38, 161–169. [Google Scholar] [CrossRef]
- Rizzuto, R.; Pozzan, T. Microdomains of intracellular Ca2+: Molecular determinants and functional consequences. Physiol. Rev. 2006, 86, 369–408. [Google Scholar] [CrossRef]
- Rizzuto, R.; Bernardi, P.; Pozzan, T. Mitochondria as all-round players of the calcium game. J. Physiol. 2000, 529, 37–47. [Google Scholar] [CrossRef]
- Nelson, O.; Tu, H.; Lei, T.; Bentahir, M.; de Strooper, B.; Bezprozvanny, I. Familial Alzheimer disease-linked mutations specifically disrupt Ca2+ leak function of presenilin 1. J. Clin. Investig. 2007, 117, 1230–1239. [Google Scholar] [CrossRef]
- Joseph, N.; Reicher, B.; Mira Barda-Saad, M. The calcium feedback loop and T cell activation: How cytoskeleton networks control intracellular calcium flux. Biochim. Biophys. Acta 2014, 1838, 557–568. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, L.G.; Hernández-Morales, M.; Nuñez, L.; Villalobos, C. Inhibition of Polyamine Biosynthesis Reverses Ca2+ Channel Remodeling in Colon Cancer Cells. Cancers 2019, 11, 83. [Google Scholar] [CrossRef] [PubMed]
- Iamshanova, O.; Pla, A.F.; Prevarskaya, N. Molecular mechanisms of tumour invasion: Regulation by calcium signals. J. Physiol. 2017, 595, 3063–3075. [Google Scholar] [CrossRef] [PubMed]
- Shu, Z.; Liu, Q.; Xing, C.; Zhang, Y.; Zhou, Y.; Zhang, J.; Liu, H.; Cao, M.; Yang, X.; Liu, G. Viridicatol Isolated from Deep-Sea Penicillium Griseofulvum Alleviates Anaphylaxis and Repairs the Intestinal Barrier in Mice by Suppressing Mast Cell Activation. Mar. Drugs 2020, 18, 517. [Google Scholar] [CrossRef] [PubMed]
- Luckner, M.; Mohammed, Y.S. About metabolic products of Penicillium viridicatum westling and Penicillium cyclopium westling; Synthesis of viridicatol, 3′-O-methylviridicatol and N-methyl-3′-O-methylviridicatol. Tetrahedron Lett. 1964, 5, 1987–1989. [Google Scholar] [CrossRef]
- Tangella, Y.; Manasa, K.L.; Krishna, N.H.; Sridhar, B.; Kamal, A.; Nagendra Babu, B. Regioselective Ring Expansion of Isatins with In Situ Generated α-Aryldiazomethanes: Direct Access to Viridicatin Alkaloids. Org. Lett. 2018, 20, 3639–3642. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Harayama, T. A Concise and Versatile Synthesis of Viridicatin Alkaloids from Cyanoacetanilides. Org. Lett. 2009, 11, 1603–1606. [Google Scholar] [CrossRef]
- Mamedov, V.A.; Mamedova, V.L.; Kadyrova, S.F.; Galimullina, V.R.; Khikmatova, G.Z.; Korshin, D.E.; Gubaidullin, A.T.; Krivolapov, D.B.; Rizvanov, I.K.; Bazanova, O.B.; et al. Synthesis of 3-Hydroxy-4-arylquinolin-2-ones Including Viridicatol via a Darzens Condensation/Friedel–Crafts Alkylation Strategy. J. Org. Chem. 2018, 83, 13132–13145. [Google Scholar] [CrossRef]
- Rho, M.C.; Nakahata, N.; Nakamura, H.; Murai, A.; Ohizumi, Y. Involvement of phospholipase C-gamma2 in activation of mitogen-activated protein kinase and phospholipase A2 by zooxanthellatoxin-A in rabbit platelets. J. Pharmacol. Exp. Ther. 1997, 282, 496–504. [Google Scholar]
