1. Introduction
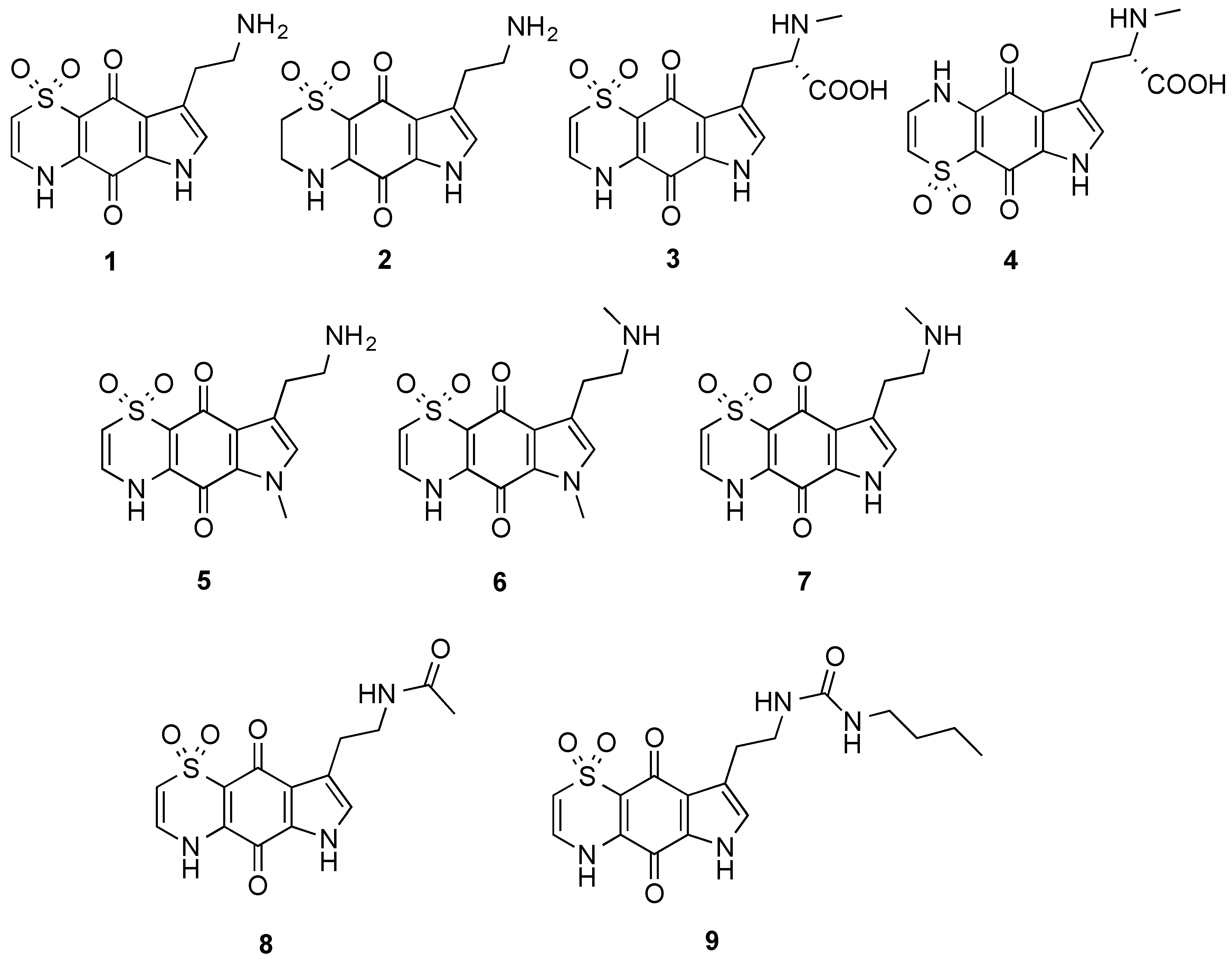
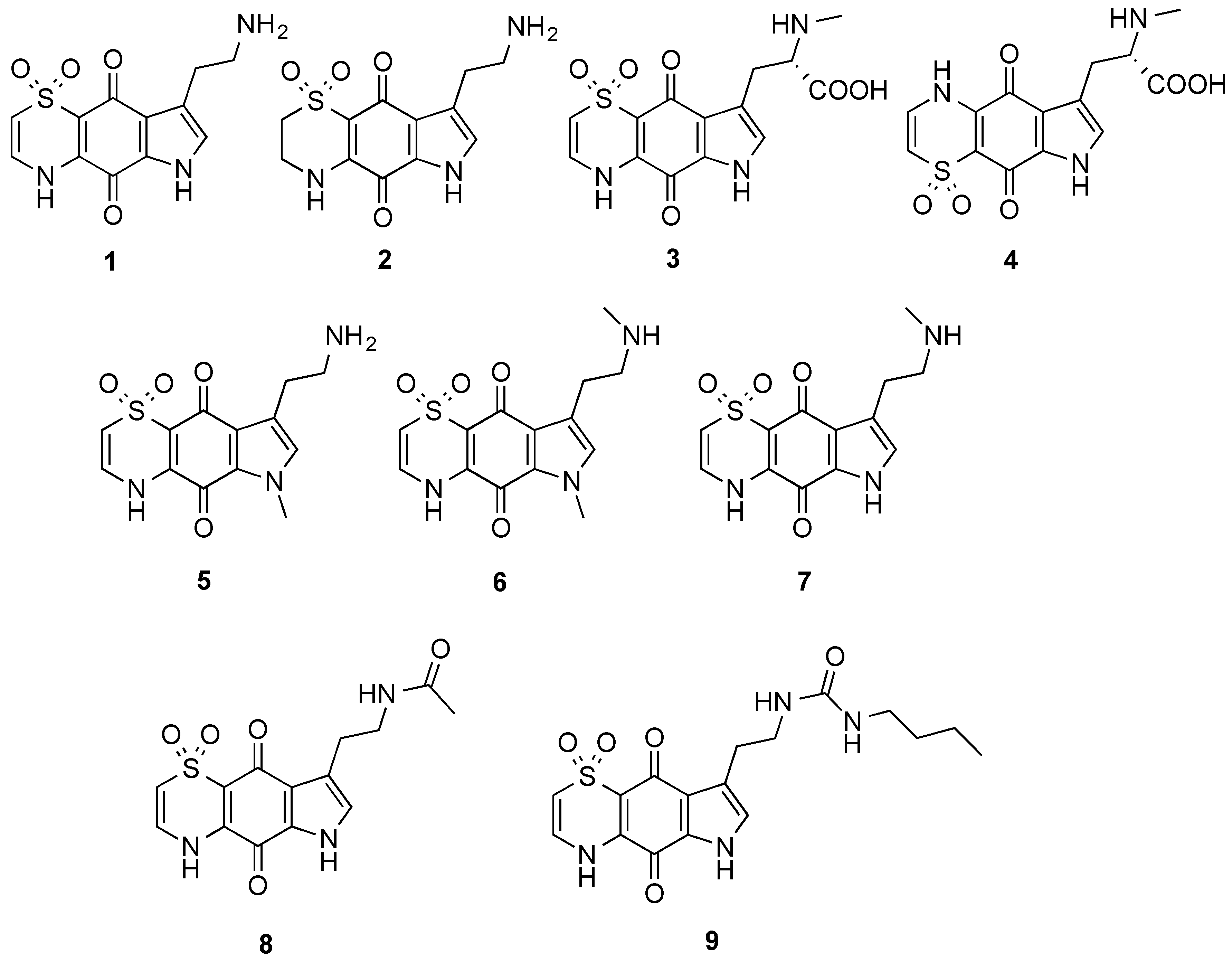
The marine natural products, thiaplakortones A–D (
1–
4), were first reported in 2013 as part of a Medicines for Malaria Venture sponsored research project that aimed to discover new antiplasmodial agents from nature (
Figure 1) [
1]. These unique thiazine-derived secondary metabolites were obtained from the organic extract from the Great Barrier Reef sponge
Plakortis lita, and all were shown to inhibit the
in vitro growth of
Plasmodium falciparum. Thiaplakortone A (
1) was the most active with
in vitro IC
50 values of 6.6 and 51 nM against multidrug-resistant (Dd2) and chloroquine-sensitive (3D7)
P. falciparum lines, respectively [
1]
. Due to supply issues initially curtailing
in vivo malaria studies, total syntheses of thiaplakortones A and B were undertaken and the first total synthesis of
1 and
2, along with a series of mono- and di-methyl analogues (
5–
7) was subsequently reported and some preliminary structure-activity relationships (SAR) ascertained (
Figure 1) [
2]. While
in vivo toxicity effects for several of the synthetic compounds indicated potential liabilities associated with this structure class, the limited number of analogues investigated made it difficult to assess their true potential as antiplasmodial leads [
2]. In order to more thoroughly explore this compound class a larger analogue library based on the thiaplakortone A scaffold was recently undertaken and reported [
