
Conformational Analysis of the Oligosaccharides Related to Side Chains of Holothurian Fucosylated Chondroitin Sulfates

Abstract
:
1. Introduction
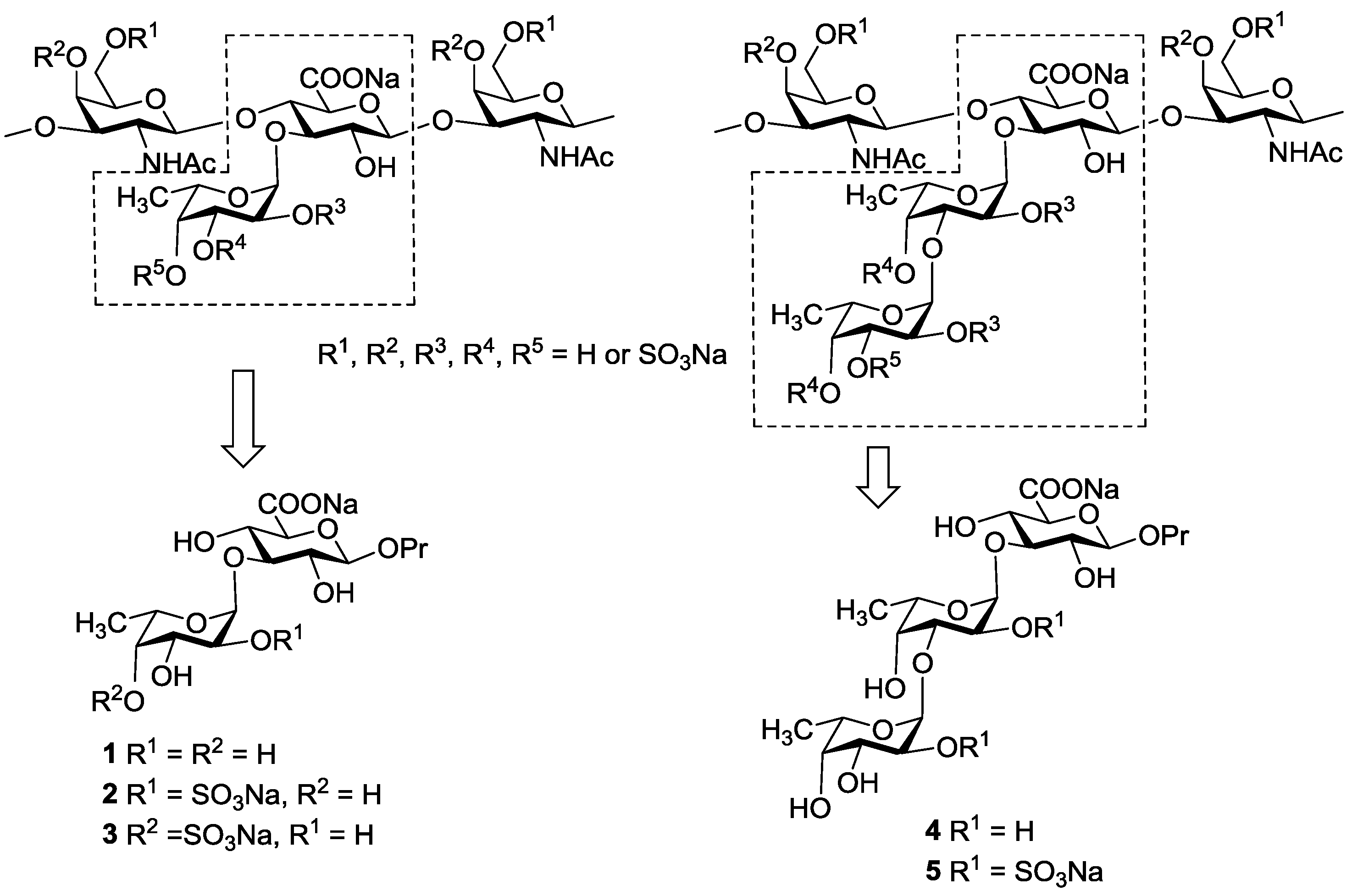
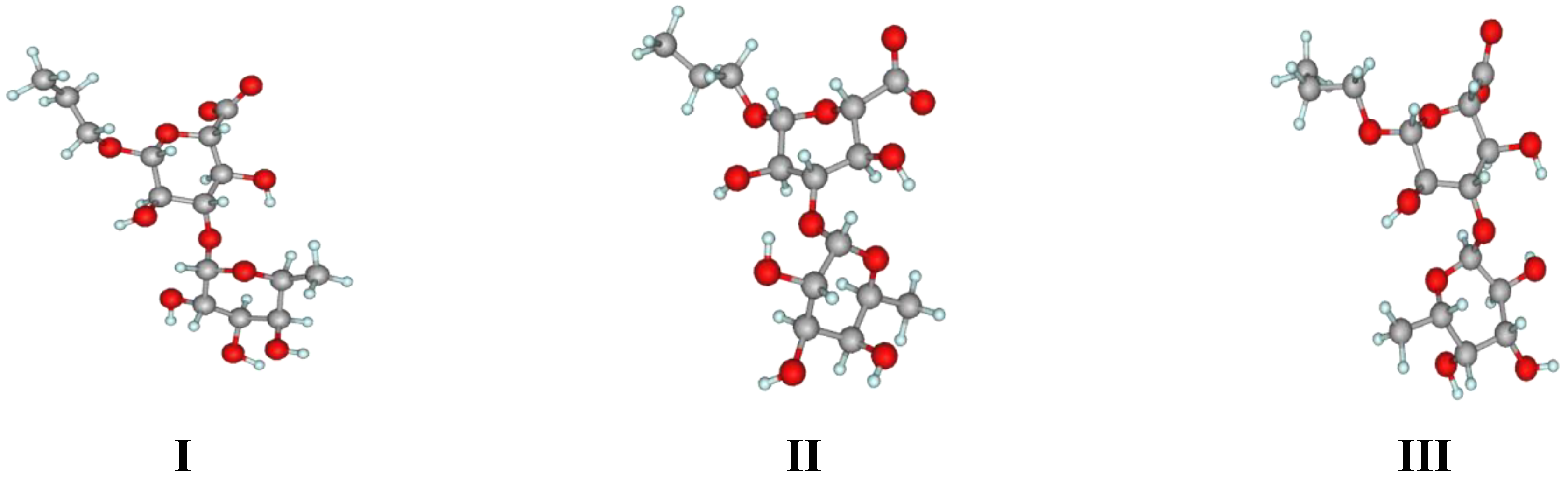