- Moriya, T.; Furukawa, K.; Nakamura, H.; Murai, A.; Ohizumi, Y. The vaso-contractile action of zooxanthellatoxin-B from a marine dinoflagellate is mediated via Ca2+ influx in the rabbit aorta. Can. J. Physiol. Pharmacol. 2001, 79, 1030–1035. [Google Scholar] [CrossRef]
- Nakamura, H.; Asari, T.; Murai, A.; Kondo, T.; Yoshida, K.; Ohizumi, Y. Structure of periodate oxidation products with characteristic partial structures of zooxanthellatoxin-A, a potent vasoconstrictive polyol from a symbiotic dinoflagellate. J. Org. Chem. 1993, 58, 313–314. [Google Scholar] [CrossRef]
- Nakamura, H.; Sato, K.; Murai, A. Absolute configuration of the spiroacetal portion of zooxanthellatoxin-A. Tetrahedron Lett. 1996, 37, 7267–7270. [Google Scholar] [CrossRef]
- Nakamura, H.; Takahashi, M.; Murai, A. Synthesis and absolute configuration of an exomethylene portion of zooxanthellatoxin-A. Tetrahedron Asymmetry 1998, 9, 2571–2574. [Google Scholar] [CrossRef]
- Nakamura, H.; Fujimaki, K.; Murai, A. Synthetic studies on the common C25 long chain acid portion of zooxanthellatoxins from a symbiotic dinoflagellate Symbiodinium sp. Tetrahedron Lett. 1996, 37, 3153–3156. [Google Scholar] [CrossRef]
- Nakamura, H.; Maruyama, K.; Fujimaki, K.; Murai, A. Absolute configuration of the common terminal acid portion of zooxanthellatoxins from a symbiotic dinoflagellate Symbiodinium sp. established by the synthesis of its ozonolysis product. Tetrahedron Lett. 2000, 41, 1927–1930. [Google Scholar] [CrossRef]
- Onodera, K.-I.; Nakamura, H.; Oba, Y.; Ohizumi, Y.; Ojika, M. Zooxanthellamide Cs: Vasoconstrictive Polyhydroxylated Macrolides with the Largest Lactone Ring Size from a Marine Dinoflagellate of Symbiodinium sp. J. Am. Chem. Soc. 2005, 127, 10406–10411. [Google Scholar] [CrossRef]
- Nishimura, S.; Matsunaga, S.; Shibazaki, M.; Suzuki, K.; Furihata, K.; van Soest, R.W.; Fusetani, N. Massadine, a novel geranylgeranyltransferase type I inhibitor from the marine sponge Stylissa aff. massa. Org. Lett. 2003, 5, 2255–2257. [Google Scholar] [CrossRef]
- Bickmeyer, U.; Grube, A.; Klings, K.W.; Köck, M. Disturbance of voltage-induced cellular calcium entry by marine dimeric and tetrameric pyrrole-imidazole alkaloids. Toxicon 2007, 50, 490–497. [Google Scholar] [CrossRef]
- Su, S.; Seiple, I.B.; Young, I.S.; Baran, P.S. Total Syntheses of (±)-Massadine and Massadine Chloride. J. Am. Chem. Soc. 2008, 130, 16490–16491. [Google Scholar] [CrossRef]
- Seiple, I.B.; Su, S.; Young, I.S.; Nakamura, A.; Yamaguchi, J.; Jørgensen, L.; Rodriguez, R.A.; O’Malley, D.P.; Gaich, T.; Köck, M.; et al. Enantioselective Total Syntheses of (−)-Palau’amine, (−)-Axinellamines, and (−)-Massadines. J. Am. Chem. Soc. 2011, 133, 14710–14726. [Google Scholar] [CrossRef]
- Chinigo, G.M.; Breder, A.; Carreira, E.M. Ugi-4-Component Reaction Enabling Rapid Access to the Core Fragment of Massadine. Org. Lett. 2011, 13, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Breder, A.; Chinigo, G.M.; Waltman, A.W.; Carreira, E.M. Enantioselective Synthesis of the Carbocyclic D-Ring Subunit of Massadine. Angew. Chem. Int. Ed. Engl. 2008, 47, 8514–8517. [Google Scholar] [CrossRef] [PubMed]