3]. This 38-membered library consisted of a series of amide and urea analogues based on the thiaplakortone A natural product scaffold. Several analogues showed potent
in vitro P. falciparum growth inhibition (IC
50 < 500 nM) and good selectivity for
P. falciparum versus human neonatal foreskin fibroblast (NFF) cells (selectivity index >100) [
3]. Furthermore, analogues
8 and
9 displayed good metabolic stability and solubility, and when administered subcutaneously to mice plasma concentrations remained >0.2 µM for 8 h. Analogues
8 and
9 were also well tolerated in mice after subcutaneous administration of 32 mg/kg twice daily for 4 d. In addition, using this dosing protocol blood stage
P. berghei parasitemia was suppressed by 52% for
8 and 26% for
9, relative to controls [
3]. In order to further investigate the thiaplakortone core, we have recently undertaken synthetic studies that resulted in the removal of the ethylamine side-chain present in thiaplakortones A and B in order to determine the biological implications of the –CH
2CH
2NH
2 moiety. Herein we report the total synthesis of several side-chain truncated regioisomers associated with the tricyclic core of thiaplakortones A–D, along with their
in vitro antiplasmodial activity and mammalian cell toxicity.
Figure 1.
Chemical structures of the natural products thiaplakortones A–D (1–4) and some of the previously synthesized thiaplakortone A analogues (5–9).
Figure 1.
Chemical structures of the natural products thiaplakortones A–D (1–4) and some of the previously synthesized thiaplakortone A analogues (5–9).
