
Design and Synthesis of Novel Xyloketal Derivatives and Their Protective Activities against H2O2-Induced HUVEC Injury


Abstract
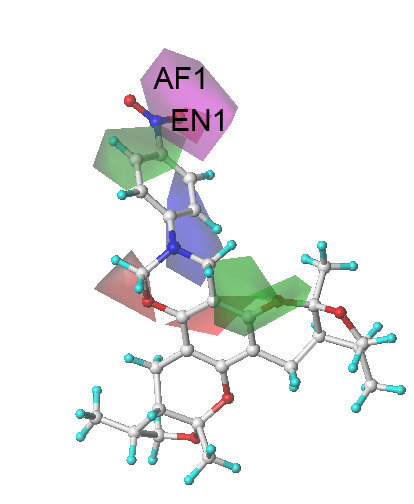
:
1. Introduction
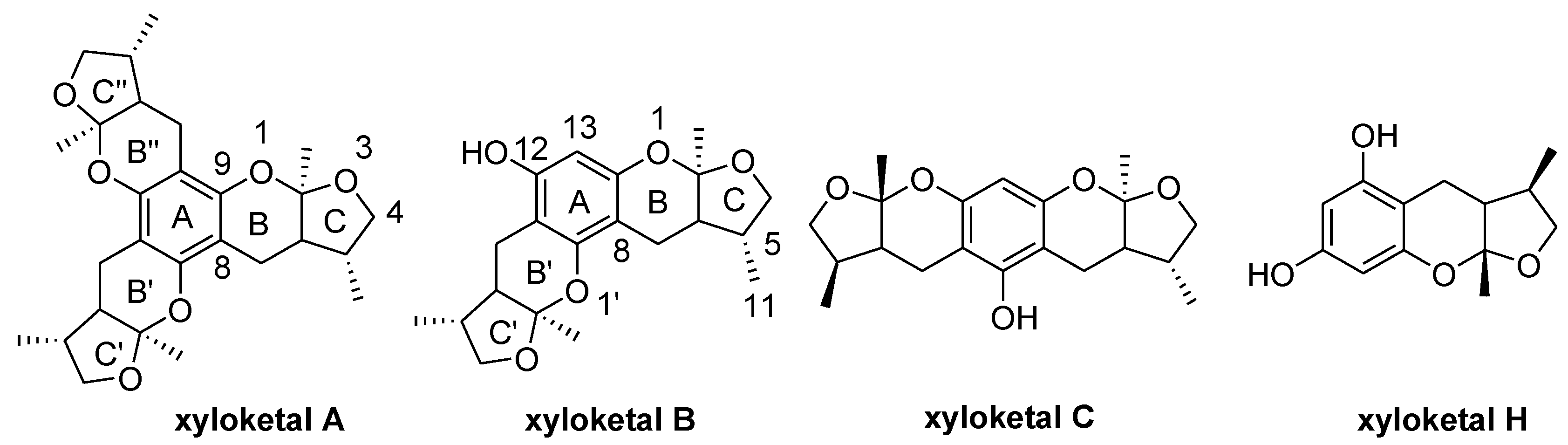
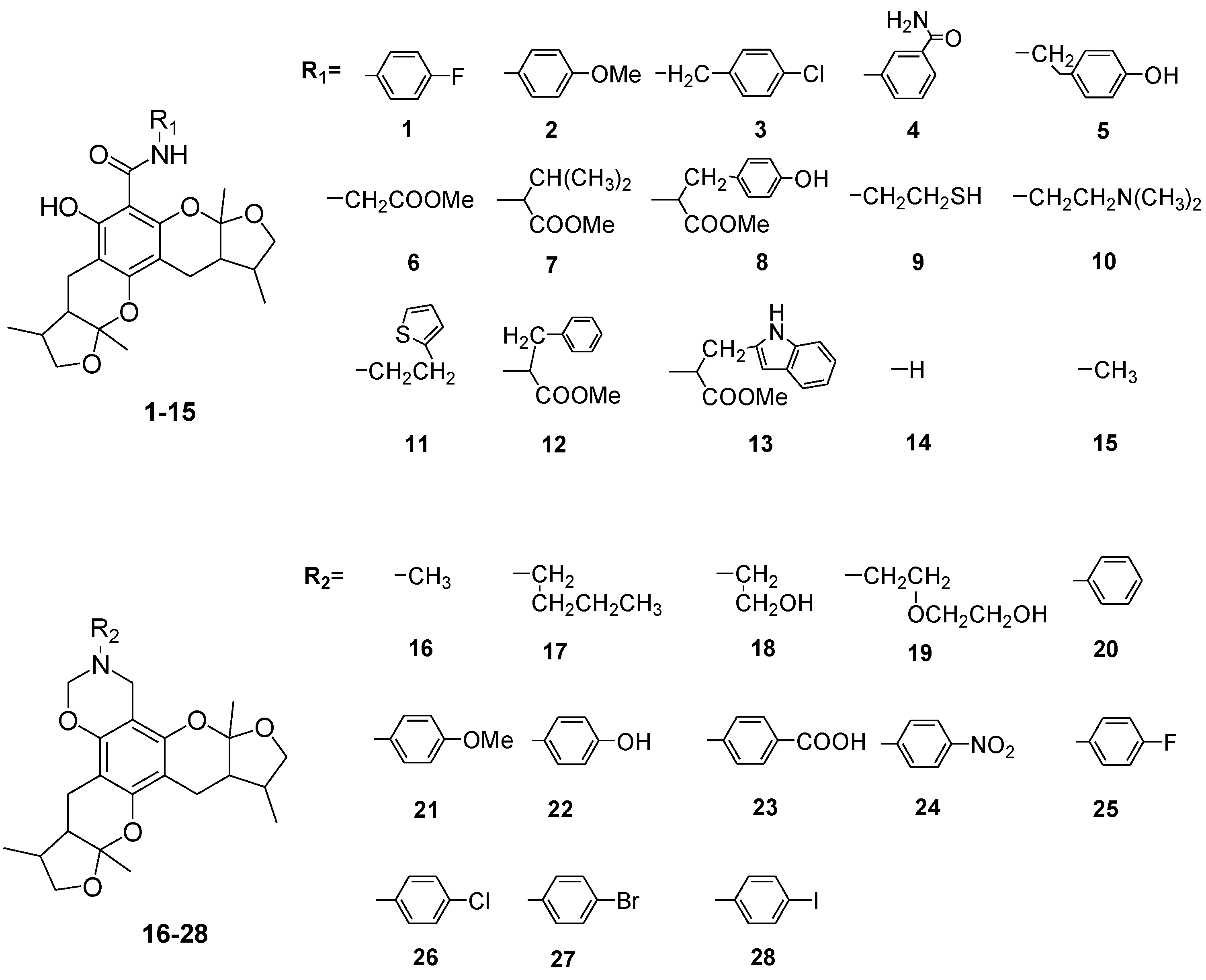
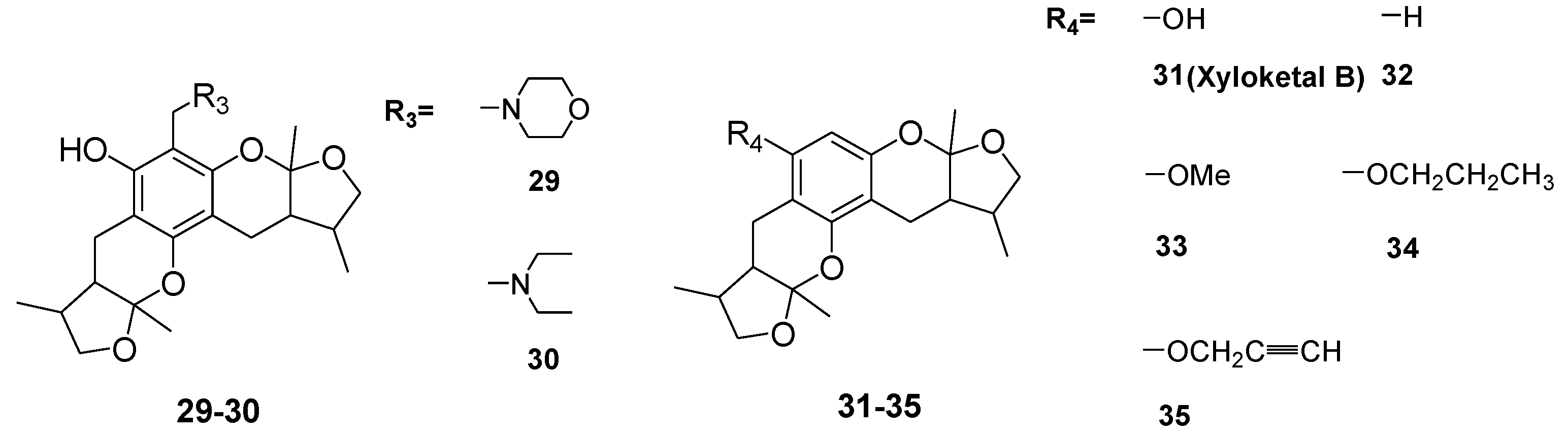
2. Results and Discussion
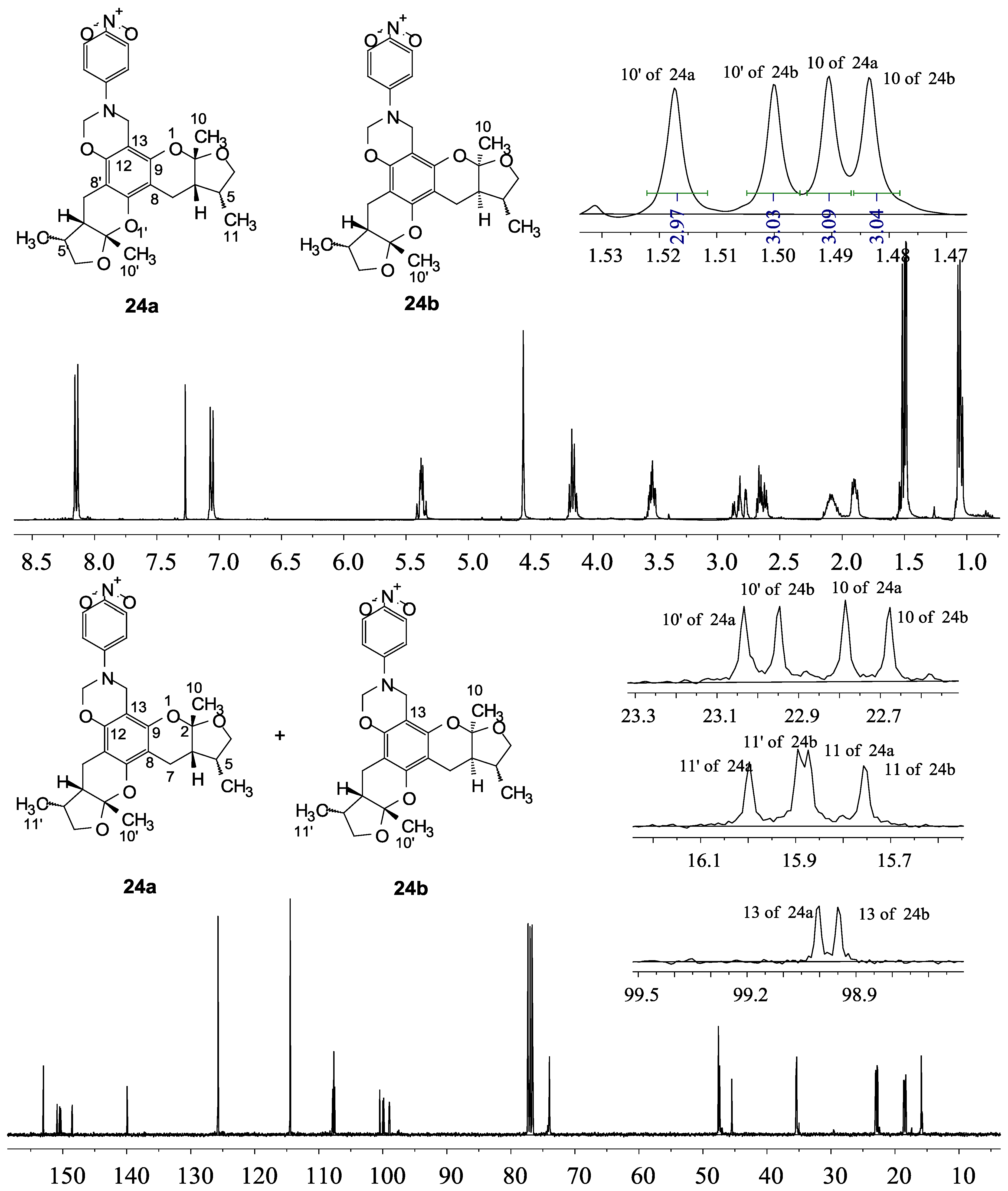
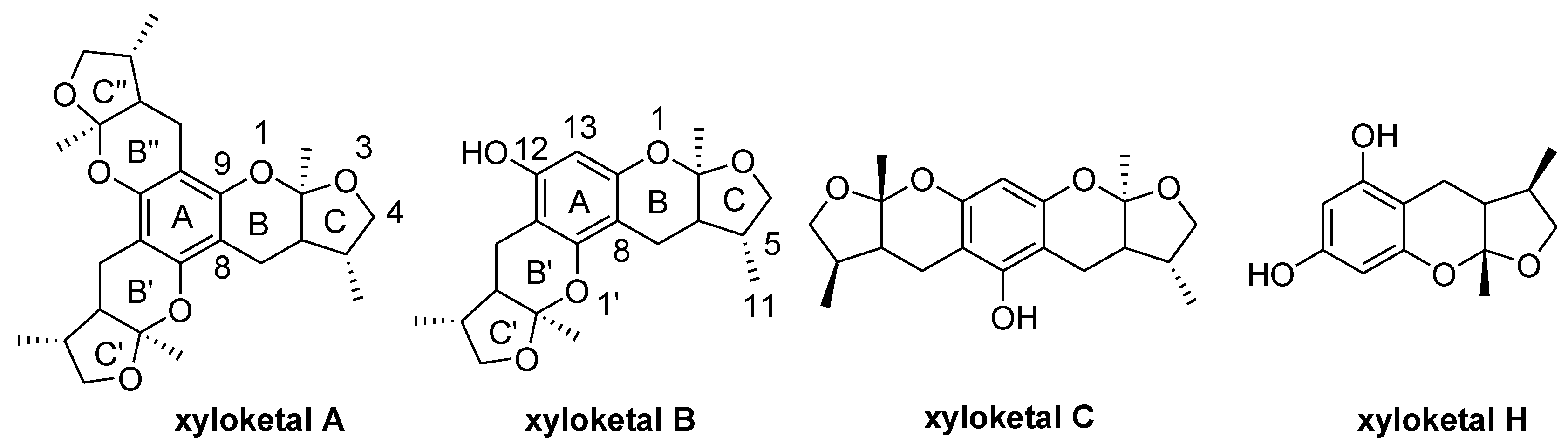
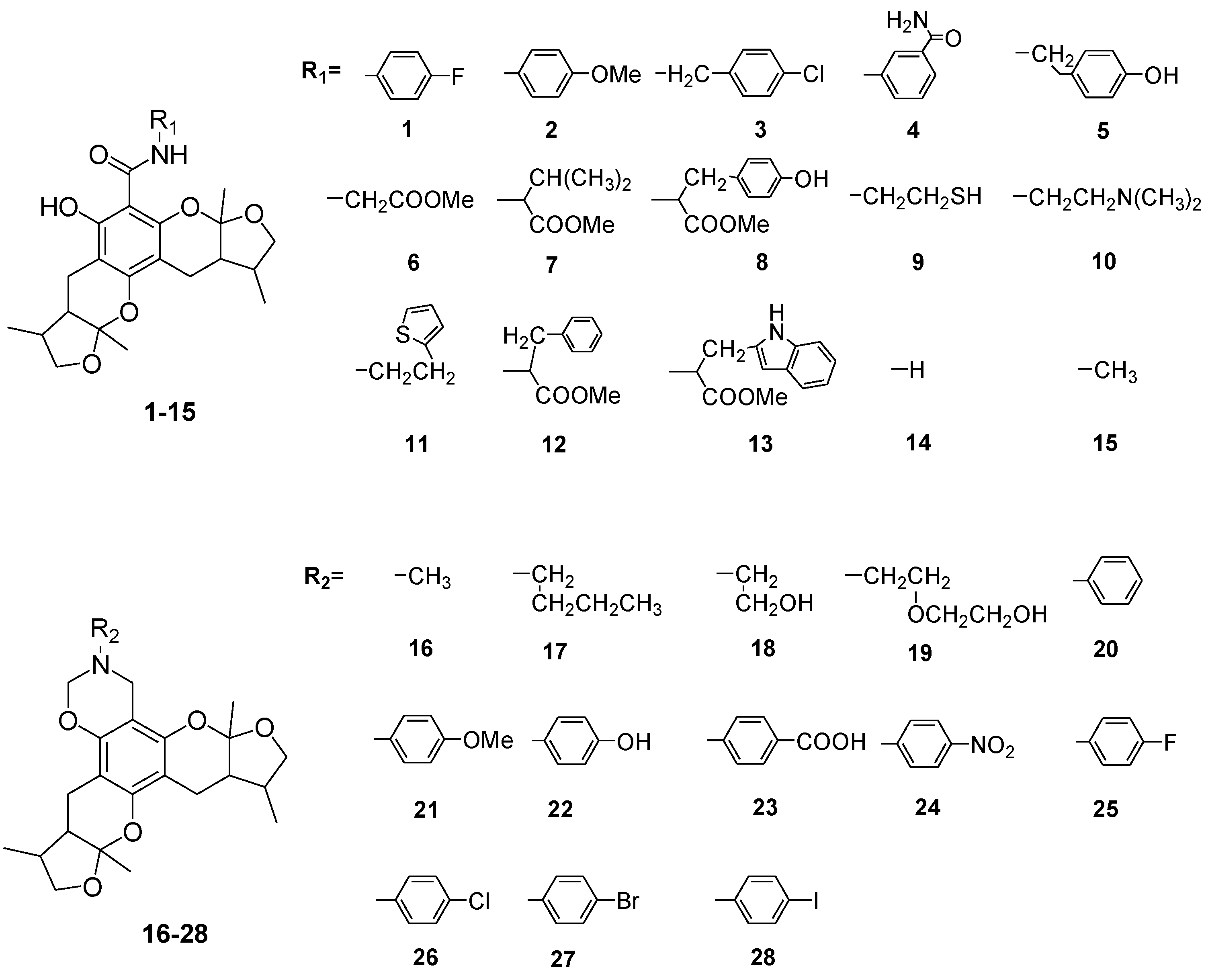
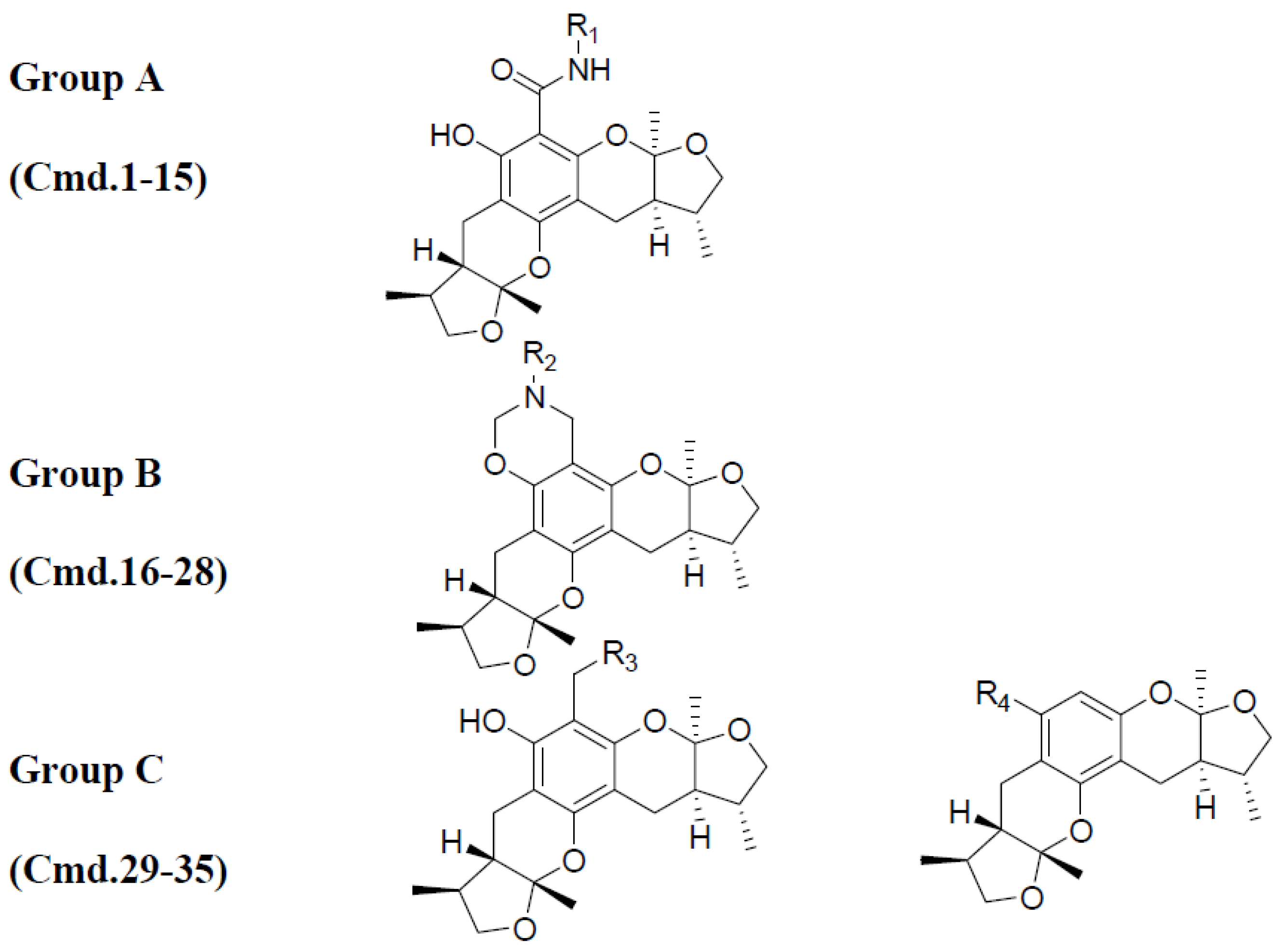
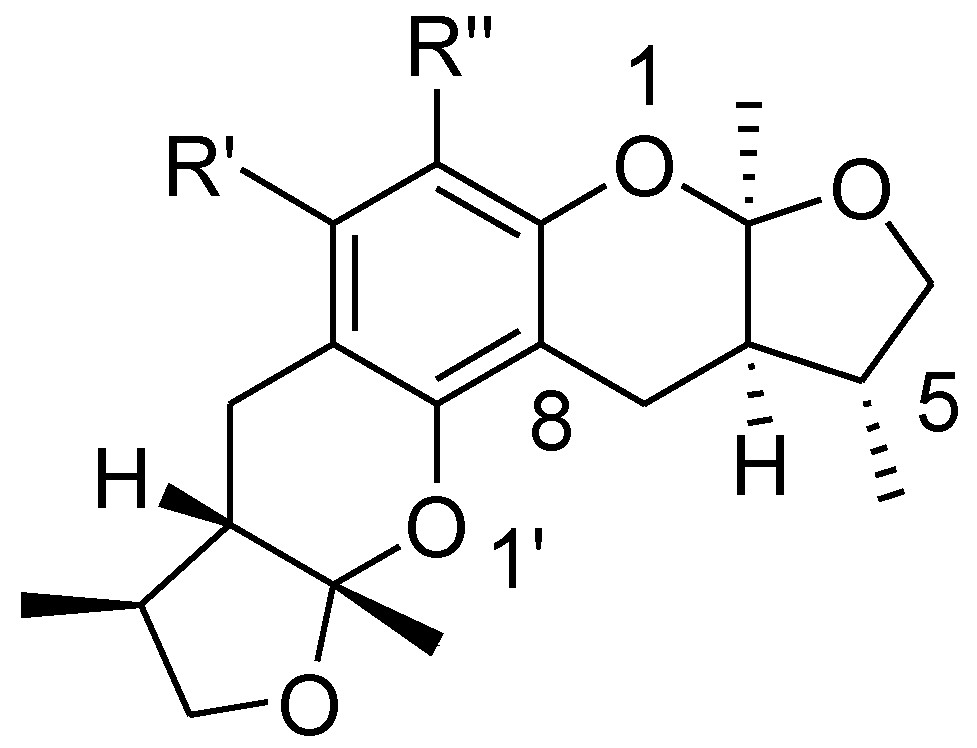
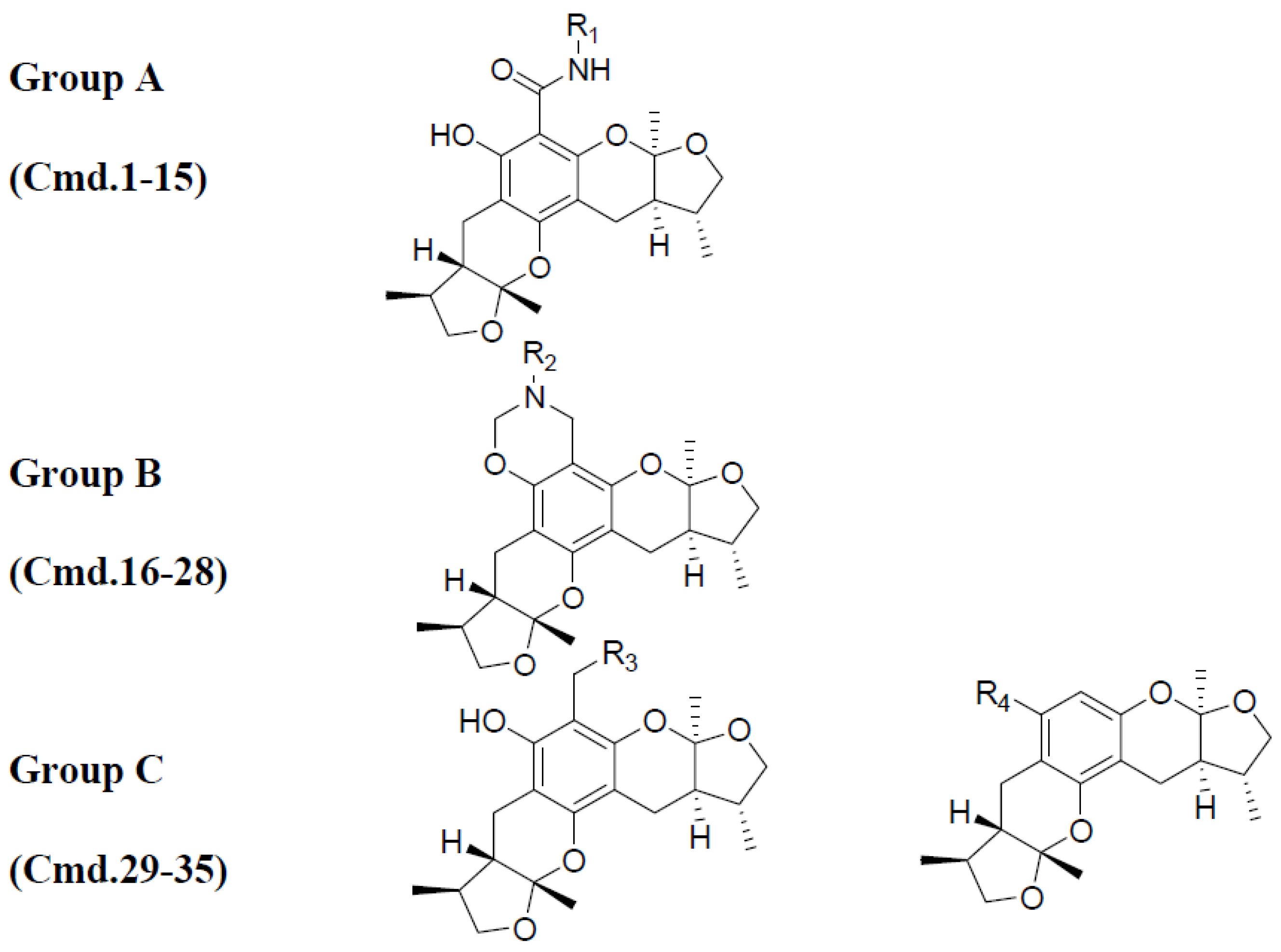
2.1. Chemistry
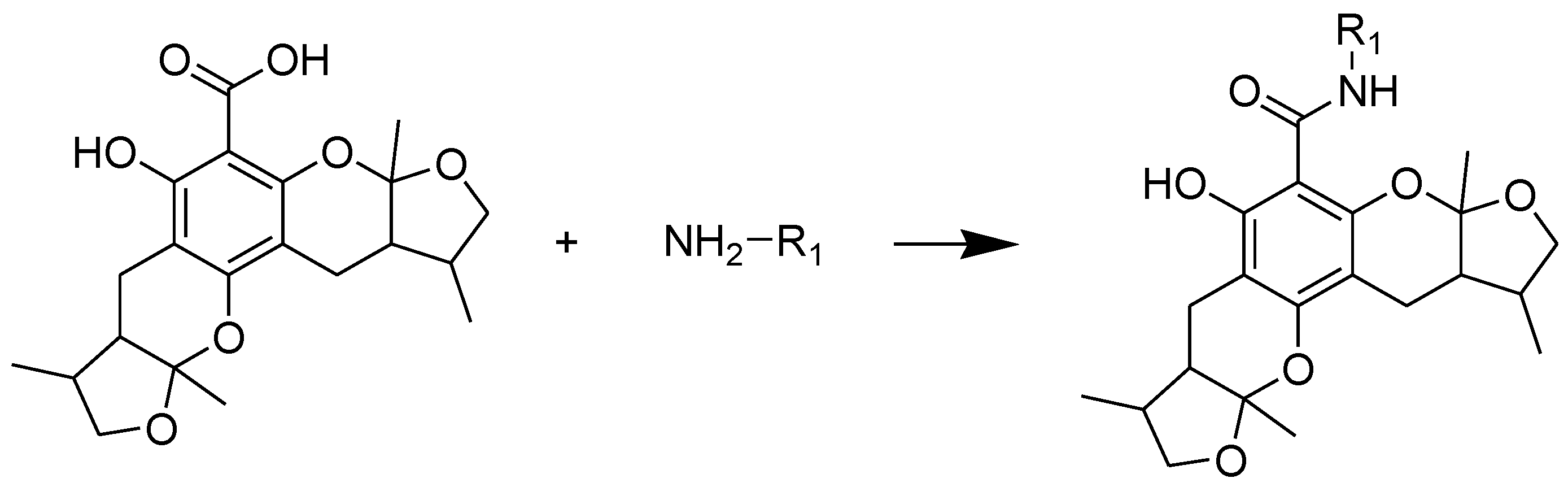
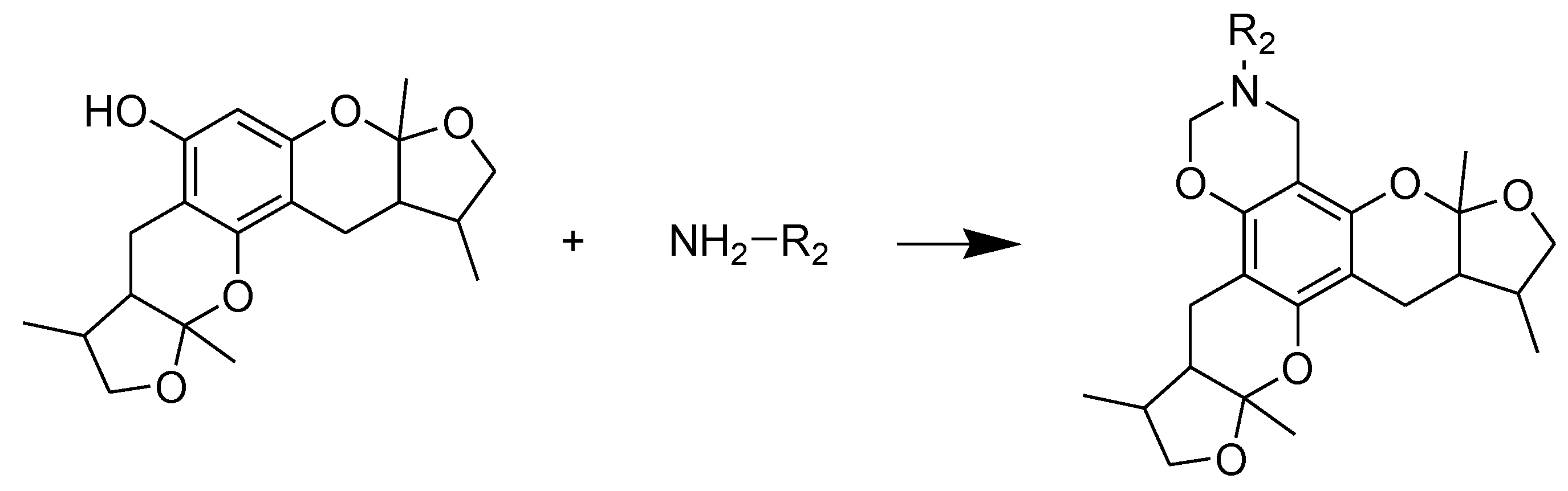
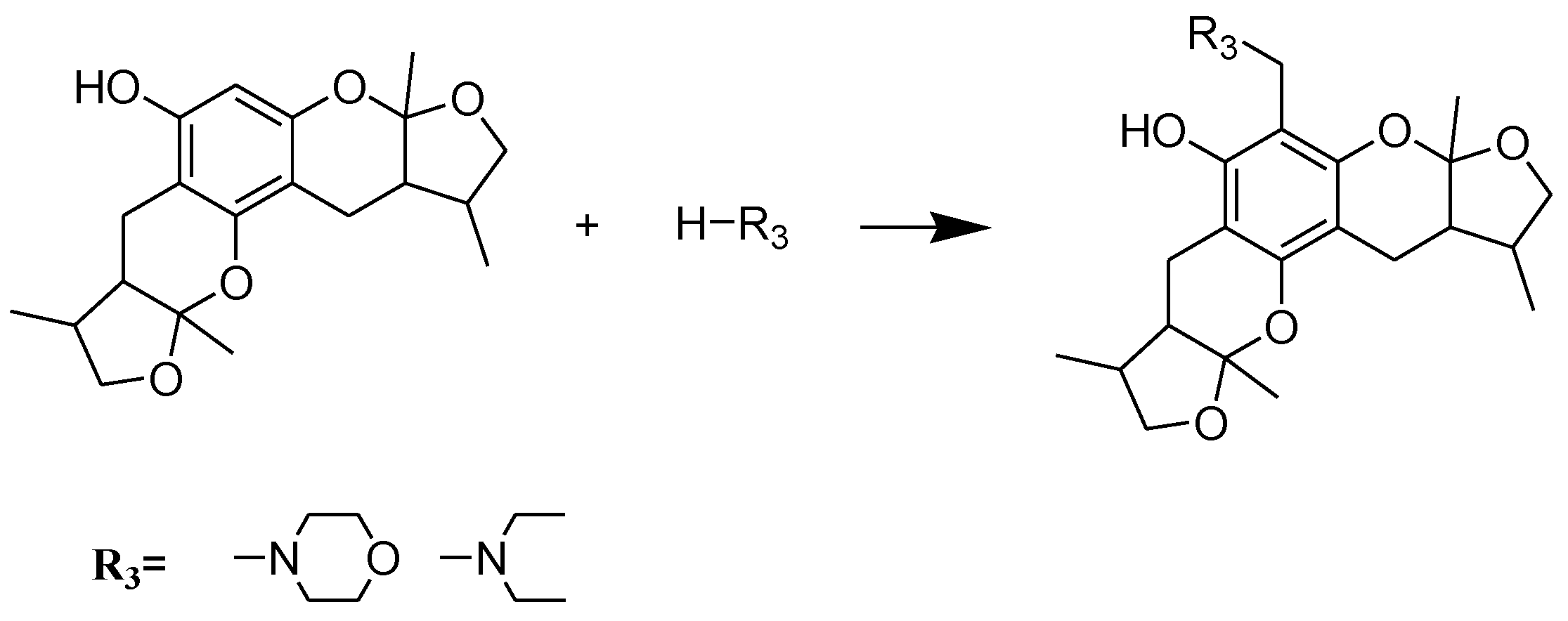
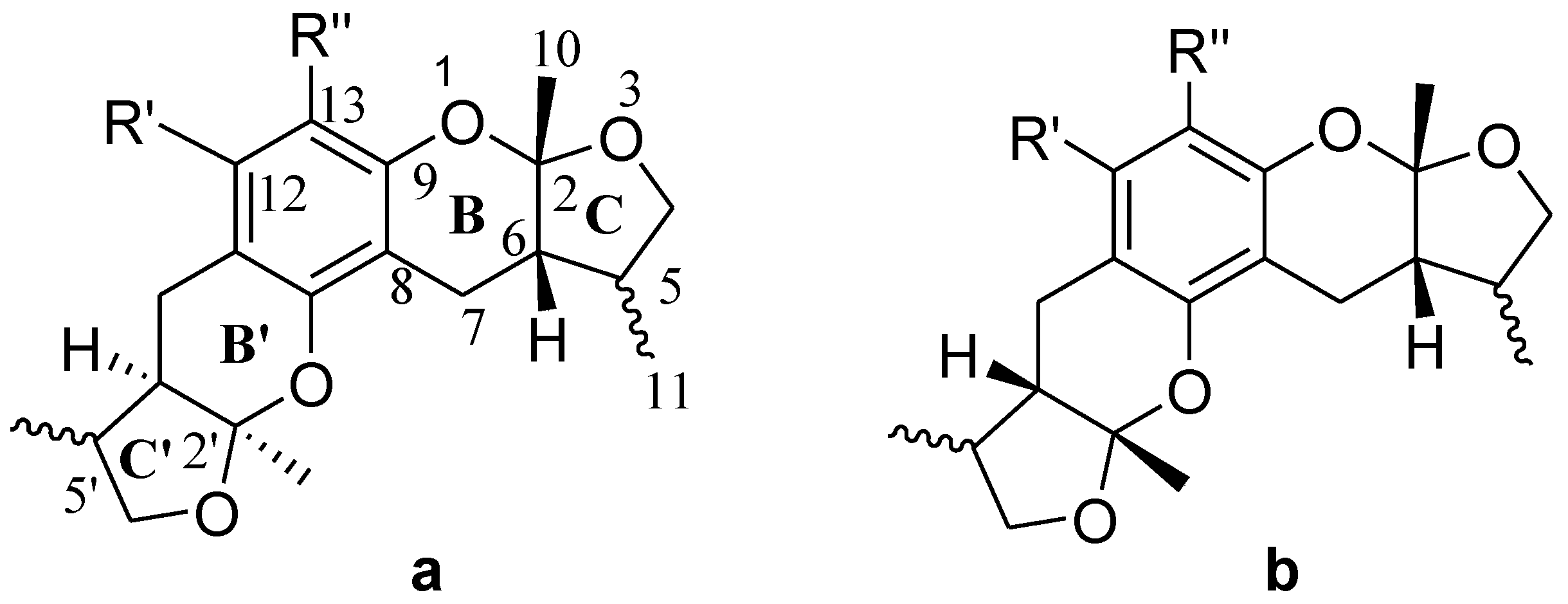

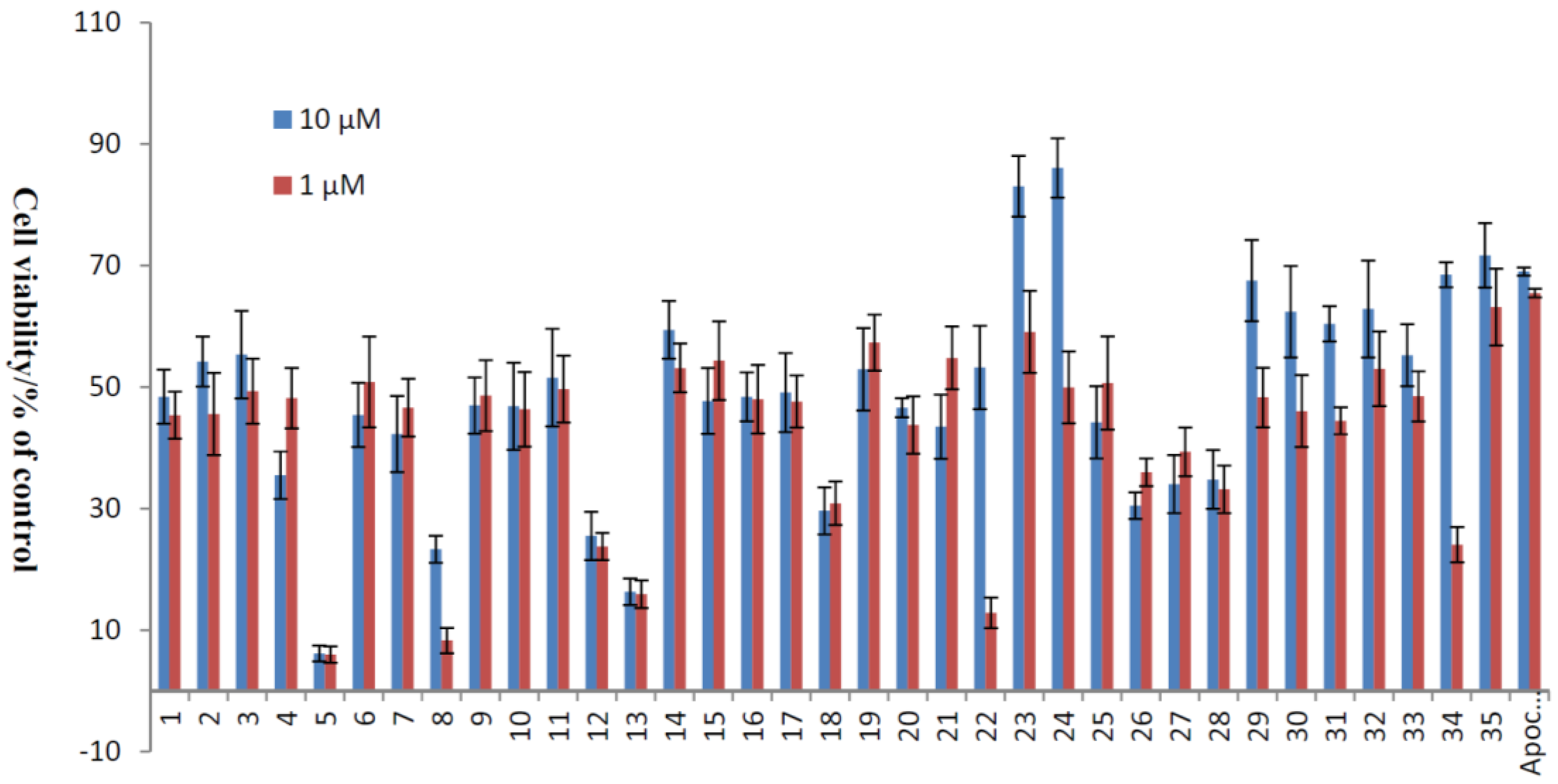
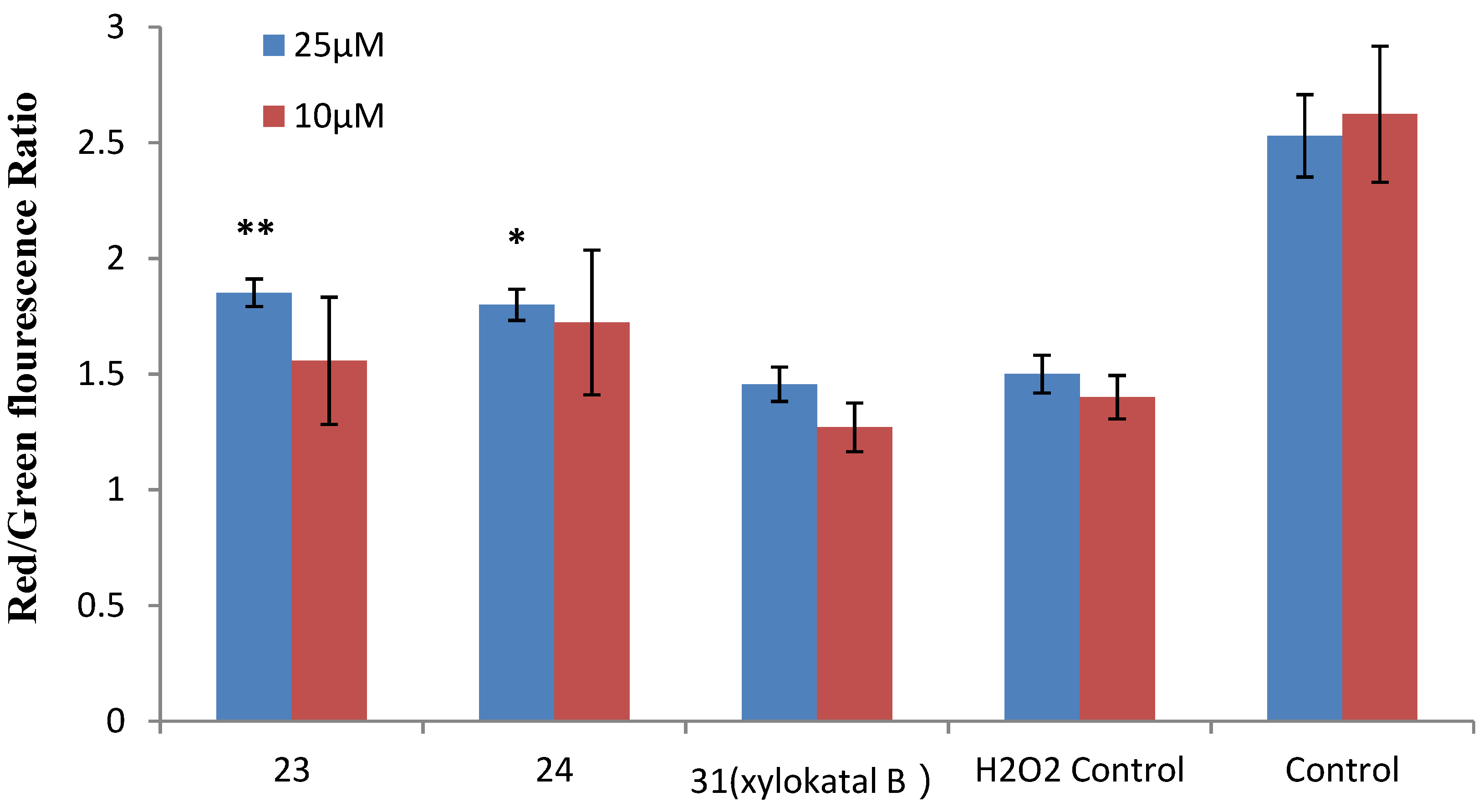
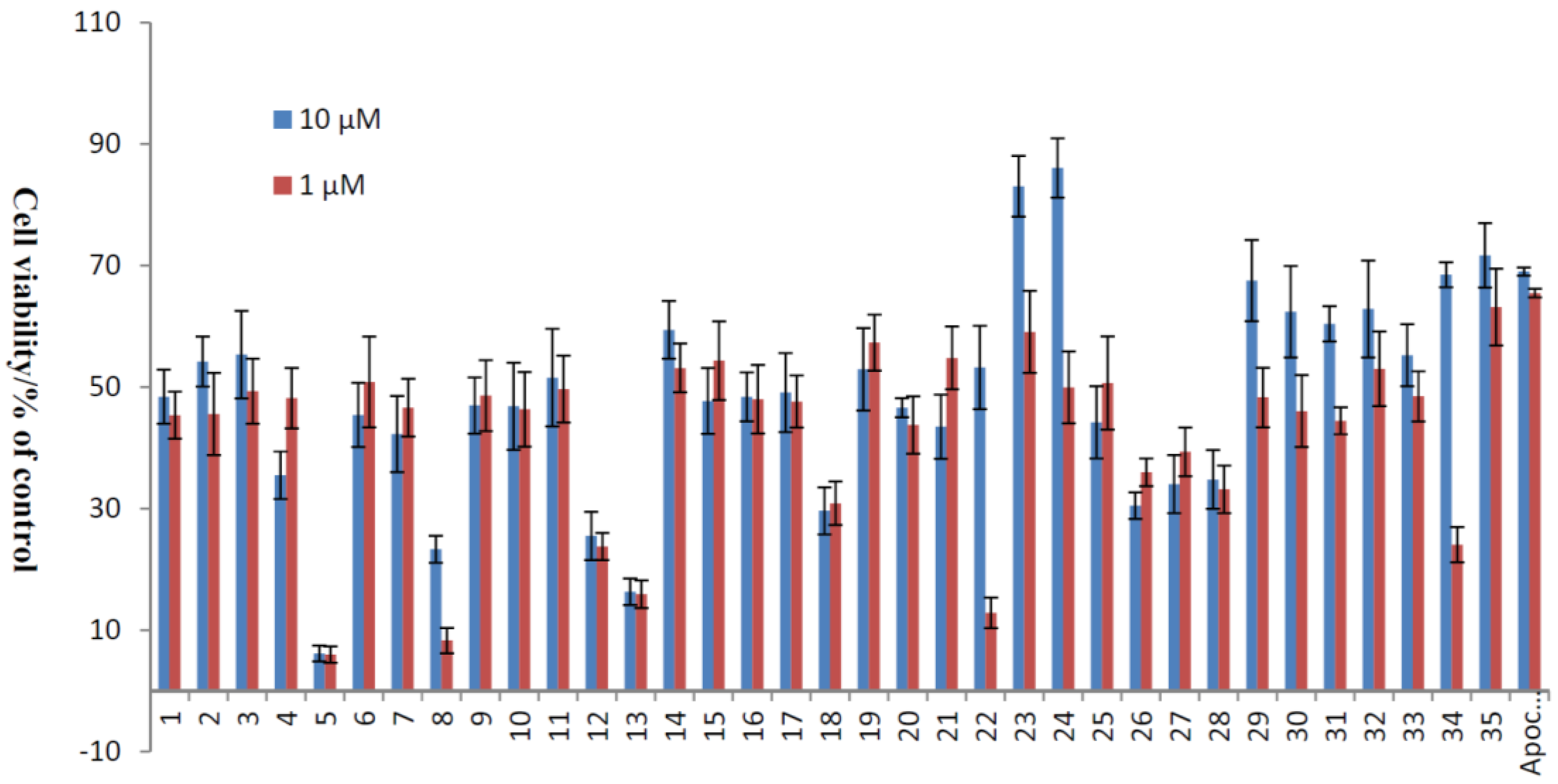
2.2. Xyloketal Derivatives Protected Endothelial Cells against H2O2-Induced Injury Assay

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
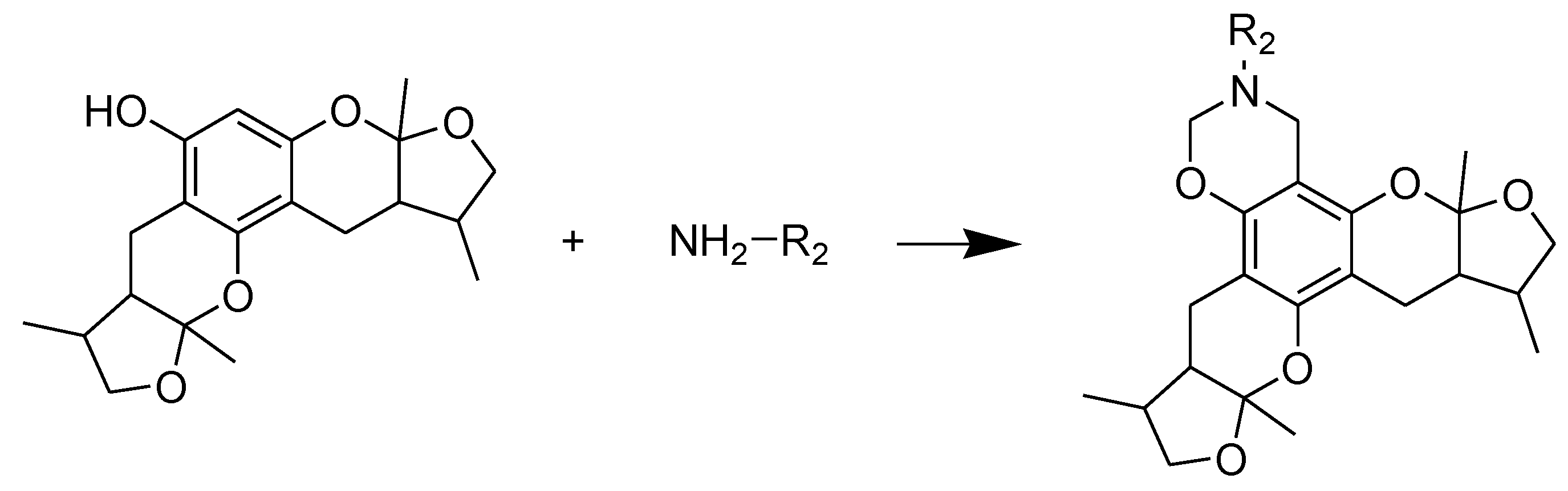{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Cell Viability/% of Control | No. | Cell Viability/% of Control | ||
|---|---|---|---|---|---|
| 10 μM | 1 μM | 10 μM | 1 μM | ||
| 1 | 48.41 ± 4.47 | 45.38 ± 3.89 | 19 | 52.94 ± 6.80 | 57.32 ± 4.59 |
| 2 | 54.19 ± 4.11 | 45.57 ± 6.78 | 20 | 46.63 ± 1.55 | 43.77 ± 4.70 |
| 3 | 55.36 ± 7.21 | 49.33 ± 5.34 | 21 | 43.49 ± 5.28 | 54.78 ± 5.16 |
| 4 | 35.49 ± 3.90 | 48.19 ± 4.96 | 22 | 53.23 ± 6.86 | 12.85 ± 2.53 |
| 5 | 6.15 ± 1.29 | 5.96 ± 1.36 | 23 | 83.07 ± 5.01 | 59.07 ± 6.76 |
| 6 | 45.41 ± 5.29 | 50.84 ± 7.46 | 24 | 86.08 ± 4.87 | 49.95 ± 5.92 |
| 7 | 42.28 ± 6.27 | 46.64 ± 4.76 | 25 | 44.20 ± 5.95 | 50.67 ± 7.66 |
| 8 | 23.32 ± 2.22 | 8.29 ± 2.08 | 26 | 30.47 ± 2.19 | 35.97 ± 2.28 |
| 9 | 46.98 ± 4.63 | 48.61 ± 5.84 | 27 | 34.02 ± 4.76 | 39.33 ± 4.00 |