- Breder, A.; Chinigo, G.M.; Waltman, A.W.; Carreira, E.M. Towards the Synthesis of Massadine: A Unified Strategy for the Stereoselective Synthesis of the Carbocyclic C,D-Ring Subunit. Chem. Eur. J. 2011, 17, 12405–12416. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Lee, H.; Lee, D. Synthesis of the Carbocyclic Core of Massadine. Org. Lett. 2015, 17, 5348–5351. [Google Scholar] [CrossRef] [PubMed]
- Cannon, J.S. A Nitrone Dipolar Cycloaddition Strategy toward an Enantioselective Synthesis of Massadine. Org. Lett. 2018, 20, 3883–3887. [Google Scholar] [CrossRef]
- Martín, V.; Vale, C.; Bondu, S.; Thomas, O.P.; Vieytes, M.R.; Botana, L.M. Differential Effects of Crambescins and Crambescidin 816 in Voltage-Gated Sodium, Potassium and Calcium Channels in Neurons. Chem. Res. Toxicol. 2013, 26, 169–178. [Google Scholar] [CrossRef]
- Berlinck, R.G.; Braekman, J.C.; Daloze, D.; Bruno, I.; Riccio, R.; Ferri, S.; Spampinato, S.; Speroni, E. Polycyclic guanidine alkaloids from the marine sponge Crambe crambe and Ca2+ channel blocker activity of crambescidin 816. J. Nat. Prod. 1993, 56, 1007–1015. [Google Scholar] [CrossRef]
- Méndez, A.G.; Juncal, A.B.; Silva, S.B.L.; Thomas, O.P.; Martín Vázquez, V.; Alfonso, A.; Vieytes, M.R.; Vale, C.; Botana, L.M. The Marine Guanidine Alkaloid Crambescidin 816 Induces Calcium Influx and Cytotoxicity in Primary Cultures of Cortical Neurons through Glutamate Receptors. ACS Chem. Neurosci. 2017, 8, 1609–1617. [Google Scholar] [CrossRef]
- Braekman, J.C.; Daloze, D.; Tavares, R.; Hajdu, E.; Van Soest, R.W.M. Novel Polycyclic Guanidine Alkaloids from Two Marine Sponges of the Genus Monanchora. J. Nat. Prod. 2000, 63, 193–196. [Google Scholar] [CrossRef]
- Moore, C.G.; Murphy, P.J.; Williams, H.L.; McGown, A.T.; Smith, N.K. Synthetic studies towards ptilomycalin A: Total synthesis of crambescidin 359. Tetrahedron 2007, 63, 11771–11780. [Google Scholar] [CrossRef]
- Nagasawa, K.; Hashimoto, Y. Synthesis of marine guanidine alkaloids and their application as chemical/biological tools. Chem. Rec. 2003, 3, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Aron, Z.D.; Overman, L.E. Total Synthesis and Properties of the Crambescidin Core Zwitterionic Acid and Crambescidin 359. J. Am. Chem. Soc. 2005, 127, 3380–3390. [Google Scholar] [CrossRef] [PubMed]
- Flores, P.L.; Rodríguez, E.; Zapata, E.; Carbó, R.; Farías, J.M.; Martínez, M. Maitotoxin Is a Potential Selective Activator of the Endogenous Transient Receptor Potential Canonical Type 1 Channel in Xenopus laevis Oocytes. Mar. Drugs 2017, 15, 198. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, K.C.; Gelin, C.F.; Seo, J.H.; Huang, Z.; Umezawa, T. Synthesis of the QRSTU Domain of Maitotoxin and Its 85-epi- and 86-epi-Diastereoisomers. J. Am. Chem. Soc. 2010, 132, 9900–9907. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Baker, T.M.; Nakamura, T. Synthesis of the WXYZA’ Domain of Maitotoxin. J. Am. Chem. Soc. 2011, 133, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, K.C.; Seo, J.H.; Nakamura, T.; Aversa, R.J. Synthesis of the C’D’E’F’ Domain of Maitotoxin. J. Am. Chem. Soc. 2011, 133, 214–219. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Aversa, R.J. Maitotoxin: An Inspiration for Synthesis. Isr. J. Chem. 2011, 51, 359–377. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Heretsch, P.; Nakamura, T.; Rudo, A.; Murata, M.; Konoki, K. Synthesis and Biological Evaluation of QRSTUVWXYZA’ Domains of Maitotoxin. J. Am. Chem. Soc. 2014, 136, 16444–16451. [Google Scholar] [CrossRef]
- Oishi, T.; Hasegawa, F.; Torikai, K.; Konoki, K.; Matsumori, N.; Murata, M. Convergent Synthesis and Biological Activity of the WXYZA’B’C’ Ring System of Maitotoxin. Org. Lett. 2008, 10, 3599–3602. [Google Scholar] [CrossRef]
- Kunitake, M.; Oshima, T.; Konoki, K.; Ebine, M.; Torikai, K.; Murata, M.; Oishi, T. Synthesis and Biological Activity of the C’D’E’F’ Ring System of Maitotoxin. J. Org. Chem. 2014, 79, 4948–4962. [Google Scholar] [CrossRef]
- Onoue, H.; Baba, T.; Konoki, K.; Torikai, K.; Ebine, M.; Oishi, T. Synthesis and Biological Activity of the QRS Ring System of Maitotoxin. Chem. Lett. 2014, 43, 1904–1906. [Google Scholar] [CrossRef]
- Onoue, H.; Marubayashi, R.; Ishikawa, E.; Konoki, K.; Torikai, K.; Ebine, M.; Murata, M.; Oishi, T. Syntheses and Biological Activities of the LMNO, ent-LMNO, and NOPQR(S) Ring Systems of Maitotoxin. J. Org. Chem. 2017, 82, 9595–9618. [Google Scholar] [CrossRef] [PubMed]
- Morita, M.; Ishiyama, S.; Koshino, H.; Nakata, T. Synthetic Studies on Maitotoxin. 1. Stereoselective Synthesis of the C’D’E’F’-Ring System Having a Side Chain. Org. Lett. 2008, 10, 1675–1678. [Google Scholar] [CrossRef] [PubMed]
- Morita, M.; Haketa, T.; Koshino, H.; Nakata, T. Synthetic Studies on Maitotoxin. 2. Stereoselective Synthesis of the WXYZA’-Ring System. Org. Lett. 2008, 10, 1679–1682. [Google Scholar] [CrossRef] [PubMed]
- Satoh, M.; Koshino, H.; Nakata, T. Synthetic Studies on Maitotoxin. 3. Stereoselective Synthesis of the BCDE-Ring System. Org. Lett. 2008, 10, 1683–1685. [Google Scholar] [CrossRef]
- Saito, T.; Morita, M.; Koshino, H.; Sodeoka, M.; Nakata, T. Convergent Synthesis of the ent-ZA’B’C’D’-Ring System of Maitotoxin. Org. Lett. 2017, 19, 3203–3206. [Google Scholar] [CrossRef]
- Pérez-Gómez, A.; Ferrero-Gutierrez, A.; Novelli, A.; Franco, J.M.; Paz, B.; Fernández-Sánchez, M.T. Potent Neurotoxic Action of the Shellfish Biotoxin Yessotoxin on Cultured Cerebellar Neurons. Toxicol. Sci. 2005, 90, 168–177. [Google Scholar] [CrossRef]
- de la Rosa, L.A.; Alfonso, A.; Vilariño, N.; Vieytes, M.R.; Botana, L.M. Modulation of cytosolic calcium levels of human lymphocytes by yessotoxin, a novel marine phycotoxin. Biochem. Pharmacol. 2001, 61, 827–833. [Google Scholar] [CrossRef]
- Pang, M.; Qu, P.; Gao, C.-L.; Tang, X.; Wang, Z.-L. Effect of yessotoxin on cytosolic calcium levels in human hepatocellular carcinoma cells in vitro. Biomed. Rep. 2014, 2, 93–96. [Google Scholar] [CrossRef]