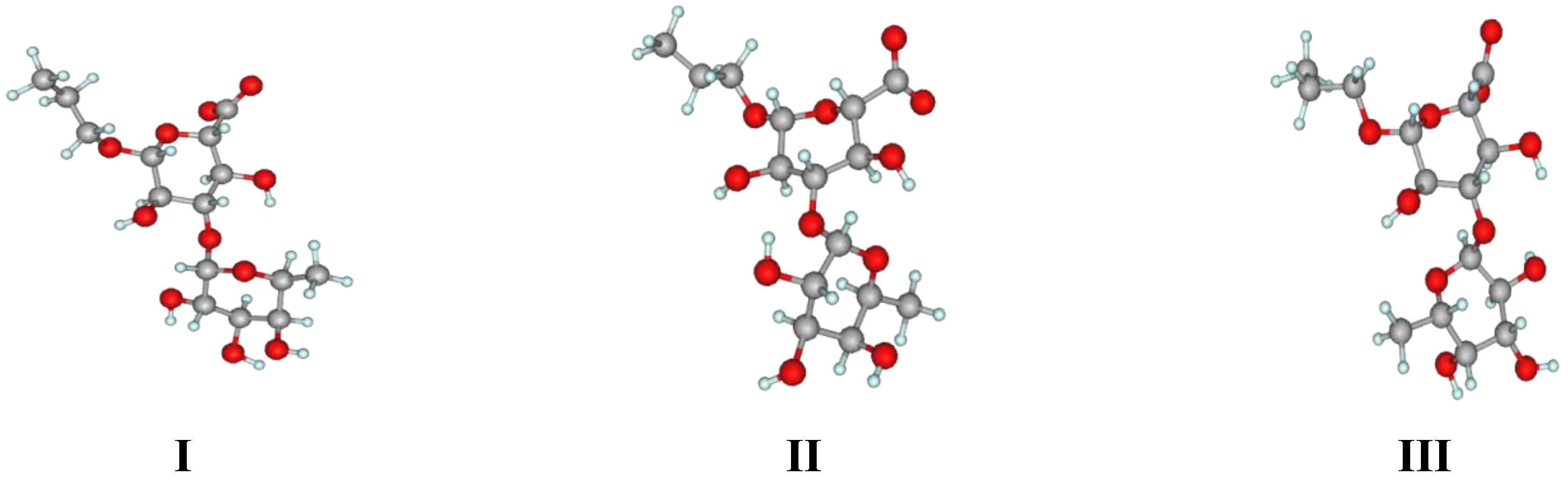
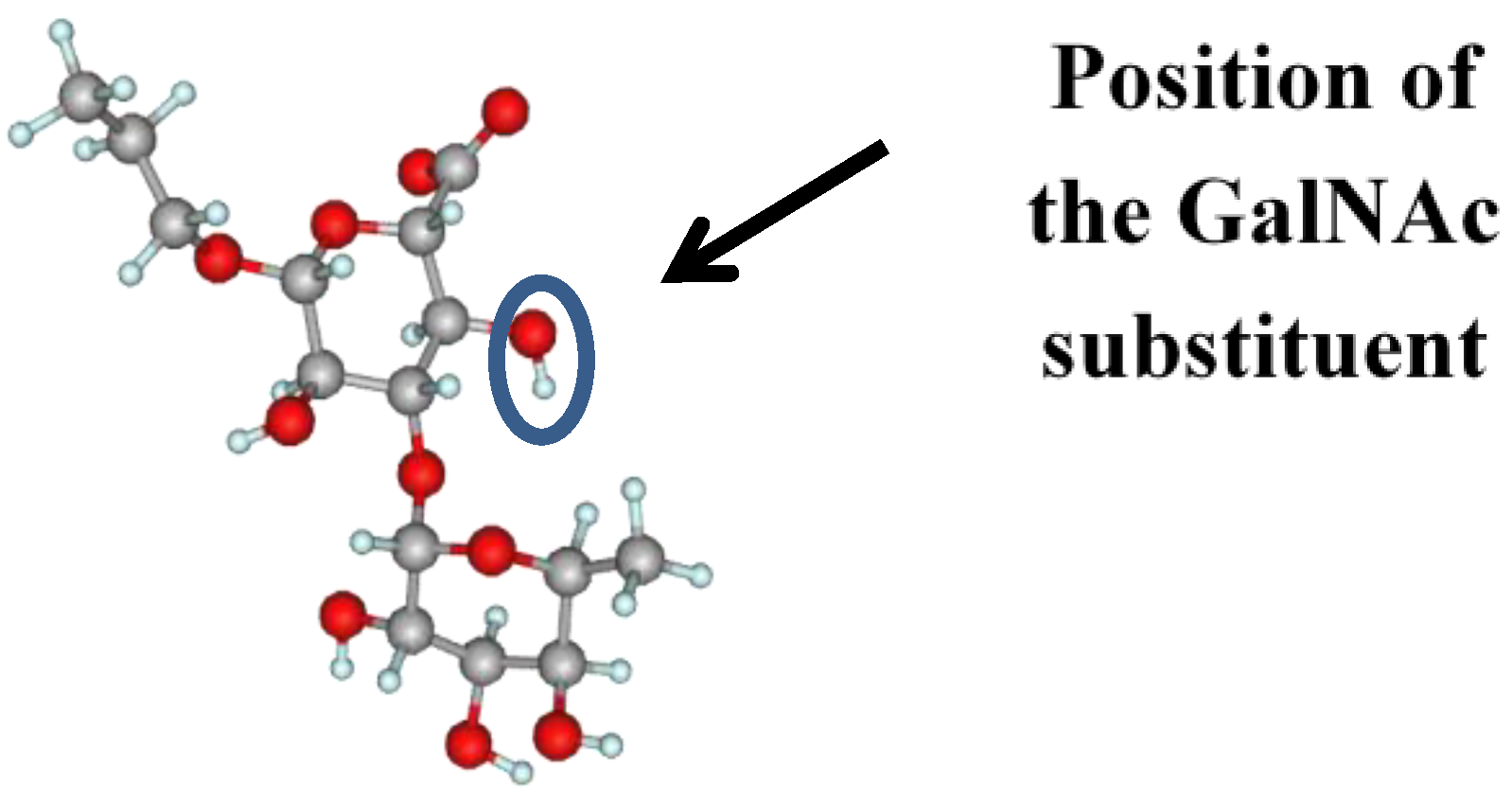
2. Results and Discussion
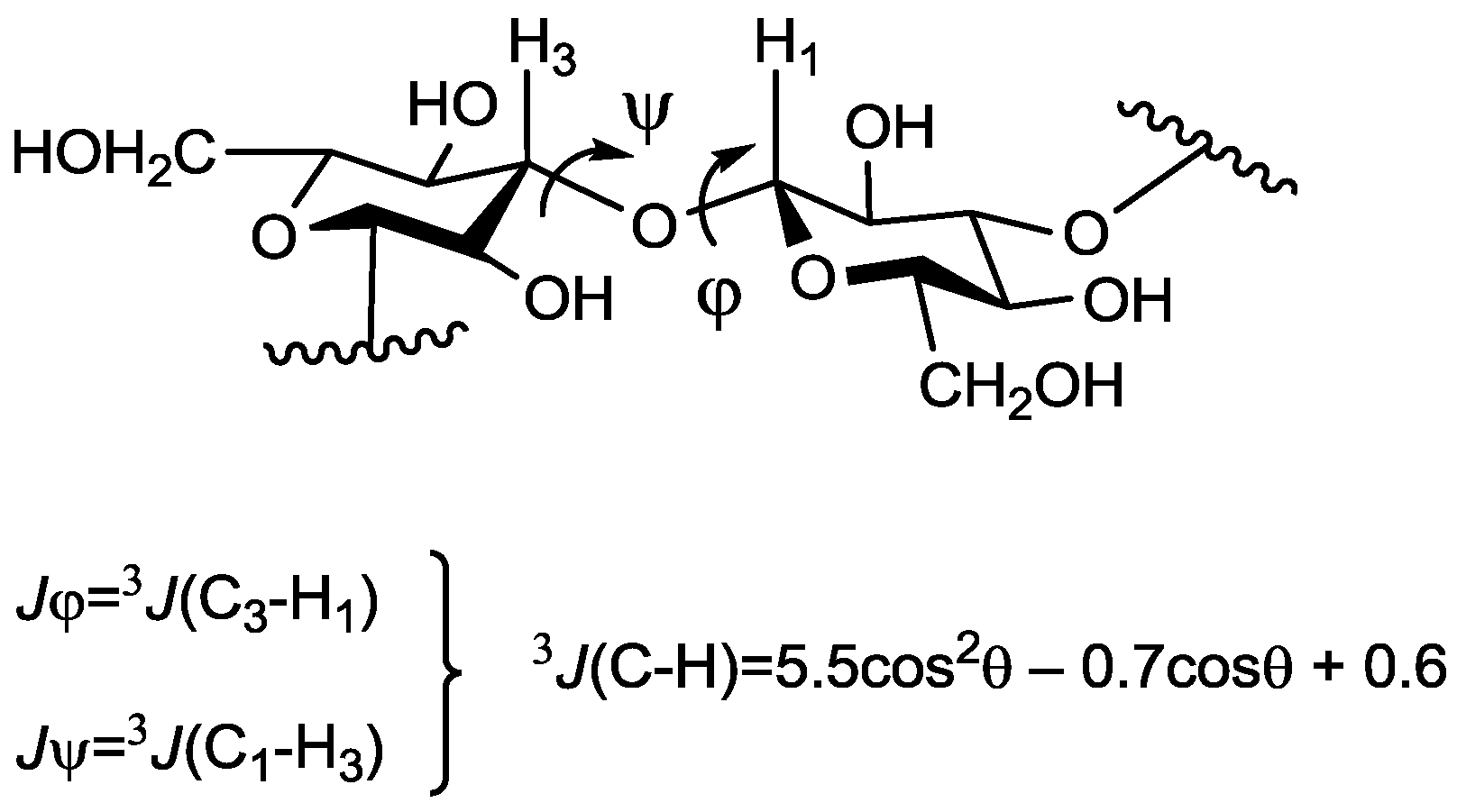

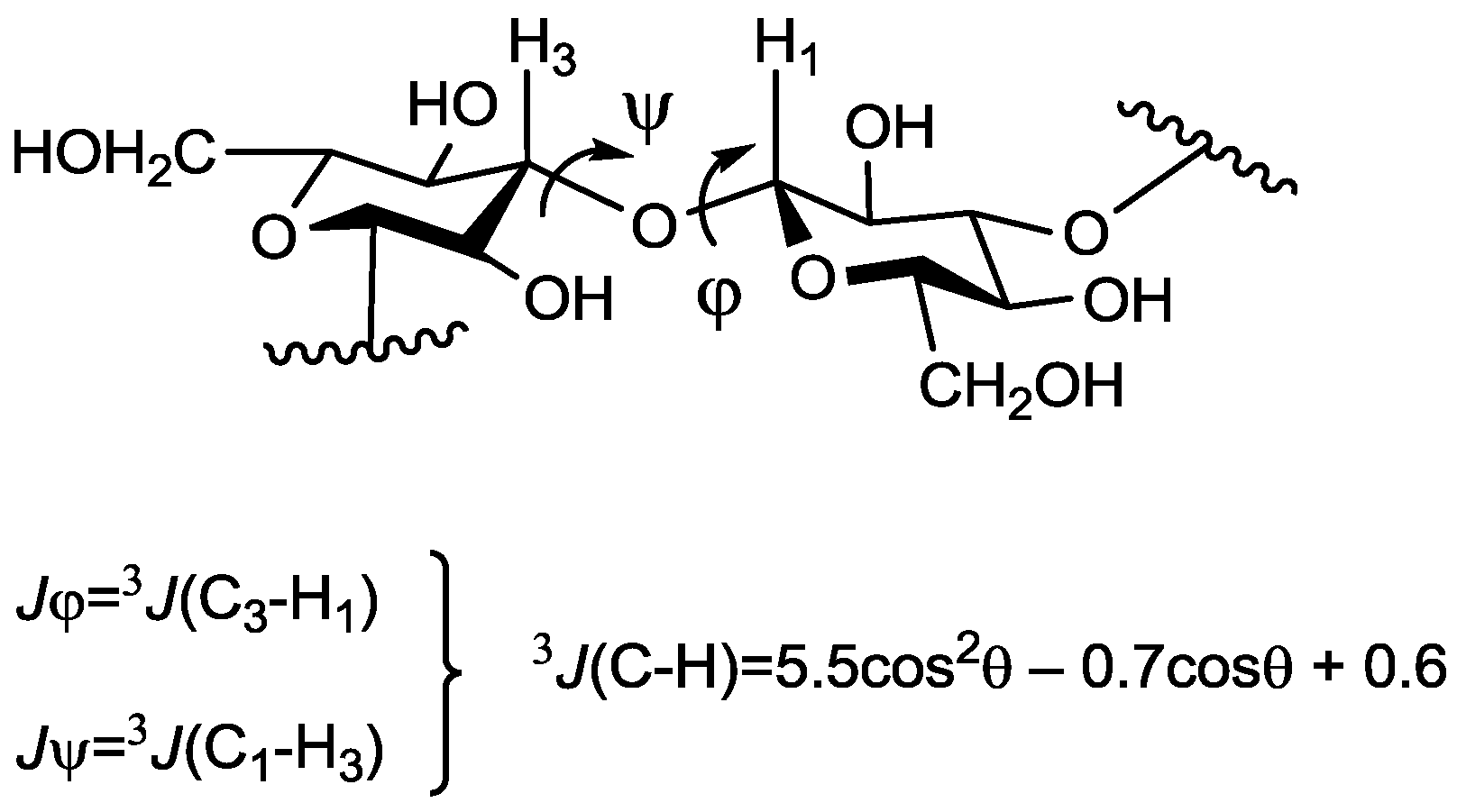
{kind=link}
{kind=link}
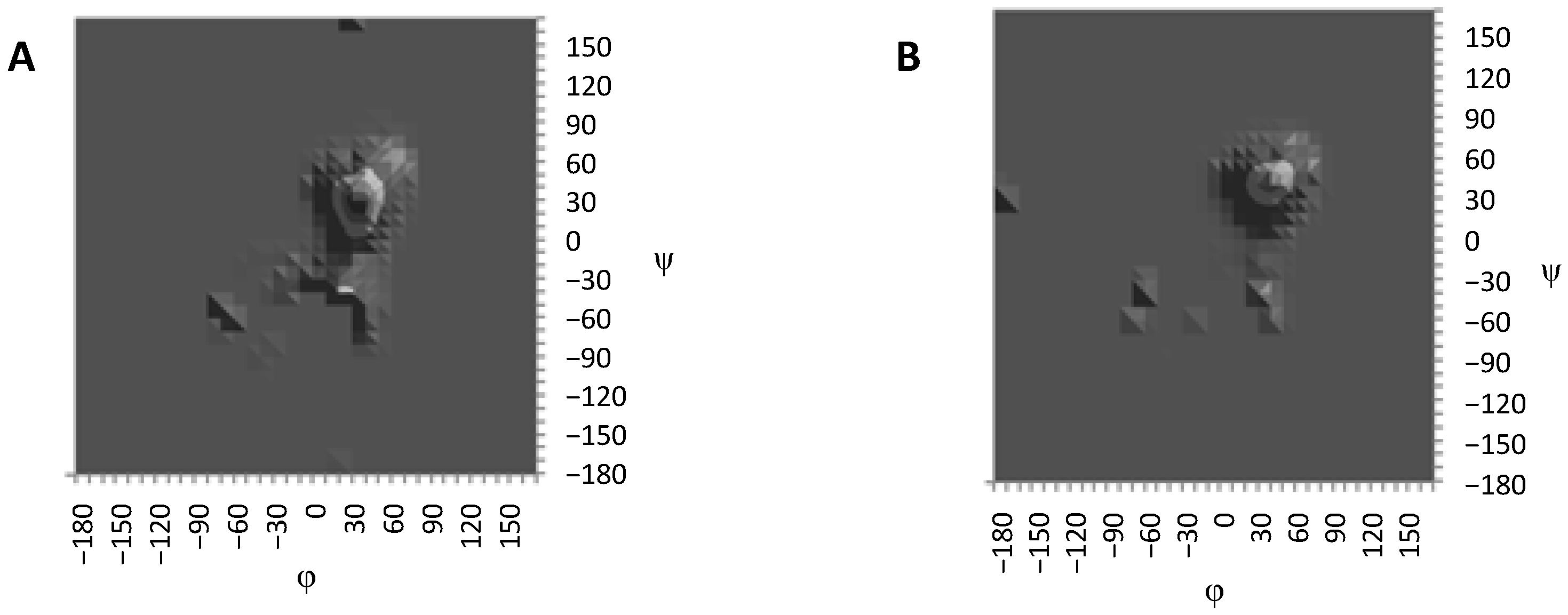