| 10 | 46.84 ± 7.17 | 46.34 ± 6.13 | 28 | 34.79 ± 4.82 | 33.18 ± 3.92 |
| 11 | 51.56 ± 8.03 | 49.66 ± 5.51 | 29 | 67.53 ± 6.68 | 48.30 ± 4.91 |
| 12 | 25.51 ± 3.94 | 23.76 ± 2.24 | 30 | 62.41 ± 7.52 | 46.04 ± 5.92 |
| 13 | 16.31 ± 2.19 | 15.91 ± 2.30 | 31 | 60.43 ± 2.89 | 44.46 ± 2.24 |
| 14 | 59.42 ± 4.76 | 53.14 ± 4.03 | 32 | 62.85 ± 7.96 | 53.00 ± 6.14 |
| 15 | 47.72 ± 5.41 | 54.35 ± 6.44 | 33 | 55.24 ± 5.09 | 48.48 ± 4.13 |
| 16 | 48.40 ± 4.02 | 48.01 ± 5.62 | 34 | 68.50 ± 2.06 | 24.06 ± 2.87 |
| 17 | 49.11 ± 6.50 | 47.62 ± 4.30 | 35 | 71.67 ± 5.28 | 63.16 ± 6.32 |
| 18 | 29.64 ± 3.88 | 30.88 ± 3.59 | Apo-cynin | 69.03 ± 0.68 | 65.48 ± 0.70 |
| The EC50 of 23, 24 and 31 (xyloketal B) | |||
| No. | 23 | 24 | 31 (xyloketal B) |
| EC50 a (μM) | 5.10 | 3.59 | 15.97 |
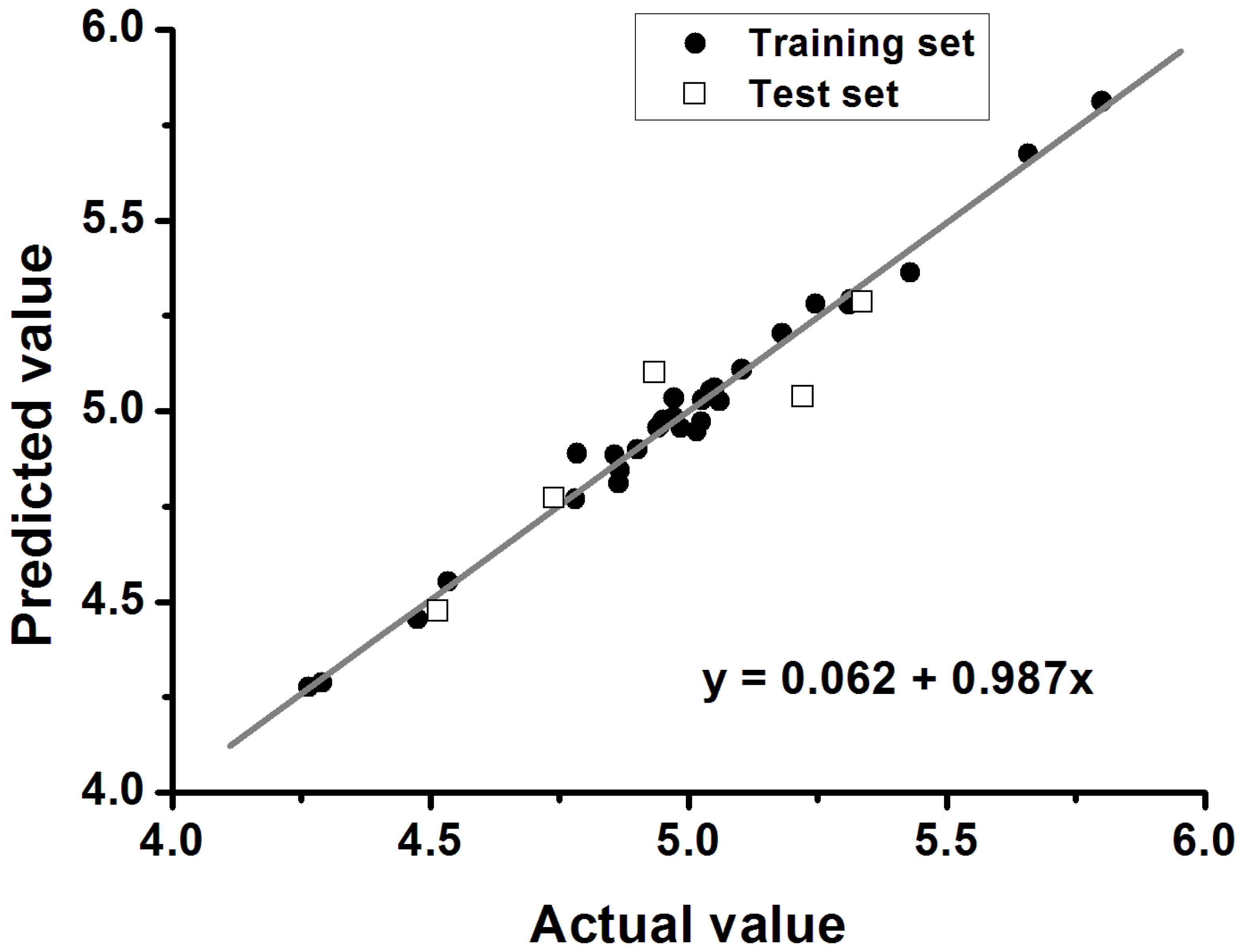
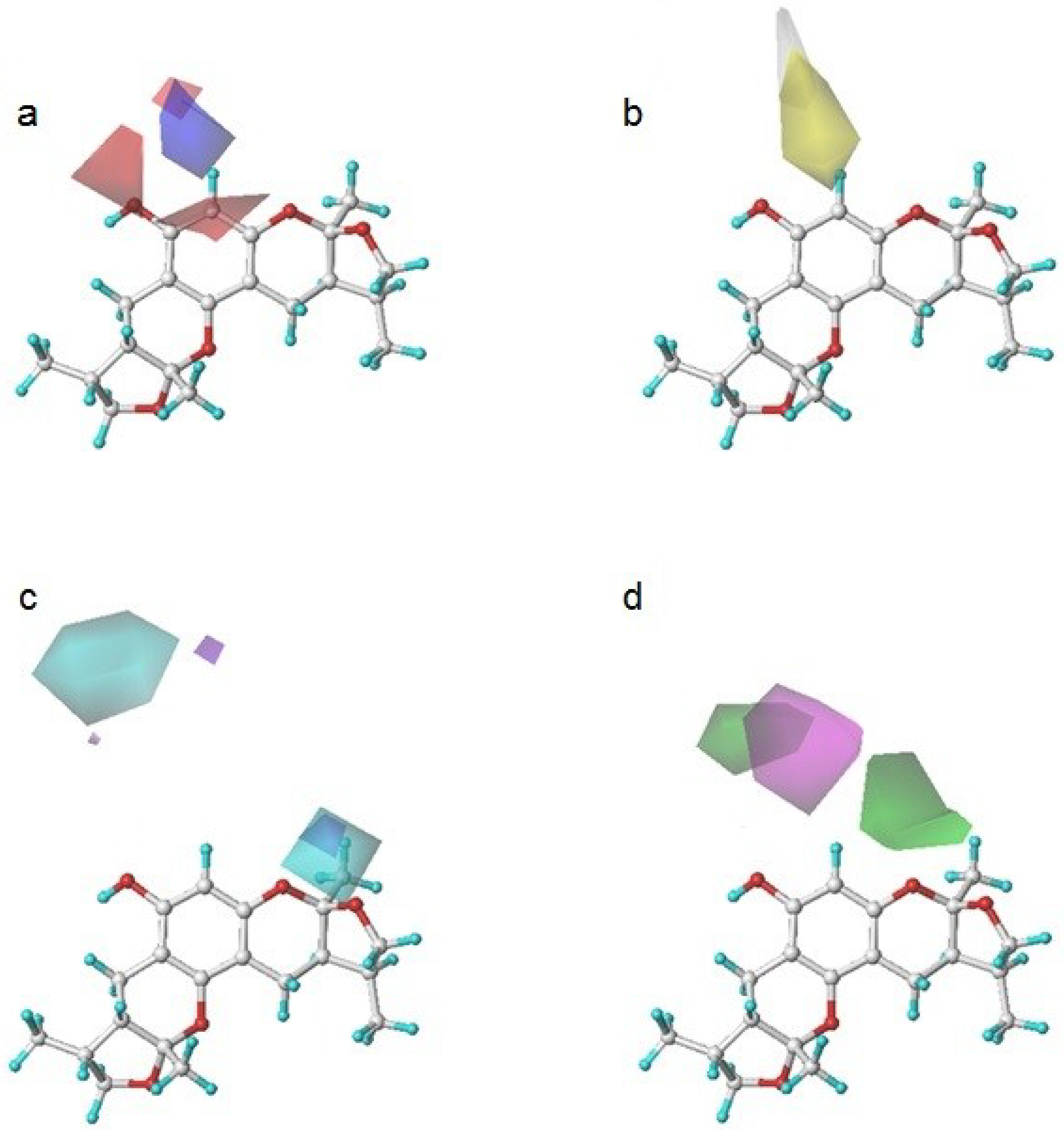
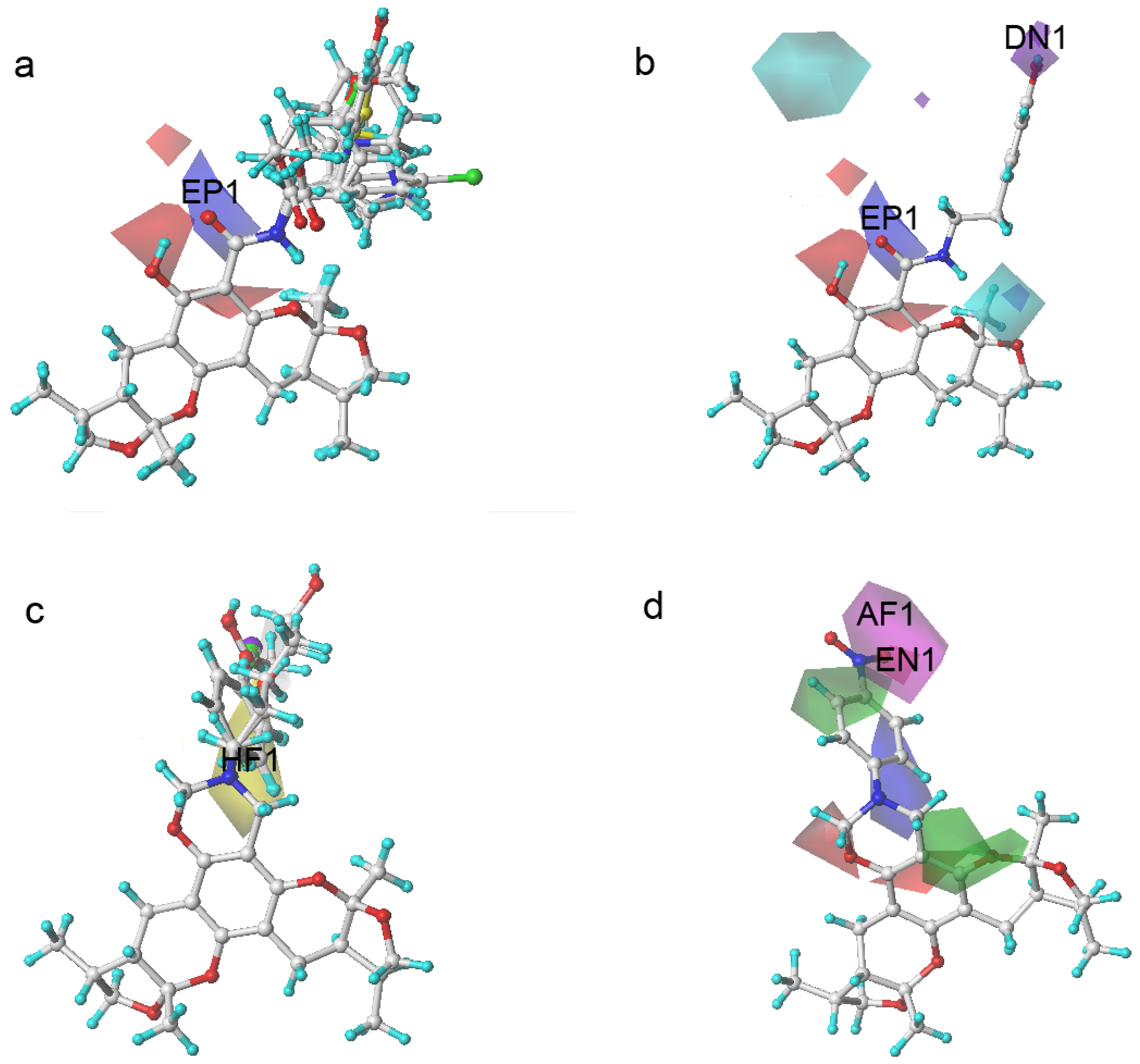
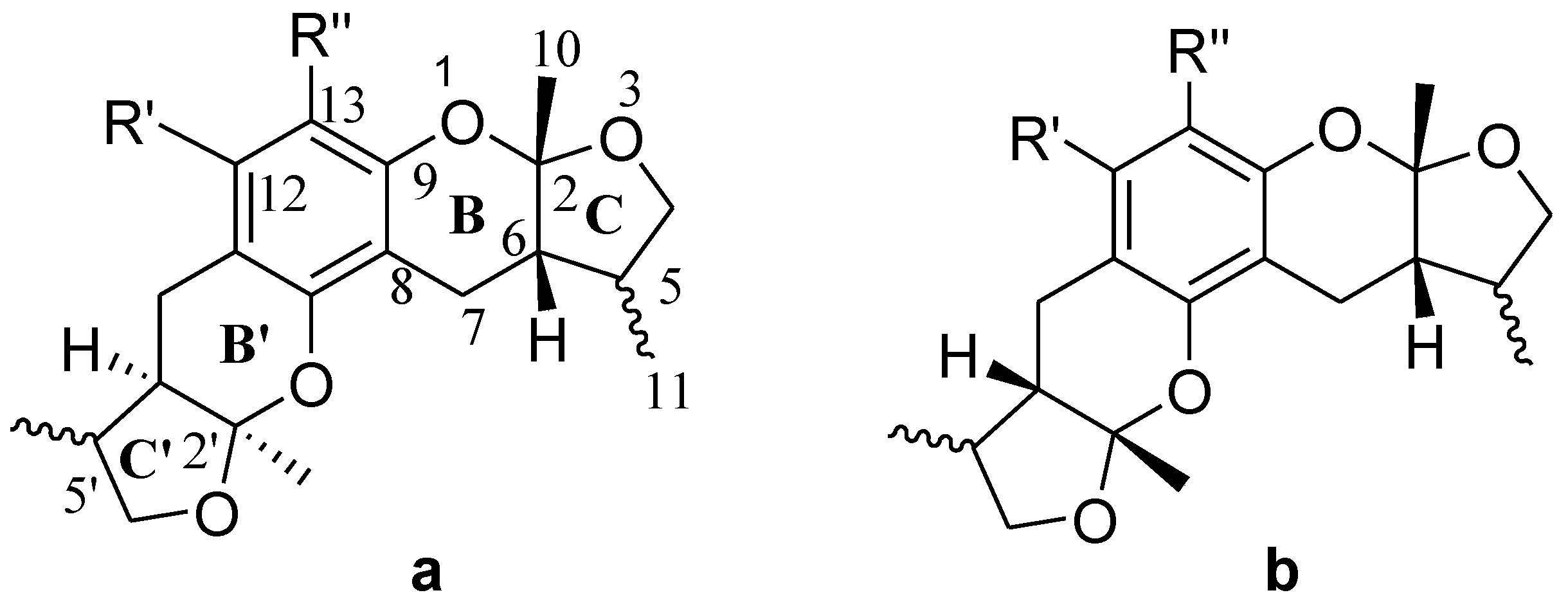
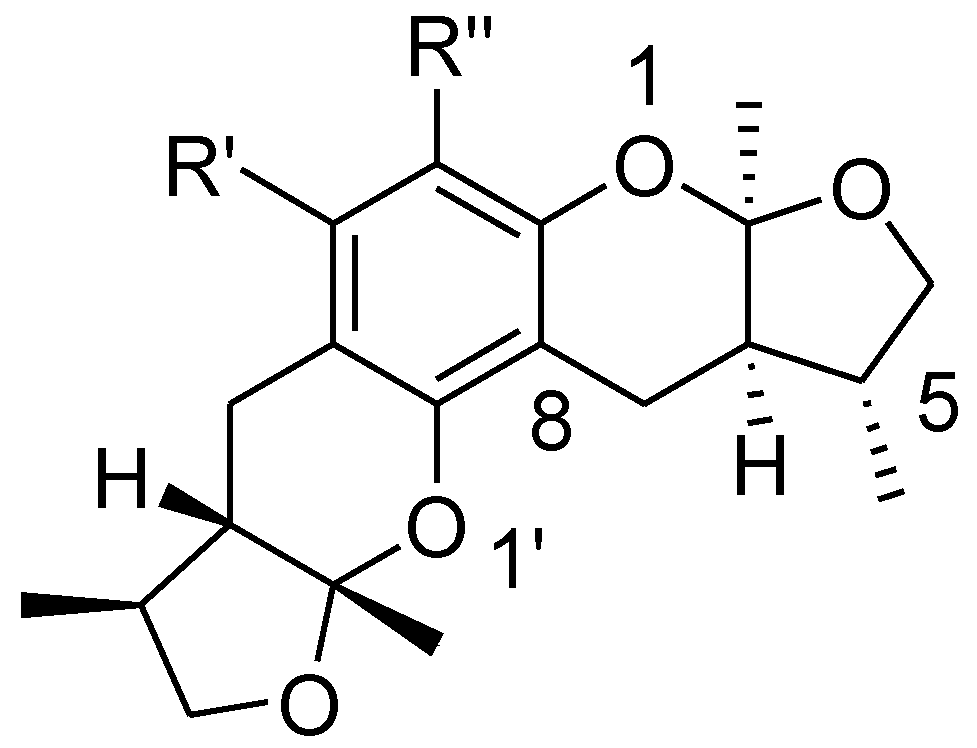
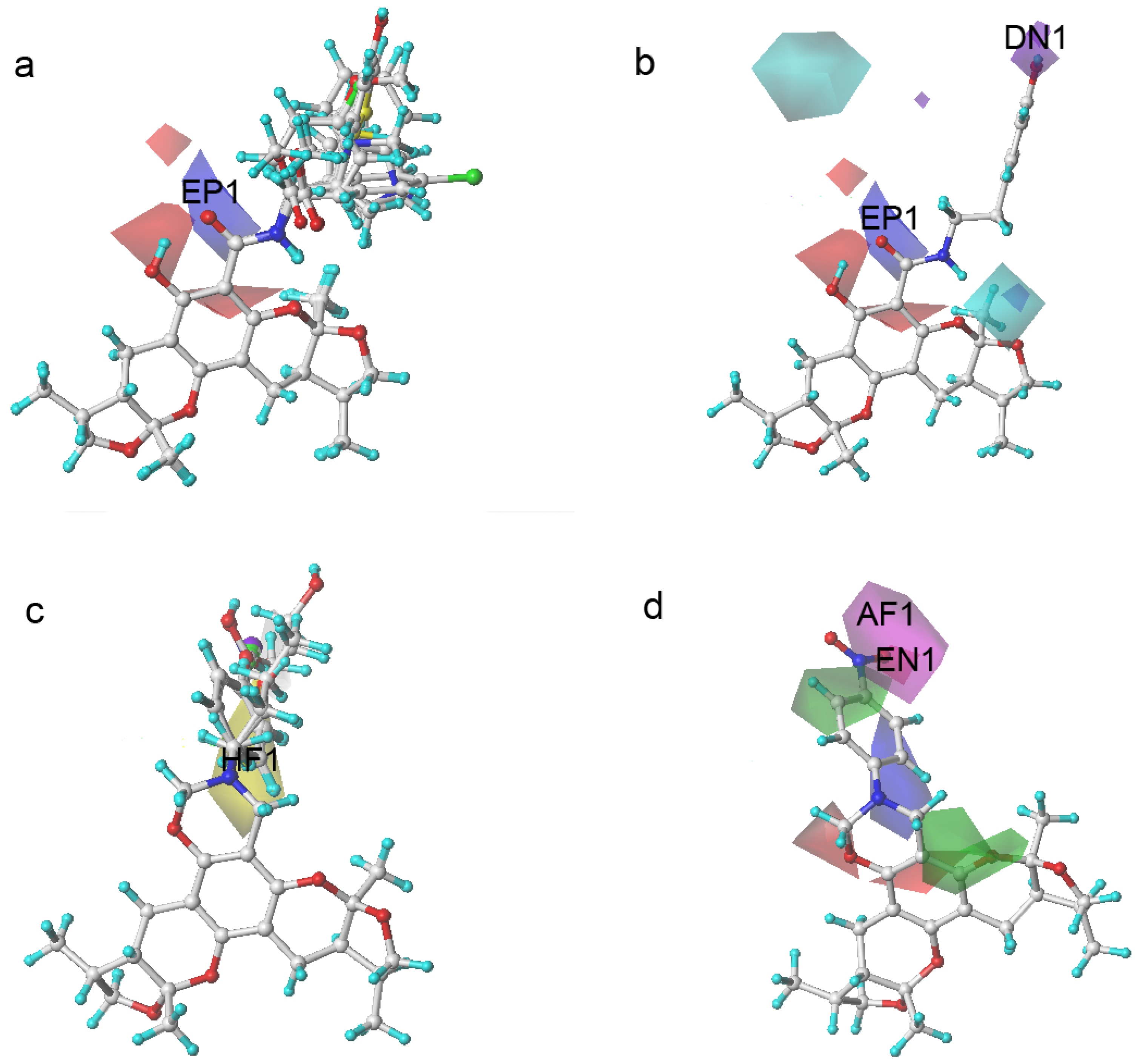
2.3. The Structural Activity Relationship of Xyloketals on a COMSIA Model
| Training Set | |
|---|---|
| q2 | 0.577 |
| r2 | 0.988 |
| SEE a | 0.041 |
| F b | 316.828 |
| Optimal components | 6 |
| Test set | |
| qtest2 | 0.648 |
| rtest2 | 0.858 |
| k | 0.987 |
| No. | Cell Viability/% of Control | Actual Value | Predicted Value | Residual Value |
|---|---|---|---|---|
| 1 | 48.41 ± 4.47 | 4.972 | 4.984 | −0.012 |
| 2 | 54.19 ± 4.11 | 5.026 | 5.029 | −0.003 |
| 3 | 55.36 ± 7.21 | 4.901 | 4.900 | 0.001 |
| 4 * | 35.49 ± 3.90 | 4.740 | 4.772 | −0.032 |
| 5 | 6.15 ± 1.29 | 4.264 | 4.276 | −0.012 |
| 6 * | 45.41 ± 5.29 | 4.935 | 5.101 | −0.166 |
| 7 | 42.28 ± 6.27 | 4.865 | 4.811 | 0.054 |
| 8 * | 23.32 ± 2.22 | 4.515 | 4.476 | 0.039 |
| 9 | 46.98 ± 4.63 | 4.947 | 4.965 | −0.018 |
| 10 | 46.84 ± 7.17 | 5.103 | 5.109 | 0.006 |