3. Experimental Section
3.1. General
Melting points were recorded on a capillary melting point apparatus and are uncorrected. Unless otherwise specified,
1H and
13C-NMR spectra were recorded at 30 °C in DMSO-
d6 on a Varian INOVA 500 or 600 NMR spectrometer. The
1H- and
13C-NMR chemical shifts were referenced to the solvent peak for DMSO-
d6 at δ
H 2.50 and δ
C 39.5. LRESIMS was obtained from LC-MS data generated using a Waters Alliance 2790 HPLC equipped with a Waters 996 photodiode array detector and an Alltech evaporative light scattering detector that was attached to a Water ZQ mass spectrometer. HRESIMS were recorded on a Bruker (Billerica, MA, USA) MicrOTof-Q spectrometer (Dionex UltiMate 3000 micro LC system, ESI mode). Analytical thin layer chromatography (TLC) was performed on aluminum-backed 0.2 mm thick silica gel 60 F
254 plates as supplied by Merck (Frankfurt, Germany). Eluted plates were visualized using a 254 nm UV lamp and/or by treatment with a suitable dip followed by heating. These dips included phosphomolybdic acid:Ce(SO
4)
2:H
2SO
4 (conc.):H
2O (37.5 g:7.5 g:37.5 g:720 mL) or KMnO
4:K
2CO
3:5% NaOH aqueous solution:H
2O (3 g:20 g:5 mL:300 mL). Flash chromatographic separations were carried out following protocols defined by Still
et al., [
11] with silica gel 60 (40–63 mm, supplied by GRACE, Baulkham Hills, NSW, Australia) or amino bonded silica gel (Davisil
®) as the stationary phase and using the AR- or HPLC-grade solvents indicated. Semi-preparative HPLC work was performed using a Waters 600 pump and 966 PDA detector, a Gilson 715 liquid handler and a C
18-bonded silica Betasil 5 μm 143 Å column (21.2 mm × 150 mm). Alltech sample preparative C
18-bonded silica (35–75 μm, 150 Å) and an Alltech stainless steel guard cartridge (10 mm × 30 mm) were used for pre-adsorption and HPLC work. A Phenomenex C
18-bonded silica Luna 3 μm 100 Å (4.6 mm × 50 mm) column was used for LC-MS studies. All compounds were analyzed for purity using LC-MS and shown to be >95% pure, unless otherwise stated. Starting materials and reagents were available from the Sigma-Aldrich (St. Louis, MO, USA), Merck (Frankfurt, Germany), AK Scientific Inc. (Union City, CA, USA), Matrix Scientific Chemical (Columbia, SC, USA) and were used as supplied. MeOH and CH