{kind=link}
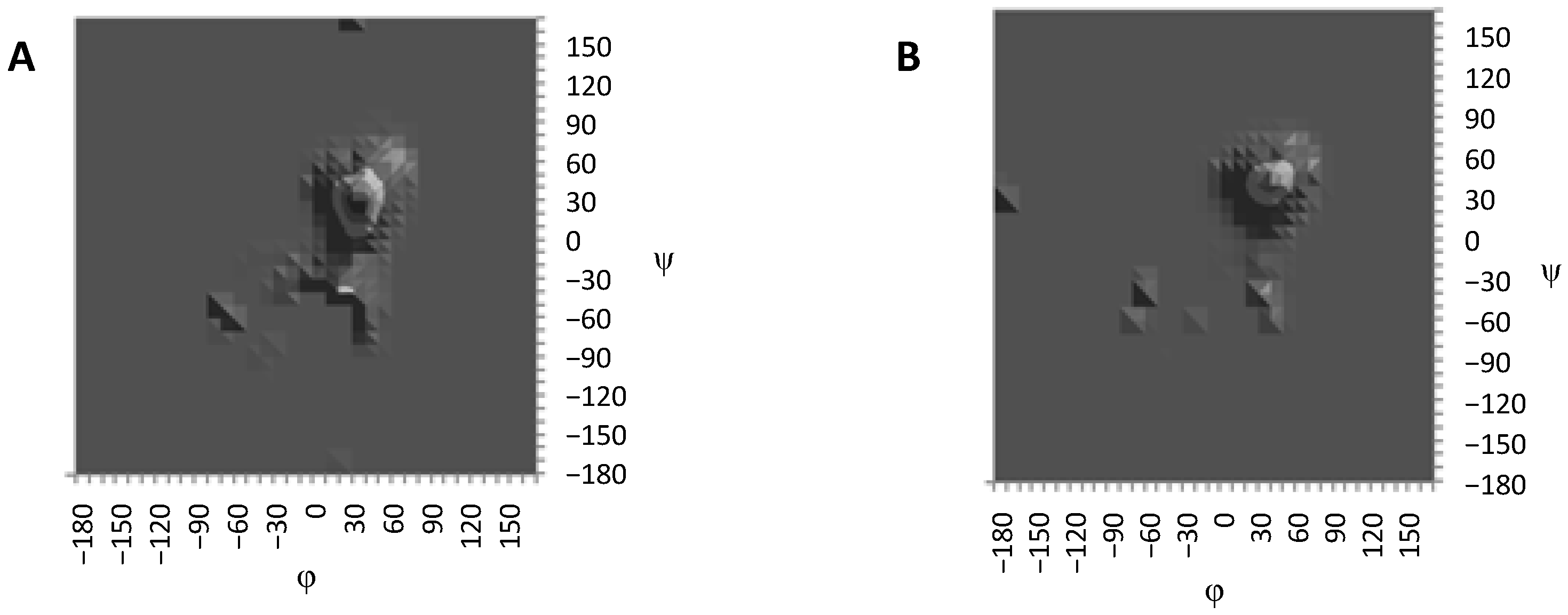{kind=link}
{kind=link}
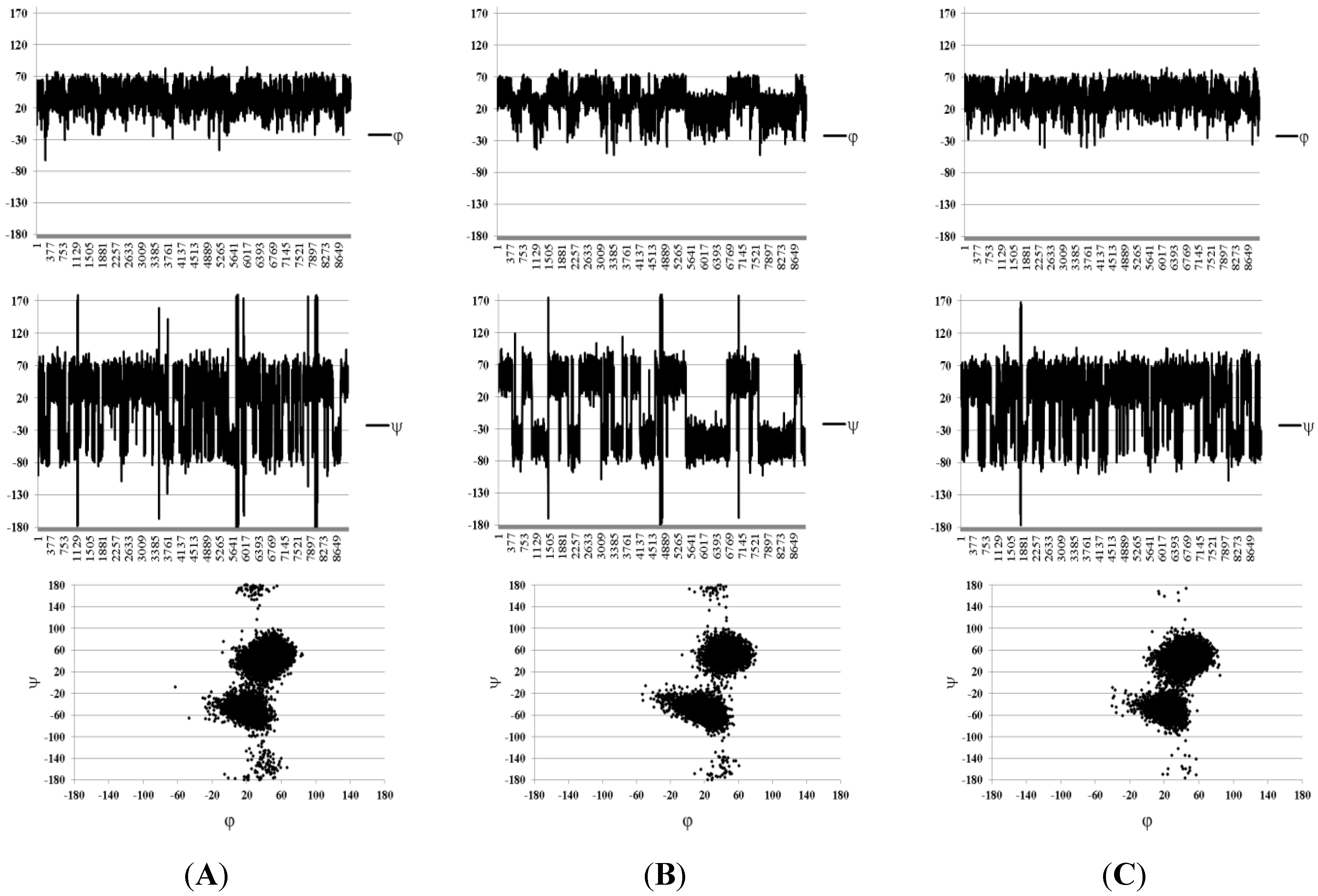
{kind=link}
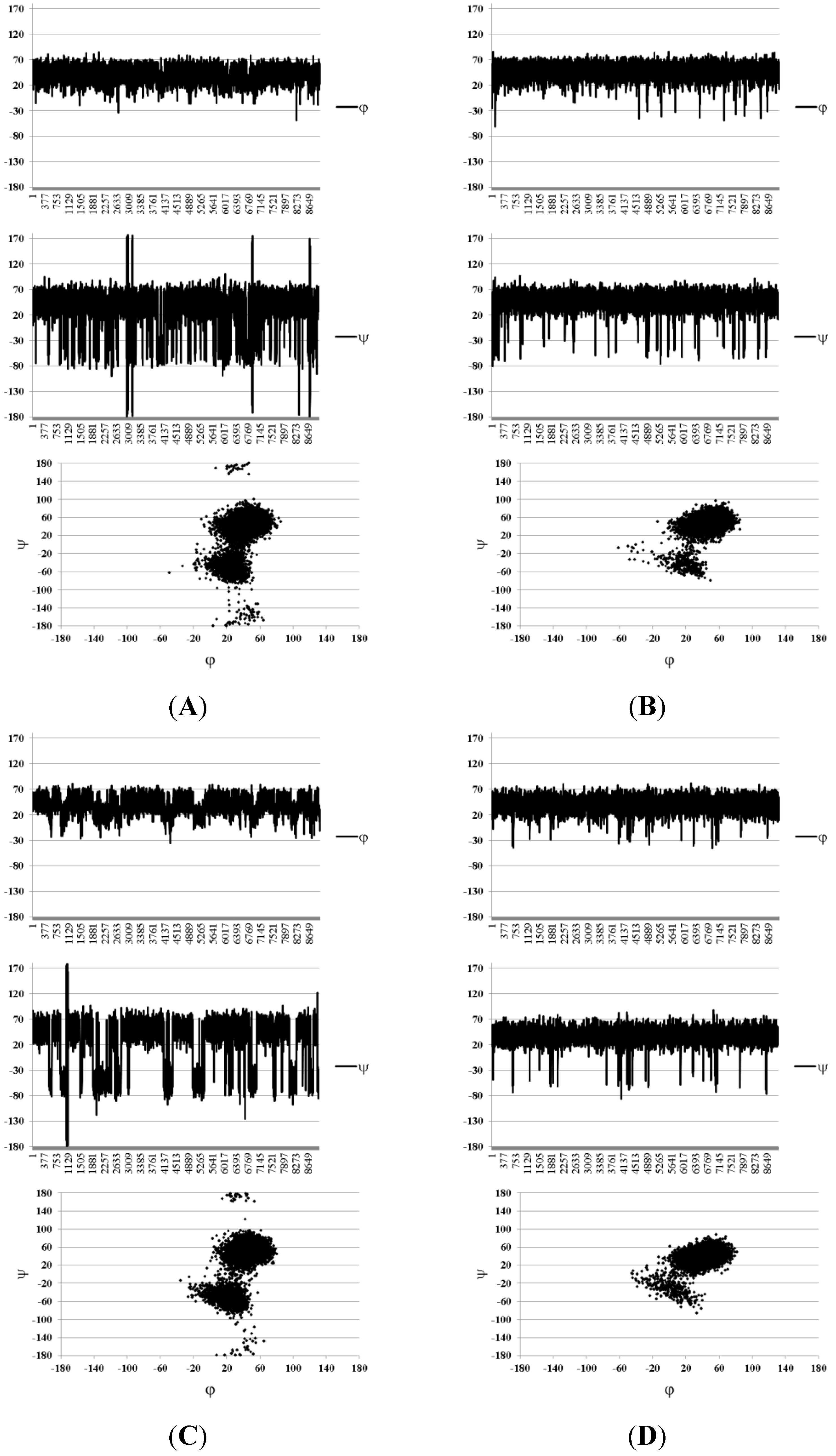
{kind=link}
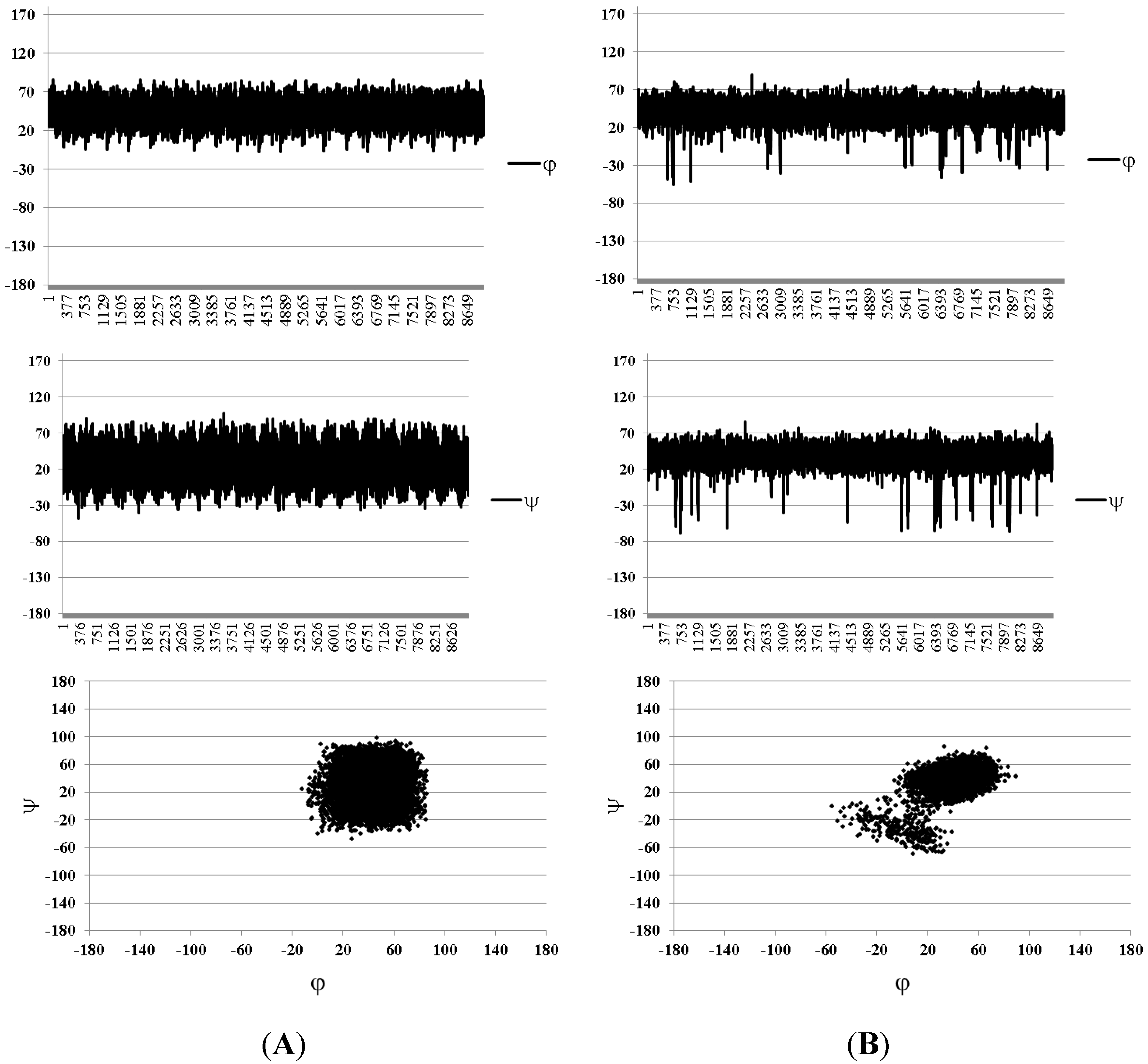{kind=link}