| 11 | 51.56 ± 8.03 | 4.857 | 4.886 | −0.029 |
| 12 | 25.51 ± 3.94 | 4.534 | 4.553 | −0.019 |
| 13 | 16.31 ± 2.19 | 4.290 | 4.288 | 0.002 |
| 14 * | 59.42 ± 4.76 | 5.222 | 5.038 | 0.184 |
| 15 | 47.72 ± 5.41 | 5.060 | 5.026 | 0.034 |
| 16 | 48.40 ± 4.02 | 4.972 | 5.034 | −0.062 |
| 17 | 49.11 ± 6.50 | 4.985 | 4.956 | 0.029 |
| 18 | 29.64 ± 3.88 | 4.475 | 4.454 | 0.021 |
| 19 | 52.94 ± 6.80 | 5.051 | 5.061 | −0.010 |
| 20 | 46.63 ± 1.55 | 4.941 | 4.957 | −0.016 |
| 21 | 43.49 ± 5.28 | 5.025 | 4.972 | 0.053 |
| 22 | 53.23 ± 6.86 | 4.951 | 4.976 | −0.025 |
| 23 | 83.07 ± 5.01 | 5.658 | 5.674 | −0.016 |
| 24 | 86.08 ± 4.87 | 5.801 | 5.811 | −0.010 |
| 25 | 44.20 ± 5.95 | 5.016 | 4.948 | 0.068 |
| 26 | 30.47 ± 2.19 | 4.784 | 4.889 | −0.105 |
| 27 | 34.02 ± 4.76 | 4.867 | 4.845 | 0.022 |
| 28 | 34.79 ± 4.82 | 4.781 | 4.770 | 0.011 |
| 29 | 67.53 ± 6.68 | 5.430 | 5.363 | 0.067 |
| 30 | 62.41 ± 7.52 | 5.042 | 5.054 | −0.012 |
| 31 | 60.43 ± 2.89 | 5.181 | 5.204 | −0.023 |
| 32 | 62.85 ± 7.96 | 5.311 | 5.280 | 0.031 |
| 33 | 55.24 ± 5.09 | 5.246 | 5.281 | −0.035 |
| 34 * | 68.50 ± 2.06 | 5.337 | 5.286 | 0.051 |
| 35 | 71.67 ± 5.28 | 5.314 | 5.293 | 0.021 |
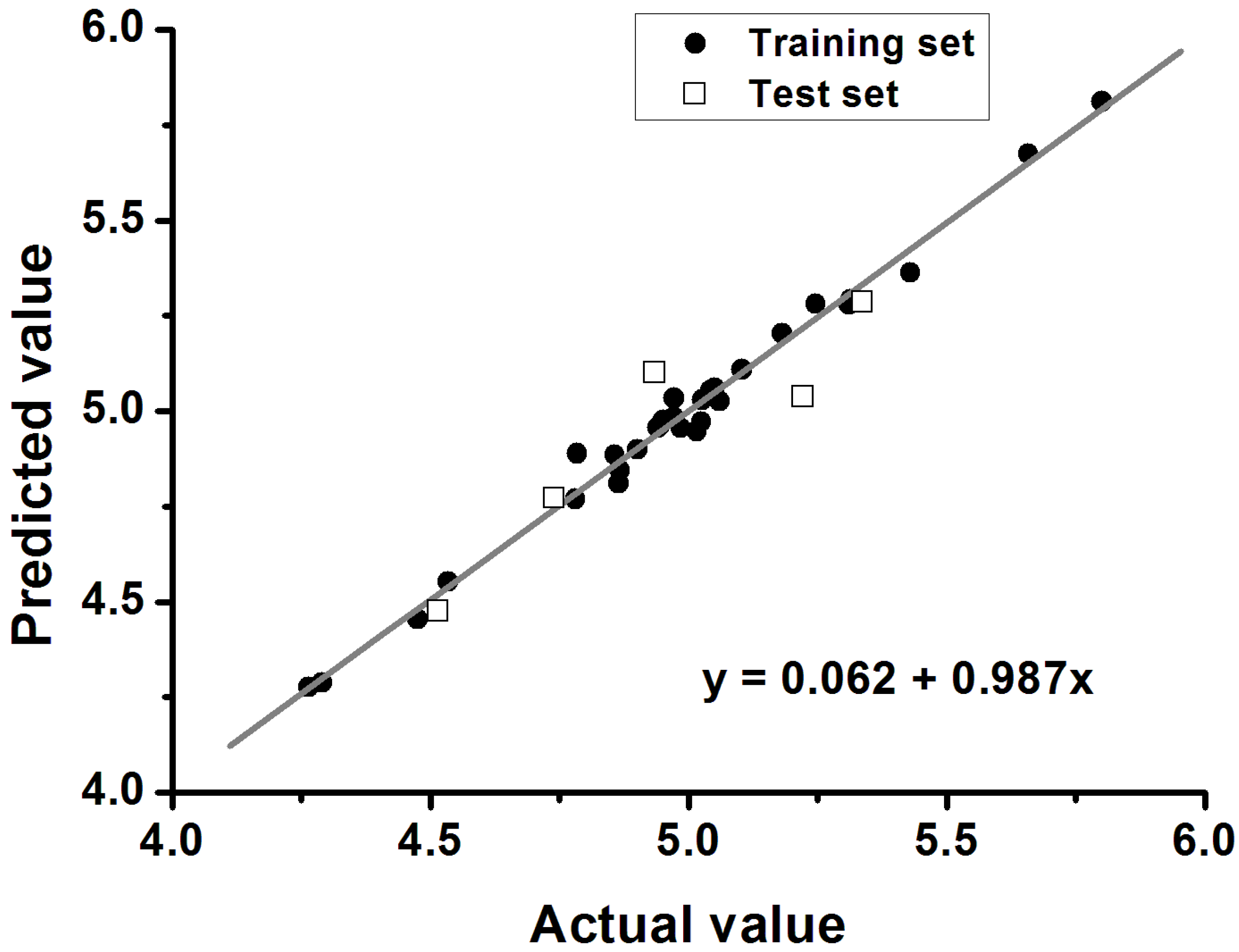

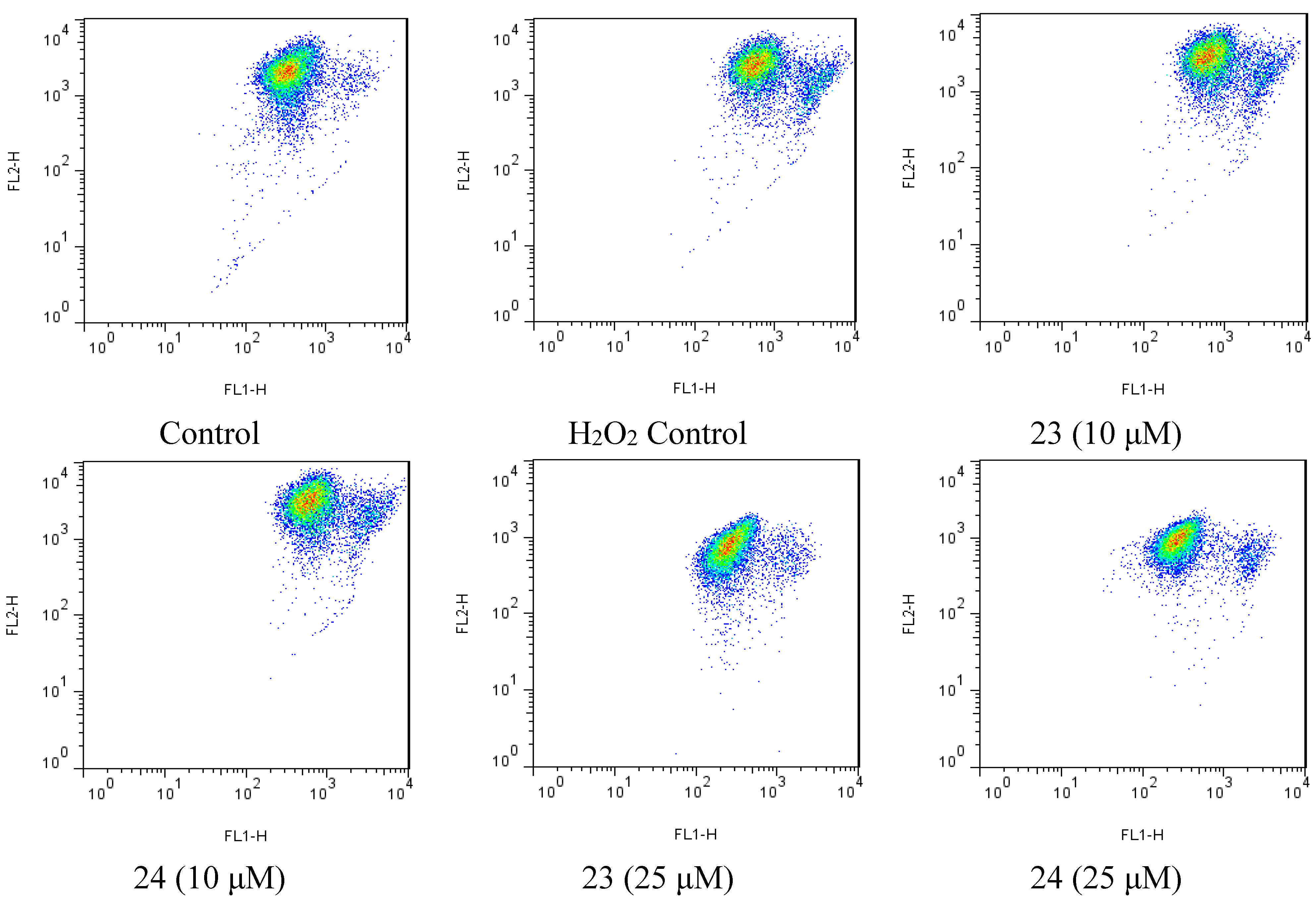
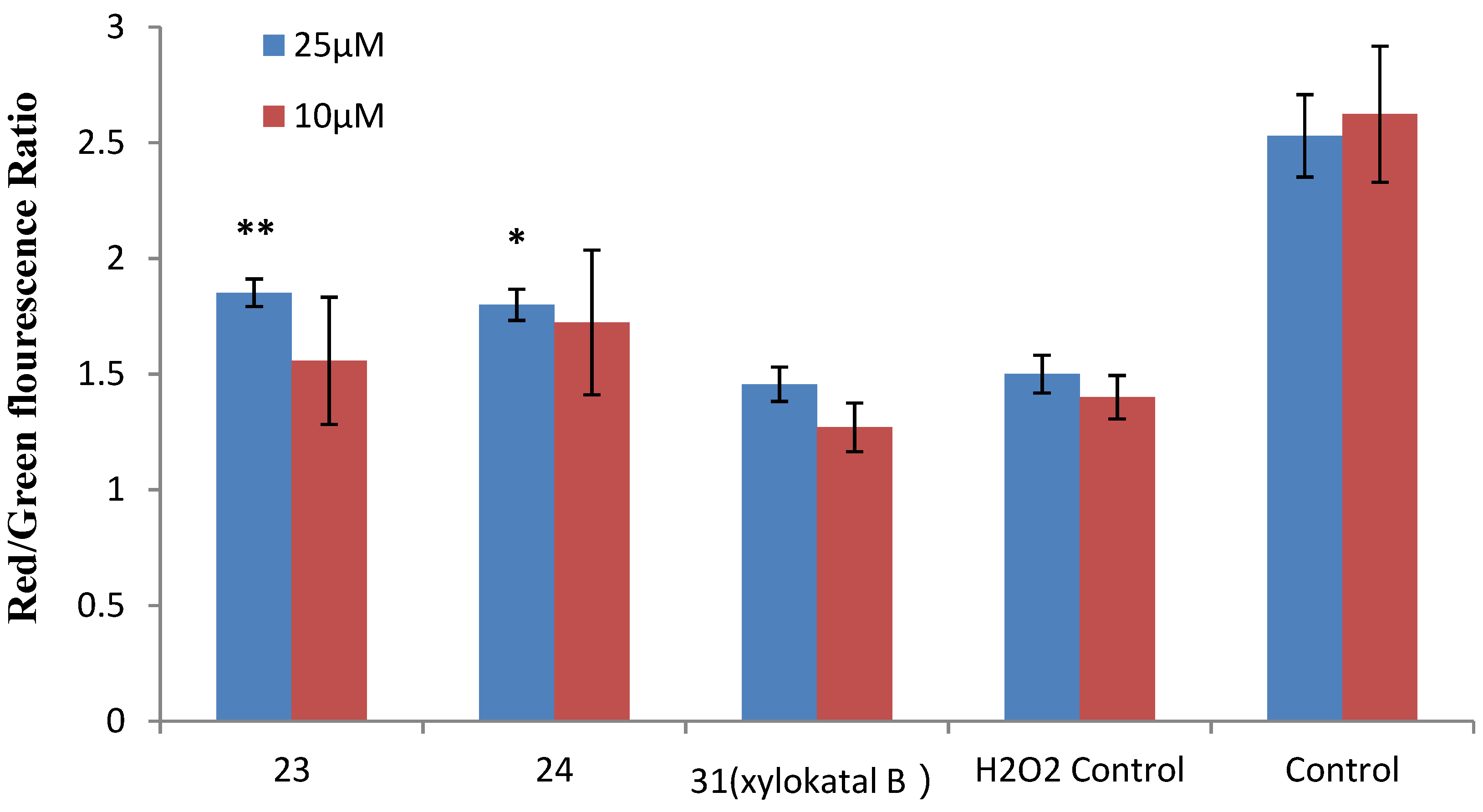
2.4. Xyloketal Derivatives Restored the H2O2-Induced Reduction of the Mitochondrial Membrane Potential (ΔΨm)
3. Experimental Section
3.1. Chemistry
3.2. General Procedure of Synthesizing Compounds
3.3. Biological Evaluation
3.3.1. Pharmacological Assays
3.3.2. Construction and Validation of the QSAR Model
3.3.3. Mitochondrial Membrane Potentials Assay
3.3.4. Statistics
4. Conclusions
Acknowledgments
Author Contributions
Abbreviations
| 3D-QSAR | three-dimensional quantitative structure-activity relationship |
| As | atherosclerosis |
| BOP | (benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate |
| CoMSIA | a comparative molecular similarity indices analysis |
| CVD | cardiovascular disease |
| DCM | dichloromethane |
| DIEA | N,N-diisopropylethylamine |
| DMF | N,N-dimethylformamide |
| DMSO | dimethyl sulfoxide |
| FACs | fluorescence activating cell sorter |
| FCM | flow cytometry |
| H2DCFDA | 2′,7′-dihydrodichlorofluorescein diacetate |
| H2O2 | hydrogen peroxide |
| HCHO | formaldehyde |
| HUVECs | human umbilical vein endothelial cells |
| LOO | leave-one-out |
| MAPK | mitogen-activated protein kinase |
| MMP | mitochondrial membrane potential |
| MPP+ | 1-methyl-4-phenylpyridinium |
| NMR | nuclear magnetic resonance |
| OGD | oxygen-glucose deprivation |
| OxLDL | oxidized low density lipoprotein |
| PLS | partial-least-squares |
| ROS | reactive oxygen species |
| SAR | structure-activity relationship |
| THF | tetrahydrofuran |
Conflicts of Interest
References
- Yang, B.H.; Oo, T.N.; Rizzo, V. Lipid rafts mediate H2O2 prosurvival effects in cultured endothelial cells. FASEB J. 2006, 20, 1501–1503. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Gu, L.; Ma, Q.; Zhu, D.; Huang, X. Resveratrol attenuates hydrogen peroxide-induced apoptosis in human umbilical vein endothelial cells. Eur. Rev. Med. Pharmacol. Sci. 2013, 17, 88–94. [Google Scholar] [PubMed]
- Kamata, H; Hirata, H. Redox regulation of cellular signalling. Cell. Signal. 1999, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Favero, T.G.; Zable, A.C.; Abramson, J.J. Hydrogen peroxide stimulates the Ca2+ release channel from skeletal muscle sarcoplasmic reticulum. J. Biol. Chem. 1995, 270, 25557–25563. [Google Scholar] [CrossRef] [PubMed]
- Suhara, T.; Fukuo, K.; Sugimoto, T.; Morimoto, S.; Nakahashi, T.; Hata, S.; Shimizu, M.; Ogihara, T. Hydrogen peroxide induces up-regulation of Fas in human endothelial cells. J. Immunol. 1998, 160, 4042–4047. [Google Scholar] [PubMed]