- Mori, Y.; Hayashi, H. Synthetic studies of yessotoxin, a polycyclic ether implicated in diarrhetic shellfish poisoning: Convergent synthesis of the BCDE ring system via an alkyne intermediate. Tetrahedron 2002, 58, 1789–1797. [Google Scholar] [CrossRef]
- Sakai, T.; Sugimoto, A.; Tatematsu, H.; Mori, Y. Divergent synthesis of trans-fused polycyclic ethers by a convergent oxiranyl anion strategy. J. Org. Chem. 2012, 77, 11177–11191. [Google Scholar] [CrossRef] [PubMed]
- Kadota, I.; Ueno, H.; Sato, Y.; Yamamoto, Y. Convergent synthesis of the FGHI ring segment of yessotoxin. Tetrahedron Lett. 2006, 47, 89–92. [Google Scholar] [CrossRef]
- Kadota, I.; Abe, T.; Sato, Y.; Kabuto, C.; Yamamoto, Y. Stereocontrolled synthesis of the IJK ring segment of yessotoxin. Tetrahedron Lett. 2006, 47, 6545–6548. [Google Scholar] [CrossRef]
- Oishi, T.; Suzuki, M.; Watanabe, K.; Murata, M. Synthesis of the ABC and IJ ring fragments of yessotoxin. Tetrahedron Lett. 2006, 47, 3975–3978. [Google Scholar] [CrossRef]
- Watanabe, K.; Suzuki, M.; Murata, M.; Oishi, T. Convergent synthesis of the FGHI ring system of yessotoxin: Stereoselective construction of the G ring. Tetrahedron Lett. 2005, 46, 3991–3995. [Google Scholar] [CrossRef]
- Torikai, K.; Watanabe, K.; Minato, H.; Imaizumi, T.; Murata, M.; Oishi, T. Convergent Synthesis of the A-J Ring System of Yessotoxin. Synlett 2008, 2008, 2368–2372. [Google Scholar] [CrossRef]
- Oishi, T.; Imaizumi, T.; Murata, M. Reductive Etherification under Microfluidic Conditions: Application to Practical Synthesis of the FGHIJ-Ring System of Yessotoxin. Chem. Lett. 2010, 39, 108–109. [Google Scholar] [CrossRef]
- Zhang, Y.; Rohanna, J.; Zhou, J.; Iyer, K.; Rainier, J.D. Total Synthesis of Brevenal. J. Am. Chem. Soc. 2011, 133, 3208–3216. [Google Scholar] [CrossRef]
- Zhang, Y.; Rainier, J.D. Synthesis of the ABCDEF and FGHI ring system of yessotoxin and adriatoxin. J. Antibiot. 2016, 69, 259–272. [Google Scholar] [CrossRef]
- Hirai, Y.; Yokoyama, H.; Kusumoto, Y.; Sumiyoshi, K.; Miyazawa, M. Synthetic Studies of Yessotoxin: Iterative Synthesis of the AB Ring System via Pd(II)-Catalyzed Cyclization of Alcohol. Heterocycles 2014, 89, 353–358. [Google Scholar] [CrossRef]
- Czabaniuk, L.C.; Jamison, T.F. Hydroxyl-Substituted Ladder Polyethers via Selective Tandem Epoxidation/Cyclization Sequence. Org. Lett. 2015, 17, 774–777. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, H.; Nishida, K.; Togawa, T.; Yamagami, M.; Miyazawa, M.; Hirai, Y. Stereoselective synthesis of the KJ ring system of yessotoxin by Pd(II)-catalyzed cyclization. Tetrahedron Lett. 2016, 57, 4379–4381. [Google Scholar] [CrossRef]
- Trost, B.M.; Rhee, Y.H. A Flexible Approach toward trans-Fused Polycyclic Tetrahydropyrans. A Synthesis of Prymnesin and Yessotoxin Units. Org. Lett. 2004, 6, 4311–4313. [Google Scholar] [CrossRef] [PubMed]
- Habermann, E. Palytoxin acts through Na+, K+-ATPase. Toxicon 1989, 27, 1171–1187. [Google Scholar] [CrossRef]