2Cl
2 were dried using a glass contour solvent purification system that is based upon a technology originally described by Grubbs
et al. [
12]. Where necessary, reactions were performed under a nitrogen atmosphere and glassware was heated in an oven at 140 °C then dried under vacuum prior to use. Compounds for biological studies were placed under high vacuum (0.05 mmHg) for several hours before testing to remove trace, residual solvents.
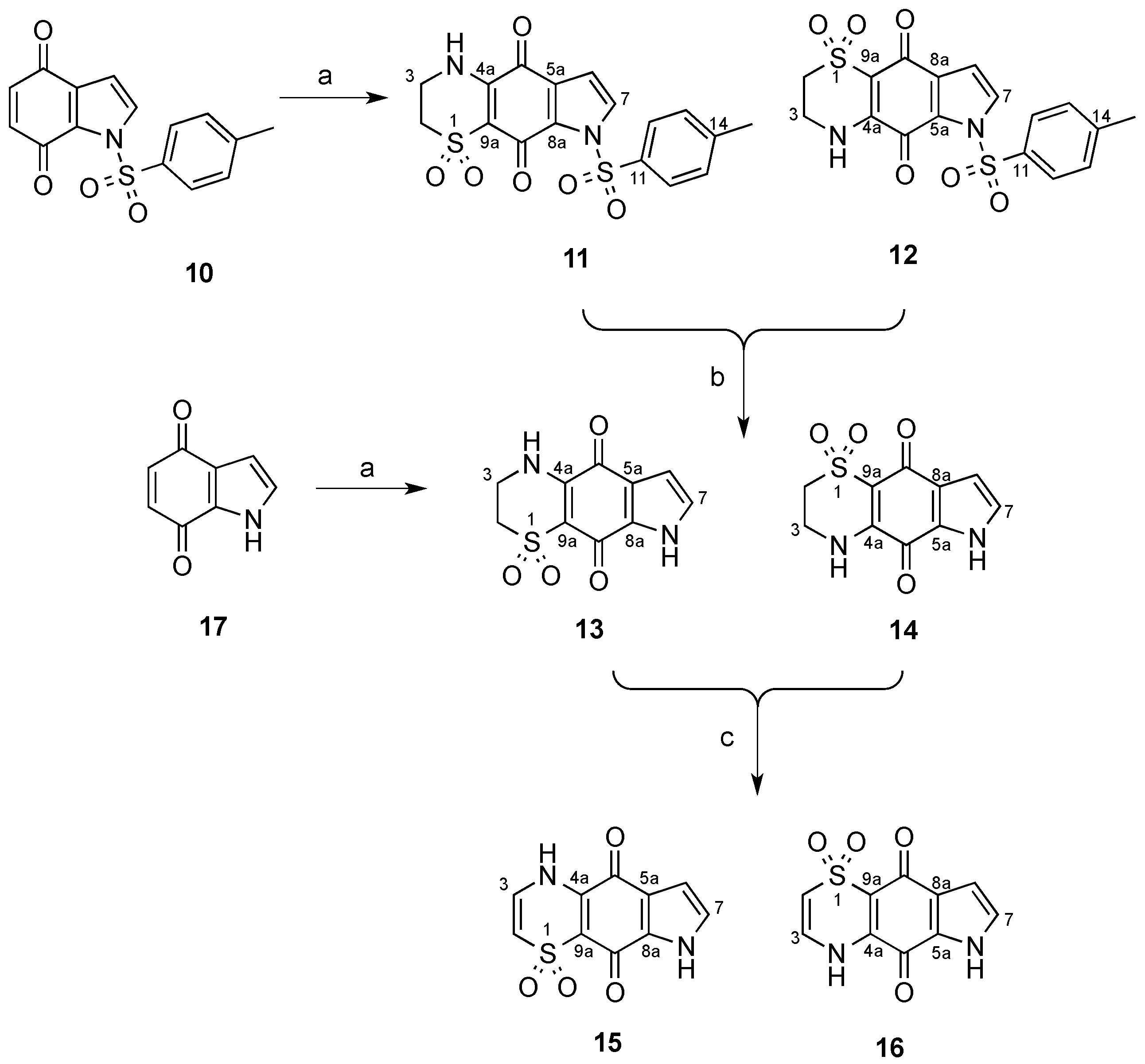
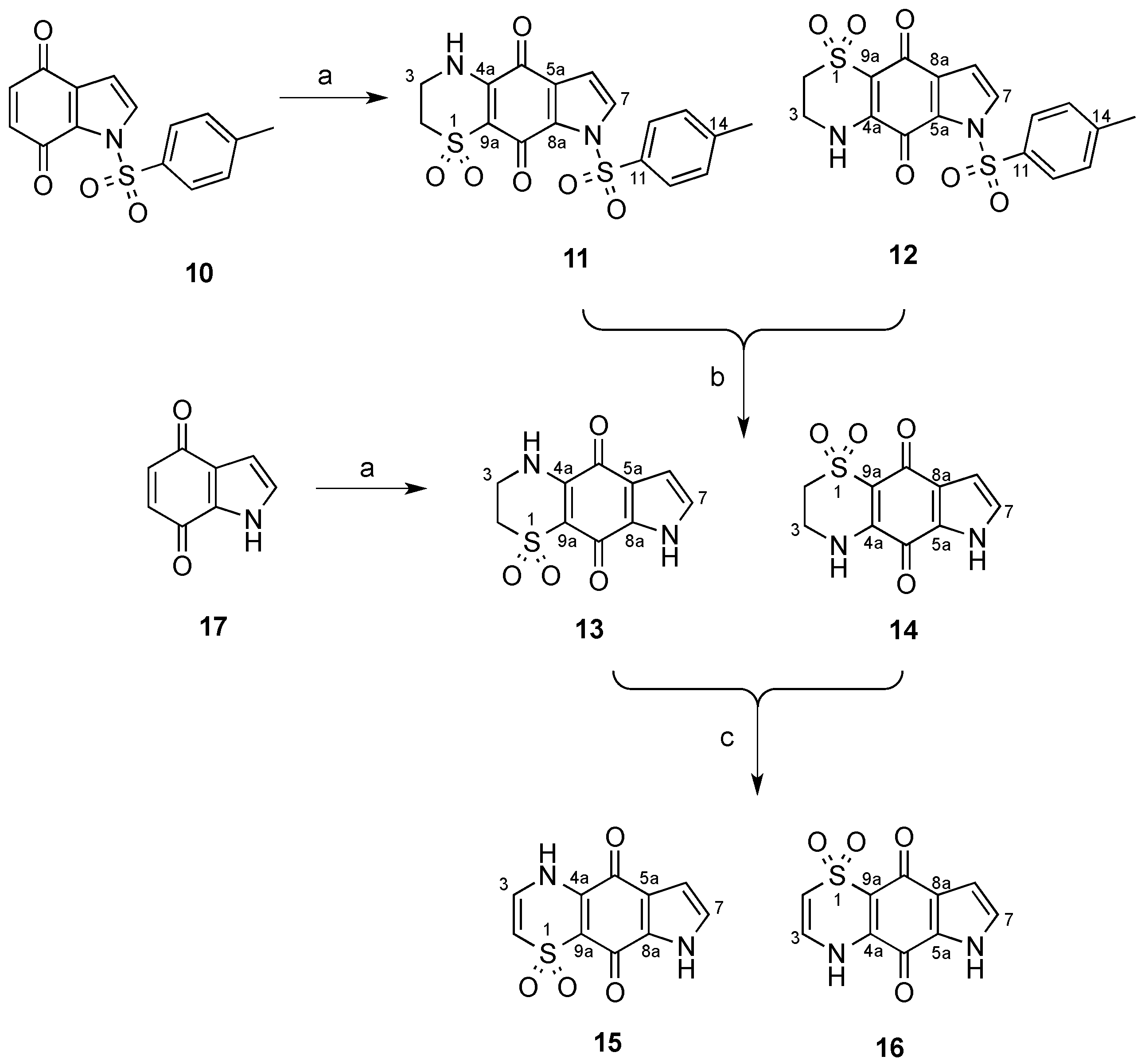
3.2. Synthesis of N-Tosyl Regioisomers 11 and 12
A solution of N-tosyl-1H-indole-4,7-dione (500 mg, 1.66 mmol) in MeCN (80 mL) was treated with a solution of 2-aminoethanesulfinic acid (236 mg, 2.16 mmol) in H2O (50 mL) in one portion. The mixture was stirred for 20 h open to the atmosphere and then H2O was removed by rotary evaporation and the resulting solid collected by vacuum filtration. The crystals were washed with H2O (20 mL) then dried to afford a crude ~1:11 mixture of regioisomers 12 and 11, respectively. This material (236 mg) was pre-adsorbed to C18-bonded silica (1 g) overnight, then packed into a guard cartridge that was subsequently attached to a C18-bonded silica semi-preparative HPLC column. Isocratic HPLC conditions of 90% H2O (0.1% TFA)/10% MeOH (0.1% TFA) were employed for the first 10 min, then a linear gradient to MeOH (0.1% TFA) was run over 40 min, followed by isocratic conditions of MeOH (0.1% TFA) for a further 10 min, all at a flow rate of 9 mL/min. Sixty fractions (60 × 1 min) were collected by time from the start of the HPLC run. All UV active fractions were analyzed by 1H-NMR spectroscopy and MS, and identical fractions were combined. This afforded 12 (7.6 mg, 1%, tR = 37.0–38.0 min) and 11 (69 mg, 10%, tR = 53.0–60.0 min). X-ray quality crystals of 11 were obtained through slow evaporation using a H2O/MeOH (1:9) mix.