- Sabri, A.; Byron, K.L.; Samarel, A.M.; Bell, J.; Lucchesi, P.A. Hydrogen peroxide activates mitogen-activated protein kinases and Na+-H+ exchange in neonatal rat cardiac myocytes. Circ. Res. 1998, 82, 1053–1062. [Google Scholar] [CrossRef] [PubMed]
- Cyrne, L.; Marques, V.O.; Marinho, H.S.; Antunes, F. H2O2 in the induction of NF-κB-dependent selective gene expression. Methods Enzymol. 2013, 528, 173–188. [Google Scholar] [PubMed]
- Xia, Z.Y.; Liu, M.; Wu, Y.; Sharma, V.; Luo, T.; Ouyang, J.P.; McNeill, J.H. N-acetylcysteine attenuates TNF-alpha-induced human vascular endothelial cell apoptosis and restores eNOS expression. Eur. J. Pharmacol. 2006, 550, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Martinou, J.C.; Green, D.R. Breaking the mitochondrial barrier. Nat. Rev. Mol. Cell Biol. 2001, 2, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Guido, K.; Bruno, D.; Michele, R.R. The mitochondrial death/life regulator in apoptosis and necrosis. Annu. Rev. Physiol. 1998, 60, 619–642. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Wu, X.Y.; Feng, S.; Jiang, G.; Luo, J.H.; Zhou, S.N.; Vrijmoed, L.L.P.; Jones, E.B.G.; Krohn, K.; Steingrover, K.; et al. Five unique compounds: Xyloketals from mangrove fungus Xylaria sp. from the South China Sea coast. J. Org. Chem. 2001, 66, 6252–6256. [Google Scholar]
- Wu, X.Y.; Liu, X.H.; Lin, Y.C.; Luo, J.H.; She, Z.G.; Li, H.J.; Chan, W.L.; Antus, S.; Kurtan, T.; Elsasser, B.; et al. Xyloketal F: A strong l-calcium channel blocker from the mangrove fungus Xylaria sp. (#2508) from the South China Sea coast. Eur. J. Org. Chem. 2005, 12, 4061–4064. [Google Scholar] [CrossRef]
- Chen, W.L.; Qian, Y.; Meng, W.F.; Pang, J.Y.; Lin, Y.C.; Guan, Y.Y.; Chen, S.P.; Liu, J.; Pei, Z.; Wang, G.L. A novel marine compound xyloketal B protects against oxidized LDL-induced cell injury in vitro. Biochem. Pharmacol. 2009, 78, 941–950. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Li, L.; Ling, C.; Li, J.; Pang, J.Y.; Lin, Y.C.; Liu, J.; Huang, R.X.; Wang, G.L.; Pei, Z.; et al. Marine compound Xyloketal B protects PC12 cells against OGD-induced cell damage. Brain Res. 2009, 1302, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.L.; Yao, X.L.; Liu, Z.Y.; Zhang, H.; Li, W.; Li, Z.X.; Wang, G.L.; Pang, J.Y.; Lin, Y.C.; Xu, Z.L.; et al. Protective effects of xyloketal B against MPP+-induced neurotoxicity in Caenorhabditiselegans and PC12 cells. Brain Res. 2010, 1332, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Li, Y.; Xiang, Q.; Pei, Z.; Liu, X.; Lu, B.; Chen, L.; Wang, G.; Pang, J.; Lin, Y. Design and synthesis of novel xyloketal derivatives and their vasorelaxing activities in rat thoracic aorta and angiogenic activities in zebrafish angiogenesis screen. J. Med. Chem. 2010, 53, 4642–4653. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.X.; Chen, J.W.; Feng, Y.; Huang, Y.Y.; Zhao, L.Y.; Li, J.; Su, H.X.; Liu, J.; Pang, J.Y.; Lin, Y.C.; et al. Xyloketal B exhibits its antioxidant activity through induction of HO-1 in vascular endothelial cells and zebrafish. Mar. Drugs 2013, 11, 504–522. [Google Scholar] [CrossRef] [PubMed]
- Li, S.C.; Shen, C.Z.; Guo, W.Y.; Zhang, X.F.; Liu, S.X.; Liang, F.Y.; Xu, Z.L.; Pei, Z.; Song, H.C.; Qiu, L.Q.; et al. Synthesis and neuroprotective action of xyloketal derivatives in Parkinson’s Disease models. Mar. Drugs 2013, 11, 5159–5189. [Google Scholar] [CrossRef] [PubMed]
- Sheng, C.Q.; Zhang, W.N.; Ji, H.T.; Zhang, M.; Song, Y.L.; Xu, H.; Zhu, J.; Miao, Z.Y.; Jiang, Q.F.; Yao, J.Z.; et al. Structure-based optimization of azole antifungal agents by CoMFA, CoMSIA, and molecular docking. J. Med. Chem. 2006, 49, 2512–2525. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.P.; Moroni, E.; Yan, B.; Colombo, G.; Blagg, B.S.J. 3D-QSAR-assisted design, synthesis, and evaluation of novobiocin analogues. Med. Chem. Lett. 2013, 4, 57–62. [Google Scholar] [CrossRef]