- Shimahara, T.; Molgó, J. Palytoxin enhances quantal acetylcholine release from motor nerve terminals and increases cytoplasmic calcium levels in a neuronal hybrid cell line. Life Sci. Adv. Pharmacol. 1990, 9, 785–792. [Google Scholar]
- Rakowski, R.F.; Artigas, P.; Palma, F.; Holmgren, M.; De Weer, P.; Gadsby, D.C. Sodium Flux Ratio in Na/K Pump-Channels Opened by Palytoxin. J. Gen. Physiol. 2007, 130, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Fujiki, H.; Suganuma, M.; Nakayasu, M.; Hakii, H.; Horiuchi, T.; Takayama, S.; Sugimura, T. Palytoxin is a non-12-O-tetradecanoylphorbol-13-acetate type tumor promoter in two-stage mouse skin carcinogenesis. Carcinogenesis 1986, 7, 707–710. [Google Scholar] [CrossRef]
- Ko, S.S.; Finan, J.M.; Yonaga, M.; Kishi, Y.; Uemura, D.; Hirata, Y. Stereochemistry of palytoxin. Part 2. C1-C6, C47-C74, and C77-C83 segments. J. Am. Chem. Soc. 1982, 104, 7364–7367. [Google Scholar] [CrossRef]
- Kishi, Y. Natural products synthesis: Palytoxin. Pure Appl. Chem. 1989, 61, 313–324. [Google Scholar] [CrossRef][Green Version]
- Suh, E.M.; Kishi, Y. Synthesis of Palytoxin from Palytoxin Carboxylic Acid. J. Am. Chem. Soc. 1994, 116, 11205–11206. [Google Scholar] [CrossRef]
- Still, W.C.; Galynker, I. Stereospecific synthesis of the C30-C43 segment of palytoxin by macrocyclically controlled remote asymmetric induction. J. Am. Chem. Soc. 1982, 104, 1774–1776. [Google Scholar] [CrossRef]
- Hodgson, R.; Nelson, A. A two-directional synthesis of the C58–C71 fragment of palytoxin. Org. Biomol. Chem. 2004, 2, 373–386. [Google Scholar] [CrossRef]
- Morton, W.M.; Ayscough, K.R.; McLaughlin, P.J. Latrunculin alters the actin-monomer subunit interface to prevent polymerization. Nat. Cell Biol. 2000, 2, 376–378. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F. Latrunculin A depolarizes starfish oocytes. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2007, 148, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Gokina, N.I.; Osol, G. Actin cytoskeletal modulation of pressure-induced depolarization and Ca2+ influx in cerebral arteries. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H1410–H1420. [Google Scholar] [CrossRef]
- Chun, J.T.; Limatola, N.; Vasilev, F.; Santella, L. Early events of fertilization in sea urchin eggs are sensitive to actin-binding organic molecules. Biochem. Biophys. Res. Commun. 2014, 450, 1166–1174. [Google Scholar] [CrossRef]
- Spector, I.; Shochet, N.R.; Blasberger, D.; Kashman, Y. Latrunculins-novel marine macrolides that disrupt microfilament organization and affect cell growth: Comparison with cytochalasin D. Cell Motil. Cytoskelet. 1989, 13, 127–144. [Google Scholar] [CrossRef]
- Fürstner, A.; Turet, L. Concise and Practical Synthesis of Latrunculin A by Ring-Closing Enyne–Yne Metathesis. Angew. Chem. Int. Ed. Engl. 2005, 44, 3462–3466. [Google Scholar] [CrossRef]
- Fürstner, A.; De Souza, D.; Parra-Rapado, L.; Jensen, J.T. Catalysis-Based Total Synthesis of Latrunculin B. Angew. Chem. Int. Ed. Engl. 2003, 42, 5358–5360. [Google Scholar] [CrossRef]