Compound 11: Dull orange crystals (H2O/MeOH); mp > 300 °C; 1H-NMR (600 MHz, DMSO-d6) δH 2.42 (3H, s, H-15), 3.27–3.29 (2H, m, H-2), 3.76–3.78 (2H, m, H-3), 6.83 (1H, d, J = 3.4 Hz, H-6), 7.50 (2H, d, J = 8.2 Hz, H-13), 7.90 (2H, d, J = 3.4 Hz, H-7), 8.01 (2H, d, J = 8.2 Hz, H-12), 9.01 (1H, br s, H-4); 13C-NMR (150 MHz, DMSO-d6) δC 21.2 (C-15), 39.3 (C-3), 48.2 (C-2), 107.5 (C-6), 108.2 (C-9a), 126.7 (C-5a), 128.6 (2C, C-12), 129.3 (C-7), 129.8 (2C, C-13), 130.8 (C-8a), 133.5 (C-11), 146.2 (C-14), 146.6 (C-4a), 167.1 (C-9), 175.1 (C-5); (+)-LRESIMS m/z (rel. int.) 407 (100) [M + H]+; (−)-LRESIMS m/z (rel. int.) 405 (100) [M − H]−; (+)-HRESIMS m/z 429.0200 [M + Na]+ (calcd for C17H14N2NaO6S2, 429.0185).
Compound 12: Bright orange amorphous solid; 1H-NMR (600 MHz, DMSO-d6) δH 2.41 (3H, s, H-15), 3.26–3.28 (2H, m, H-2), 3.75–3.77 (2H, m, H-3), 6.81 (1H, d, J = 3.2 Hz, H-8), 7.48 (2H, d, J = 8.4 Hz, H-13), 7.98 (2H, d, J = 8.4 Hz, H-12), 8.12 (2H, d, J = 3.2 Hz, H-7), 9.13 (1H, br s, H-4); 13C-NMR (150 MHz, DMSO-d6) δC 21.1 (C-15), 39.2 (C-3), 47.9 (C-2), 108.0 (C-9a), 108.8 (C-8), 125.9 (C-8a), 128.3 (2C, C-12), 130.0 (2C, C-13), 133.1 (C-11), 133.2 (C-7), 133.3 (C-5a), 146.4 (C-14), 147.4 (C-4a), 166.3 (C-5), 172.9 (C-9); (−)-LRESIMS m/z (rel. int.) 405 (100) [M − H]−; (+)-LRESIMS m/z (rel. int.) 407 (100) [M + H]+; (+)-HRESIMS m/z 429.0167 [M + Na]+ (calcd for C17H14N2NaO6S2, 429.0185).
3.3. Deprotection of the N-Tosyl Regioisomer Mixture to Yield 13
A 1:11 mixture of
12 and
11 (140 mg, 0.35 mmol) in a saturated solution of NaHCO
3 (5 mL) and MeOH (50 mL) was heated to reflux for 2.5 h. The mixture was acidified with HCl (32% aqueous) to pH 6 then concentrated and subjected to flash chromatography (silica, 1:10
v/
v MeOH/CH
2Cl
2 elution) to afford a 1:26 mixture of compounds
14 and
13 (59 mg, 68%). This material (59 mg) was pre-adsorbed to C
18-bonded silica (1 g) overnight, then packed into a guard cartridge that was attached to a C
18-bonded silica semi-preparative HPLC column. Application of the same reversed-phase HPLC purification method described above (
Section 3.2) afforded
13 (15 mg, 11%,
tR = 23.0–24.0 min) as an orange powder.
Compound 13: Orange amorphous solid; 1H-NMR (500 MHz, DMSO-d6) δH 3.31–3.33 (2H, m, H-2), 3.81–3.84 (2H, m, H-3), 6.57 (1H, d, J = 2.8 Hz, H-6), 7.13 (1H, d, J = 2.8 Hz, H-7), 9.05 (1H, brs, H-4), 12.76 (1H, brs, H-8); 13C-NMR (125 MHz, DMSO-d6) δC 39.4 (C-3), 48.2 (C-2), 107.5 (C-6), 120.7 (C-5a), 125.4 (C-7), 132.9 (C-8a), 148.2 (C-4a), 169.8 (C-9), 174.1 (C-5); (+)-LRESIMS m/z (rel. int.) 253 (100) [M + H]+; (−)-LRESIMS m/z (rel. int.) 251 (100) [M − H]−; (+)-HRESIMS m/z 275.0104 [M + Na]+ (calcd for C10H8N2NaO4S, 275.0097).