- Fleury, C.; Mignotte, B.; Vayssière, J.L. Mitochondrial reactive oxygen species in cell death signaling. Biochimie 2002, 8, 131–141. [Google Scholar] [CrossRef]
- Chen, Q.; Vazquez, E.J.; Moghaddas, S.; Hoppel, C.L.; Lesnefsky, E.J. Production of reactive oxygen species by mitochondria. J. Biol. Chem. 2003, 278, 36027–36031. [Google Scholar] [CrossRef] [PubMed]
- Pettigrew, J.D.; Bexrud, J.A.; Freeman, R.P.; Wilson, P.D. Total synthesis of (+/−)-xyloketal D and model studies towards the total synthesis of (−)-xyloketal A. Heterocycles 2004, 62, 445–452. [Google Scholar] [CrossRef]
- Rodriguez, R.; Adlington, R.M.; Moses, J.E.; Cowley, A.; Baldwin, J.E. A new and efficient method for o-ouinone methide intermediate generation: application to the biomimetic synthesis of (±)-alboatrin. Org. Lett. 2004, 6, 3617–3619. [Google Scholar] [CrossRef] [PubMed]
- Krohn, K.; Riaz, M. Total synthesis of (+)-xyloketal D, a secondary metabolite from the mangrove fungus Xylaria sp. Tetrahedron Lett. 2004, 45, 293–294. [Google Scholar] [CrossRef]
- Pettigrew, J.D.; Freeman, R.P.; Wilson, P.D. Total synthesis of (−)-xyloketal D and its enantiomer—Confirmation of absolute stereochemistry. Can. J. Chem. 2004, 82, 1640–1648. [Google Scholar] [CrossRef]
- Pettigrew, J.D.; Wilson, P.D. Synthesis of xyloketal A, B, C, D and G analogues. J. Org. Chem. 2006, 71, 1620–1625. [Google Scholar] [CrossRef] [PubMed]
- Pettigrew, J.D.; Wilson, P.D. Total synthesis of (−)-xyloketal A. Org. Lett. 2006, 8, 1427–1429. [Google Scholar] [CrossRef] [PubMed]
- Krohn, K.; Riaz, M.; Flörke, U. Synthesis of xyloketals, natural products from the mangrove fungus Xylaria sp. Eur. J. Org. Chem. 2004, 1261–1270. [Google Scholar] [CrossRef]