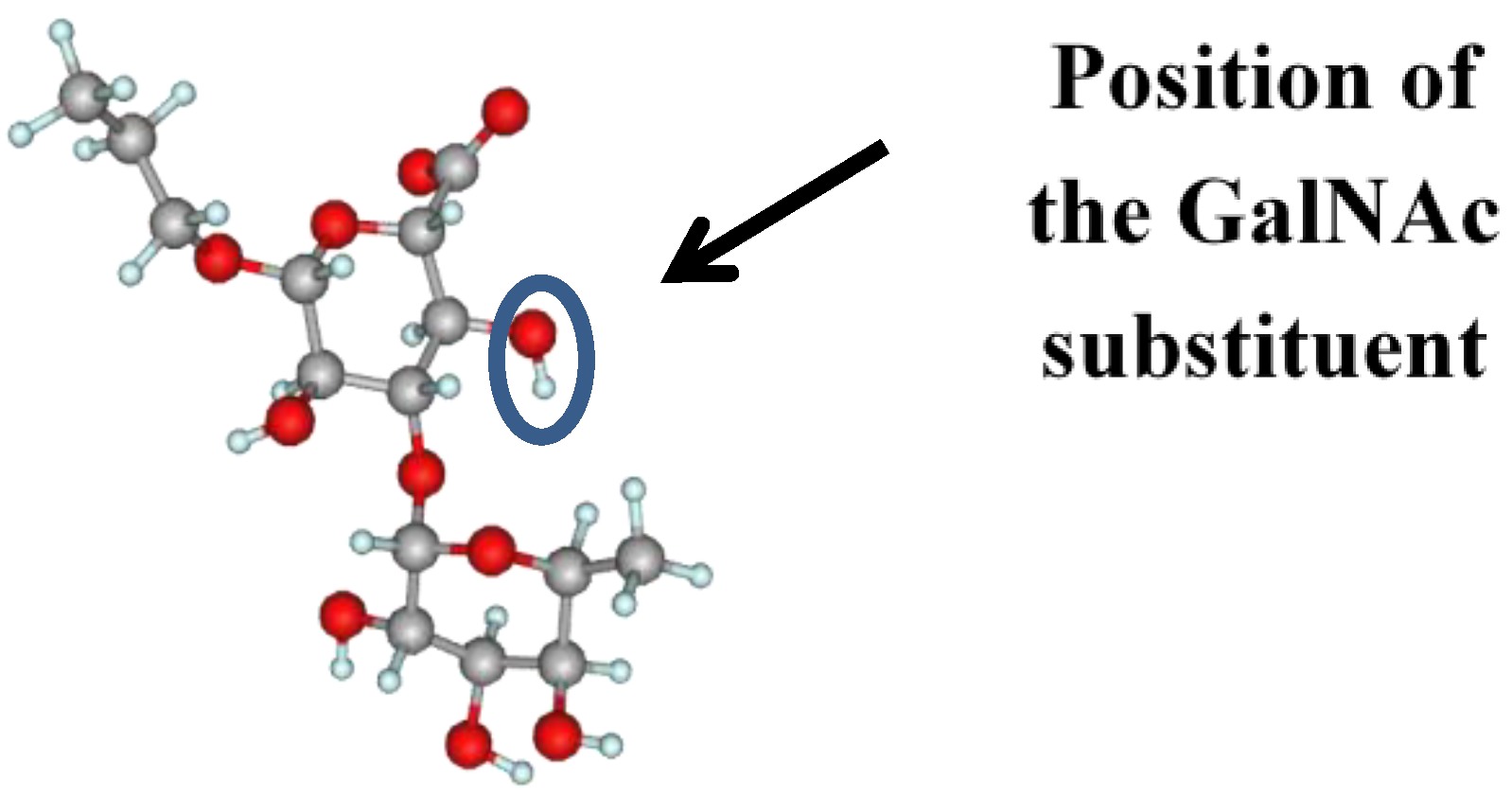{kind=link}
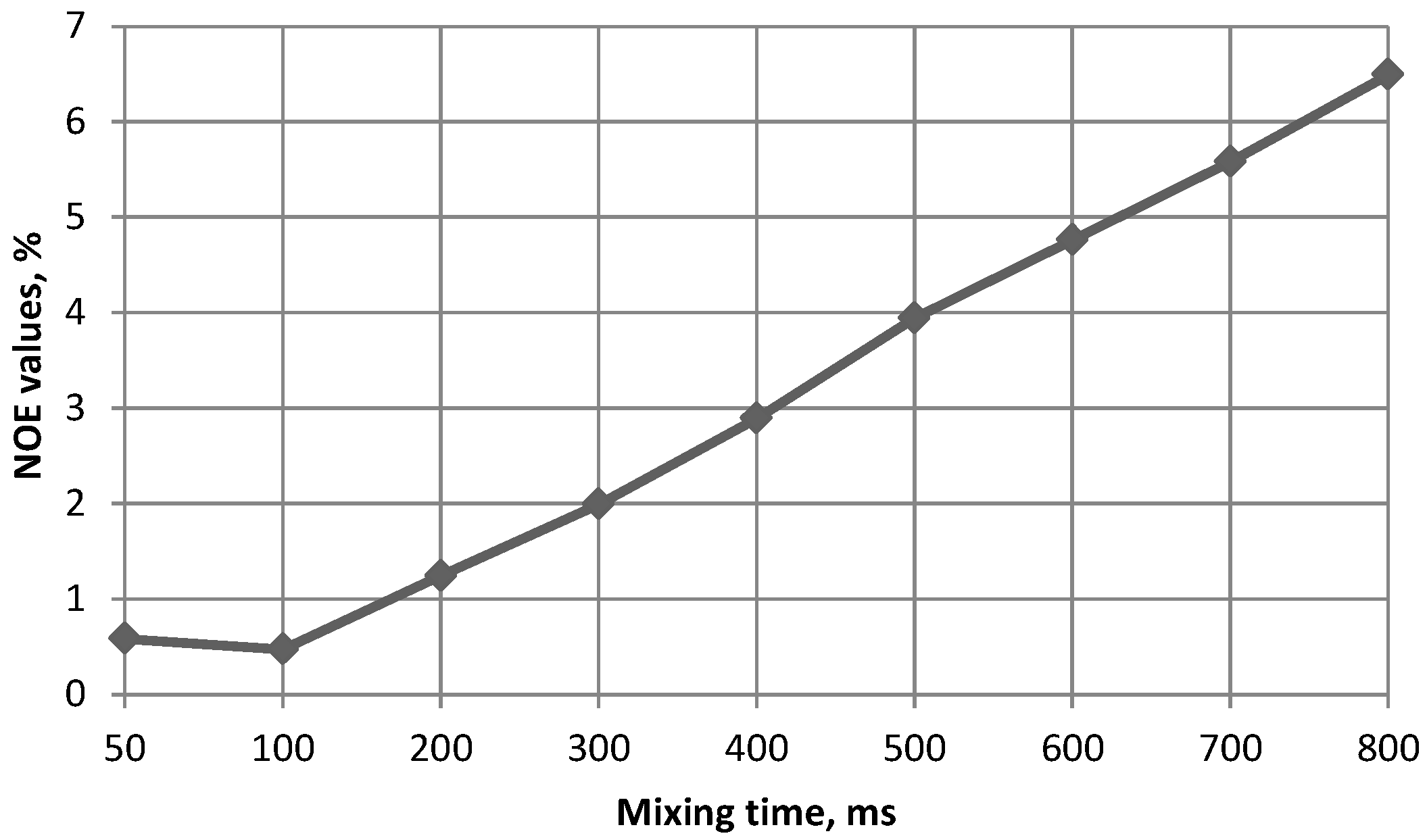{kind=link}
| Compound Number | Jφ (Fuc–GlA) | Jψ (Fuc–GlA) | Jφ (Fuc–Fuc) | Jψ (Fuc–Fuc) |
|---|---|---|---|---|
| 1 | 3.8 (exp) | 2.5 (exp) | n.a.* | n.a.* |