- Fürstner, A.; Souza, D.; Turet, L.; Fenster, M.; Parra-Rapado, L.; Wirtz, C.; Mynott, R.; Lehmann, C. Total Syntheses of the Actin-Binding Macrolides Latrunculin A, B, C, M, S and 16-epi-Latrunculin B. Chem. Eur. J. 2007, 13, 115–134. [Google Scholar] [CrossRef]
- White, J.D.; Kawasaki, M. Total synthesis of (+)-latrunculin A, an ichthyotoxic metabolite of the sponge Latrunculia magnifica and its C-15 epimer. J. Org. Chem. 1992, 57, 5292–5300. [Google Scholar] [CrossRef]
- Smith, A.B.; Noda, I.; Remiszewski, S.W.; Liverton, N.J.; Zibuck, R. Total synthesis of (+)-latrunculin A. J. Org. Chem. 1990, 55, 3977–3979. [Google Scholar] [CrossRef]
- Smith, A.B.; Leahy, J.W.; Noda, I.; Remiszewski, S.W.; Liverton, N.J.; Zibuck, R. Total synthesis of the latrunculins. J. Am. Chem. Soc. 1992, 114, 2995–3007. [Google Scholar] [CrossRef]
- White, J.D.; Kawasaki, M. Total synthesis of (+)-latrunculin A. J. Am. Chem. Soc. 1990, 112, 4991–4993. [Google Scholar] [CrossRef]
- Sikorska, J.; Hau, A.M.; Anklin, C.; Parker-Nance, S.; Davies-Coleman, M.T.; Ishmael, J.E.; McPhail, K.L. Mandelalides A-D, cytotoxic macrolides from a new Lissoclinum species of South African tunicate. J. Org. Chem. 2012, 77, 6066–6075. [Google Scholar] [CrossRef]
- Nazari, M.; Serrill, J.D.; Wan, X.; Nguyen, M.H.; Anklin, C.; Gallegos, D.A.; Smith, A.B.; Ishmael, J.E.; McPhail, K.L. New Mandelalides Expand a Macrolide Series of Mitochondrial Inhibitors. J. Med. Chem. 2017, 60, 7850–7862. [Google Scholar] [CrossRef]
- Villalobos, C.; Gutiérrez, L.G.; Hernández-Morales, M.; del Bosque, D.; Nuñez, L. Mitochondrial control of store-operated Ca2+ channels in cancer: Pharmacological implications. Pharmacol. Res. 2018, 135, 136–143. [Google Scholar] [CrossRef]
- Hamada, Y.; Shibata, M.; Shioiri, T. New methods and reagents in organic synthesis. 55. Total syntheses of patellamides B and C, cytotoxic cyclic peptides from a tunicate 1. Their proposed structures should be corrected. Tetrahedron Lett. 1985, 26, 5155–5158. [Google Scholar] [CrossRef]
- Williams, A.B.; Jacobs, R.S. A marine natural product, patellamide D, reverses multidrug resistance in a human leukemic cell line. Cancer Lett. 1993, 71, 97–102. [Google Scholar] [CrossRef]
- Hamada, Y.; Shibata, M.; Shioiri, T. New methods and reagents in organic synthesis. 56. Total syntheses of patellamides B and C, cytotoxic cyclic peptides from a tunicate 2. Their real structures have been determined by their syntheses. Tetrahedron Lett. 1985, 26, 5159–5162. [Google Scholar] [CrossRef]
- Hamada, Y.; Shibata, M.; Shioiri, T. New methods and reagents in organic synthesis. 58: A synthesis of patellamide a, a cytotoxic cyclic peptide from a tunicate. Revision of its proposed structure. Tetrahedron Lett. 1985, 26, 6501–6504. [Google Scholar] [CrossRef]
- Schmidt, U.; Griesser, H. Total synthesis and structure determination of patellamide B. Tetrahedron Lett. 1986, 27, 163–166. [Google Scholar] [CrossRef]
- García-Reynaga, P.; VanNieuwenhze, M.S. A New Total Synthesis of Patellamide A. Org. Lett. 2008, 10, 4621–4623. [Google Scholar] [CrossRef]