3.4. Synthesis of Regioisomers 13 and 14
A solution of 1H-indole-4,7-dione (10, 504 mg, 3.4 mmol) in MeCN (160 mL) was treated with a solution of 2-aminoethanesulfinic acid (482 mg, 4.42 mmol) in H2O (50 mL) in one portion and the mixture stirred for 20 h under an atmosphere of O2. The MeCN and H2O were removed by rotary evaporation to afford an orange solid, which was purified by flash chromatography (silica, 1:5 v/v MeOH/CH2Cl2 elution) to afford a 3.3:1 mixture of regioisomers 14 and 13, respectively (307 mg, 36%). A portion of this material (40 mg) was pre-adsorbed to C18-bonded silica (1 g) overnight, then packed into a guard cartridge that was attached to a C18-bonded silica semi-preparative HPLC column. Isocratic HPLC conditions of 95% H2O (0.1% TFA)/5% MeOH (0.1% TFA) were employed for the first 10 min, then a linear gradient to 50% H2O (0.1% TFA)/50% MeOH (0.1% TFA) was run over 40 min, followed by a linear gradient to MeOH (0.1% TFA) in 1 min, then isocratic conditions of MeOH (0.1% TFA) for a further 9 min, all at a flow rate of 9 mL/min. Sixty fractions (60 × 1 min) were collected by time from the start of the HPLC run. All UV active fractions were analyzed by 1H-NMR spectroscopy and MS, and identical fractions were combined. This yielded 14 (5.0 mg, 1%, tR = 25.0–27.0 min) as a bright orange powder.
Compound 14: Bright orange amorphous solid; 1H-NMR (500 MHz, DMSO-d6) δH 3.28–3.33 (2H, m, H-2), 3.79–3.82 (2H, m, H-3), 6.53 (1H, d, J = 2.5 Hz, H-8), 7.40 (1H, d, J = 2.5 Hz, H-7), 8.89 (1H, br s, H-4), 12.81 (1H, br s, H-6); 13C-NMR (125 MHz, DMSO-d6) δC 39.3 (C-3), 48.4 (C-2), 108.4 (2C, C-9a, C-8), 127.2 (C-8a), 128.2 (C-5a), 130.1 (C-7), 147.5 (C-4a), 167.9 (C-5), 174.7 (C-9); (+)-LRESIMS m/z (rel. int.) 253 (100) [M + H]+; (−)-LRESIMS m/z (rel. int.) 251 (100) [M − H]−; (+)-HRESIMS m/z 275.0087 [M + Na]+ (calcd for C10H8N2NaO4S, 275.0097).
3.5. Synthesis of Regioisomers 15 and 16
A 3.3:1 regioisomeric mixture of
14 and
13 (90 mg, 0.36 mmol) from the synthesis described above (
Section 3.4) in a solution of MeOH (10 mL) was treated with an aqueous KOH solution (6 mL, 12 M). The magnetically stirred reaction mixture was purged with O
2, and maintained at 60 °C under a balloon of O
2 for 4 h. The reaction mixture was cooled to 0 °C, carefully neutralized by the addition of an aqueous solution of HCl (1 M) and then the mixture was concentrated
in vacuo to afford a residue that was subjected to flash chromatography through a small plug of silica (1:10
v/
v MeOH/CH
2Cl
2 elution) and after concentration of the eluent
in vacuo, the residue (40 mg) was pre-adsorbed to C
18-bonded silica (1 g) overnight, then packed into a guard cartridge that was attached to a C
18-bonded silica semi-preparative HPLC column. Application of same reversed-phase HPLC purification method described above (
section 3.4) resulted in the purification of compounds
16 (15 mg, 17%,
tR = 28.0–30.0 min) and
15 (5 mg, 6%,
tR = 32.0–34.0 min) as yellow and orange powders, respectively.
Compound 15: Orange amorphous solid; 1H-NMR (500 MHz, DMSO-d6) δH 6.49 (1H, d, J = 8.8 Hz, H-2), 6.66 (1H, d, J = 2.8 Hz, H-6), 7.07 (1H, d, J = 8.8 Hz, H-3), 7.27 (1H, d, J = 2.8 Hz, H-7), 11.07 (1H, br s, H-4), 12.99 (1H, br s, H-8); 13C-NMR (125 MHz, DMSO-d6) δC 108.0 (C-6), 111.7 (C-2), 113.4 (C-9a), 121.8 (C-5a), 126.8 (C-7), 130.2 (C-3), 131.3 (C-8a), 140.5 (C-4a), 172.2 (C-9), 174.1 (C-5); (+)-LRESIMS m/z (rel. int.) 251 (100) [M + H]+; (−)-LRESIMS m/z (rel. int.) 249 (100) [M − H]−; (+)-HRESIMS m/z 272.9939 [M + Na]+ (calcd for C10H6N2NaO4S, 272.9940).
Compound 16: Yellow amorphous solid; 1H NMR (500 MHz, DMSO-d6) δH 6.42 (1H, d, J = 8.8 Hz, H-2), 6.60 (1H, d, J = 2.6 Hz, H-8), 7.04 (1H, d, J = 8.8 Hz, H-3), 7.43 (1H, d, J = 2.6 Hz, H-7), H-4 and H-6 not observed; 13C-NMR (125 MHz, DMSO-d6) δC 108.5 (C-8), 111.6 (C-2), 113.9 (C-9a), 126.9 (C-8a), 127.6 (C-5a), 129.8 (C-7), 130.1 (C-3), 140.2 (C-4a), 167.9 (C-5), 177.3 (C-9); (+)-LRESIMS m/z (rel. int.) 251 (100) [M + H]+; (−)-LRESIMS m/z (rel. int.) 249 (100) [M − H]−; (+)-HRESIMS m/z 272.9933 [M + Na]+ (calcd for C10H6N2NaO4S, 272.9940).