| 4.0 (in vacuo) | 3.8 (in vacuo) | |||
| 3.4 (SASA) | 2.7 (SASA) | |||
| 3.2 (water) | 2.5 (water) | |||
| 2 | 2.7 (exp) | 2.9 (exp) | n.a.* | n.a.* |
| 4.0 (in vacuo) | 3.7 (in vacuo) | |||
| 3.6 (SASA) | 2.3 (SASA) | |||
| 3.2 (water) | 2.4 (water) | |||
| 3 | 3.0 (exp) | 2.7 (exp) | n.a.* | n.a.* |
| 4.0 (in vacuo) | 3.8 (in vacuo) | |||
| 3.3 (SASA) | 2.7 (SASA) | |||
| 3.2 (water) | 2.6 (water) | |||
| 4 | 3.6 (exp) | 3.0 (exp) | 2.8 (exp) | 2.5 (exp) |
| 4.0 (in vacuo) | 3.8 (in vacuo) | 3.5 (in vacuo) | 3.2 (in vacuo) | |
| 3.2 (SASA) | 2.9 (SASA) | 2.7 (SASA) | 2.6 (SASA) | |
| 3.3 (water) | 3.4 (water) | 2.5 (water) | 2.5 (water) | |
| 5 | 2.7 (exp) | 2.5 (exp) | 2.6 (exp) | 2.9 (exp) |