- Bailly, C. Anticancer Properties of Lamellarins. Mar. Drugs 2015, 13, 1105–1123. [Google Scholar] [CrossRef]
- Ballot, C.; Kluza, J.; Lancel, S.; Martoriati, A.; Hassoun, S.M.; Mortier, L.; Vienne, J.-C.; Briand, G.; Formstecher, P.; Bailly, C.; et al. Inhibition of mitochondrial respiration mediates apoptosis induced by the anti-tumoral alkaloid lamellarin D. Apoptosis 2010, 15, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Andersen, R.J.; Faulkner, D.J.; He, C.H.; Van Duyne, G.D.; Clardy, J. Metabolites of the marine prosobranch mollusk Lamellaria sp. J. Am. Chem. Soc. 1985, 107, 5492–5495. [Google Scholar] [CrossRef]
- Pla, D.; Marchal, A.; Olsen, C.A.; Albericio, F.; Alvarez, M. Modular total synthesis of lamellarin D. J. Org. Chem. 2005, 70, 8231–8234. [Google Scholar] [CrossRef]
- Manjappa, K.B.; Lin, J.-M.; Yang, D.-Y. Construction of Pentacyclic Lamellarin Skeleton via Grob Reaction: Application to Total Synthesis of Lamellarins H and D. J. Org. Chem. 2017, 82, 7648–7656. [Google Scholar] [CrossRef]
- Li, Q.; Jiang, J.; Fan, A.; Cui, Y.; Jia, Y. Total Synthesis of Lamellarins D, H, and R and Ningalin B. Org. Lett. 2011, 13, 312–315. [Google Scholar] [CrossRef]
- Ploypradith, P.; Petchmanee, T.; Sahakitpichan, P.; Litvinas, N.D.; Ruchirawat, S. Total Synthesis of Natural and Unnatural Lamellarins with Saturated and Unsaturated D-Rings. J. Org. Chem. 2006, 71, 9440–9448. [Google Scholar] [CrossRef]
- Liermann, J.C.; Opatz, T. Synthesis of Lamellarin U and Lamellarin G Trimethyl Ether by Alkylation of a Deprotonated α-Aminonitrile. J. Org. Chem. 2008, 73, 4526–4531. [Google Scholar] [CrossRef] [PubMed]
- Colligs, V.C.; Dialer, C.; Opatz, T. Synthesis of Lamellarin G Trimethyl Ether by von Miller–Plöchl-Type Cyclocondensation. Eur. J. Org. Chem. 2018, 2018, 4064–4070. [Google Scholar] [CrossRef]
- Imbri, D.; Tauber, J.; Opatz, T. A High-Yielding Modular Access to the Lamellarins: Synthesis of Lamellarin G Trimethyl Ether, Lamellarin η and Dihydrolamellarin η. Chem. Eur. J. 2013, 19, 15080–15083. [Google Scholar] [CrossRef] [PubMed]
- Dialer, C.; Imbri, D.; Hansen, S.P.; Opatz, T. Synthesis of Lamellarin D Trimethyl Ether and Lamellarin H via 6π-Electrocyclization. J. Org. Chem. 2015, 80, 11605–11610. [Google Scholar] [CrossRef]
- Lade, D.M.; Pawar, A.B.; Mainkar, P.S.; Chandrasekhar, S. Total Synthesis of Lamellarin D Trimethyl Ether, Lamellarin D, and Lamellarin H. J. Org. Chem. 2017, 82, 4998–5004. [Google Scholar] [CrossRef]
- Kumar, V.; Awasthi, A.; Salam, A.; Khan, T. Scalable Total Syntheses of Some Natural and Unnatural Lamellarins: Application of a One-Pot Domino Process for Regioselective Access to the Central 1,2,4-Trisubstituted Pyrrole Core. J. Org. Chem. 2019, 84, 11596–11603. [Google Scholar] [CrossRef]
- Klintworth, R.; de Koning, C.B.; Michael, J.P. Demethylative Lactonization Provides a Shortcut to High-Yielding Syntheses of Lamellarins. J. Org. Chem. 2020, 85, 1054–1061. [Google Scholar] [CrossRef]
























































Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).