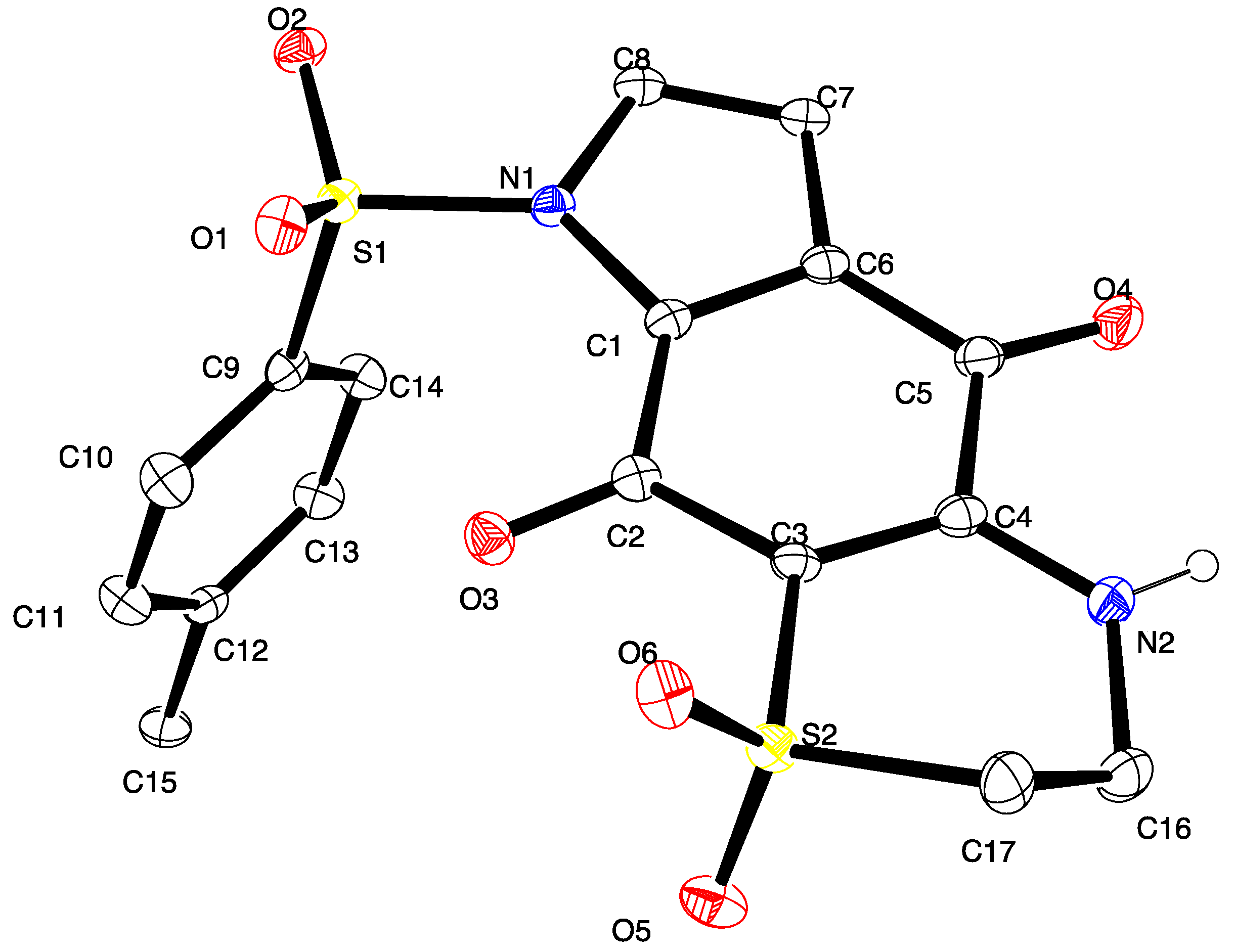
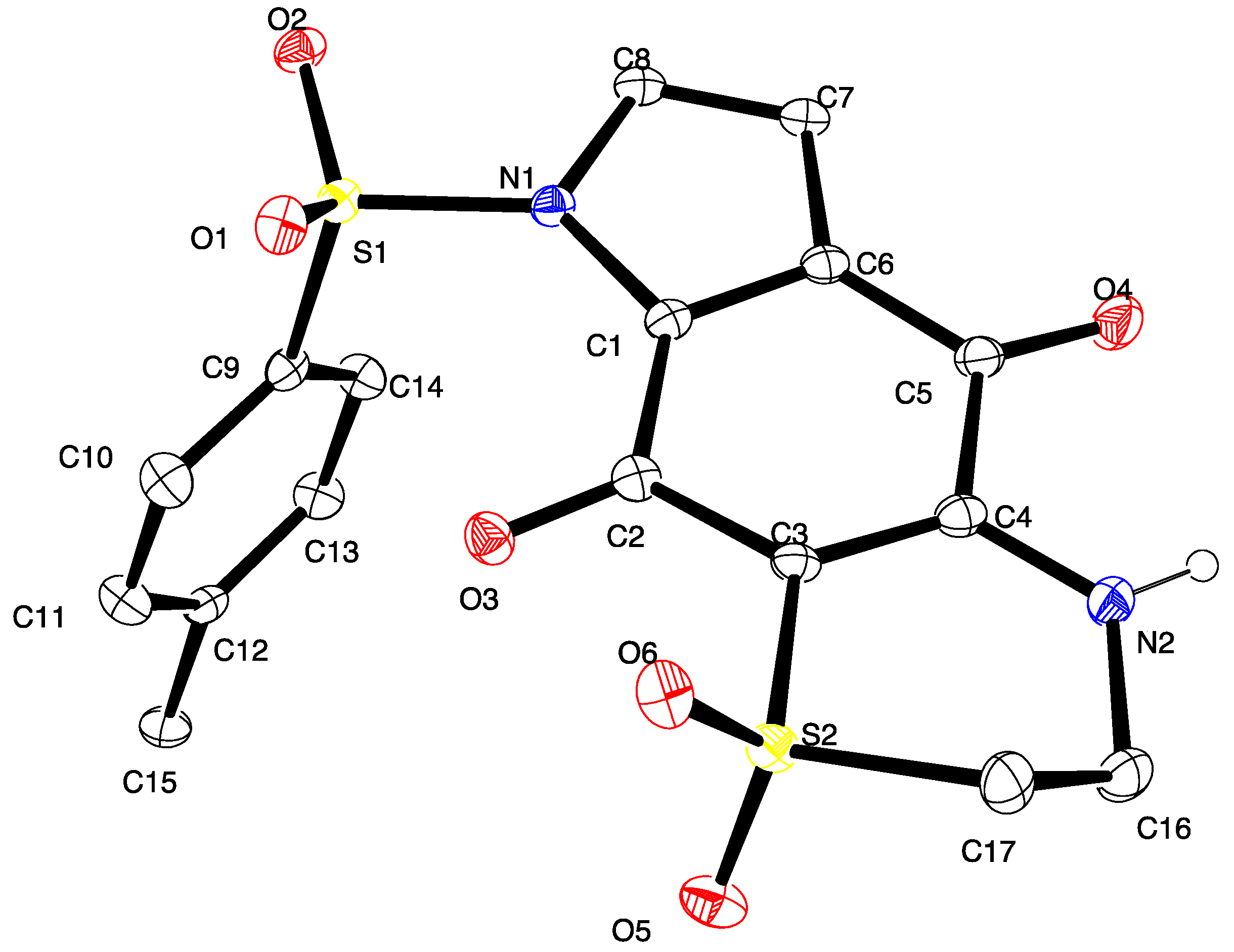
3.6. X-ray Crystallography Studies on Compound 11
Intensity data were collected with an Oxford Diffraction SuperNova CCD diffractometer using Cu-Kα radiation, the temperature during data collection was maintained at 100.0(1) using an Oxford Cryosystems cooling device. The structure was solved by direct methods and difference Fourier Synthesis [
13]. Hydrogen atoms bound to the carbon atom were placed at their idealized positions using appropriate HFIX instructions in SHELXL, and included in subsequent refinement cycles. Hydrogen atoms attached to nitrogen were located from difference Fourier maps and refined freely with isotropic displacement parameters. Thermal ellipsoid plots were generated using the program ORTEP-3 [
14] integrated within the WINGX suite of programs [
15]. Full details of the data collection and refinement and tables of atomic coordinates, bond lengths and angles, and torsion angles have been deposited with the Cambridge Crystallographic Data Centre (CCDC 1416796). Copies can be obtained free of charge on application at the following address:
http://www.ccdc.cam.ac.uk.
Crystal data for compound 11: C17H14N2O6S2, M = 406.42, T = 100.0(2) K, λ = 1.5418 Å, Triclinic, space group P21/c, a = 11.6802(7), b = 28.0975(14), c = 10.3047(6) Å, β = 91.539(5)° V = 3380.6(3) Å3, Z = 8, Z′ = 2, Dc = 1.597 Mg·M−3, μ = 3.230 mm−1, F(000) = 1680, crystal size 0.49 mm × 0.38 mm × 0.31 mm. θmax = 67.6°, 10,831 reflections measured, 5907 independent reflections (Rint = 0.051) the final R = 0.0559 [I > 2σ(I), 5134 data] and wR(F2) = 0.1577 (all data) GOOF = 1.027.
3.7. P. Falciparum Growth Inhibition Assay
P. falciparum growth inhibition assays were carried out using an isotopic microtest, as previously described [
16]. Briefly,
in vitro cultured
P. falciparum infected erythrocytes (1.0% parasitemia and 1.0% hematocrit) were seeded into triplicate wells of 96 well tissue culture plates containing vehicle control (DMSO), positive control [chloroquine (Sigma-Aldrich, St. Louis, MO, USA), catalogue #C6628, >98%] or test compounds and incubated under standard
P. falciparum culture conditions with 0.5 μCi [
3H]-hypoxanthine. The final concentration of DMSO vehicle was <0.5% in all assay wells (non-toxic). After 48 h cells were harvested onto 1450 MicroBeta filter mats (PerkinElmer, Waltham, Massachusetts, USA) and [
3H] incorporation determined using a 1450 MicroBeta liquid scintillation counter. Percentage inhibition of growth compared to matched DMSO controls was determined and IC
50 values were calculated using linear interpolation of inhibition curves [
17]. The mean IC
50 or % inhibition (±SD) was calculated for three independent experiments, each carried out in triplicate.




{kind=link}
{kind=link}
{kind=link}