- Xu, Z.L.; Li, Y.Y.; Lu, B.T.; Pang, J.Y.; Lin, Y.C. An expedient approach to the benzopyran core: application to synthesis of the natural products (±)-xyloketals and (±)-alboatrin. Chin. J. Chem. 2010, 28, 2441–2446. [Google Scholar] [CrossRef]
- Gong, G.H.; Qin, Y.; Huang, W.; Zhou, S.; Yang, X.H.; Li, D. Rutin inhibits hydrogen peroxide-induced apoptosis through regulating reactive oxygen species mediated mitochondrial dysfunction pathway in human umbilical vein endothelial cells. Eur. J. Pharmacol. 2010, 628, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Bresgen, N.; Karlhuber, G.; Krizbai, I.; Bauer, H.; Bauer, H.C.; Eckl, P.M. Oxidative stress in cultured cerebral endothelial cells induces chromosomal aberrations, micronuclei, and apoptosis. J. Neurosci. Res. 2003, 72, 327–333. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, S.; Luo, R.; Xiang, Q.; Xu, X.; Qiu, L.; Pang, J. Design and Synthesis of Novel Xyloketal Derivatives and Their Protective Activities against H2O2-Induced HUVEC Injury. Mar. Drugs 2015, 13, 948-973. https://doi.org/10.3390/md13020948
Liu S, Luo R, Xiang Q, Xu X, Qiu L, Pang J. Design and Synthesis of Novel Xyloketal Derivatives and Their Protective Activities against H2O2-Induced HUVEC Injury. Marine Drugs. 2015; 13(2):948-973. https://doi.org/10.3390/md13020948
Chicago/Turabian StyleLiu, Shixin, Rong Luo, Qi Xiang, Xianfang Xu, Liqin Qiu, and Jiyan Pang. 2015. "Design and Synthesis of Novel Xyloketal Derivatives and Their Protective Activities against H2O2-Induced HUVEC Injury" Marine Drugs 13, no. 2: 948-973. https://doi.org/10.3390/md13020948
APA StyleLiu, S., Luo, R., Xiang, Q., Xu, X., Qiu, L., & Pang, J. (2015). Design and Synthesis of Novel Xyloketal Derivatives and Their Protective Activities against H2O2-Induced HUVEC Injury. Marine Drugs, 13(2), 948-973. https://doi.org/10.3390/md13020948