| 4.0 (in vacuo) | 3.7 (in vacuo) | 3.3 (in vacuo) | 3.1 (in vacuo) | |
| 3.2 (SASA) | 2.4 (SASA) | 3.2 (SASA) | 3.4 (SASA) | |
| 3.2(water) | 2.4 (water) | 2.8 (water) | 2.6 (water) |
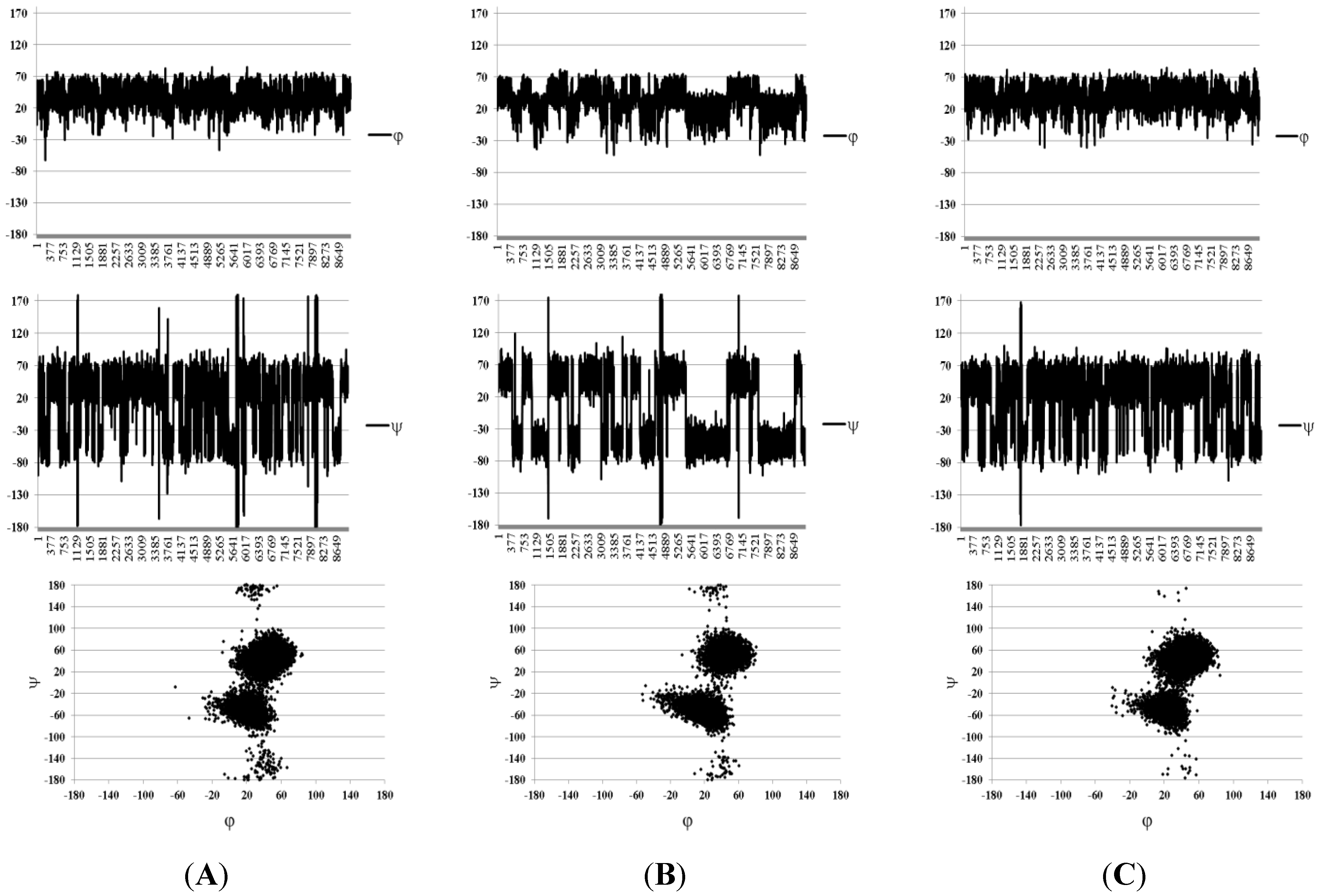
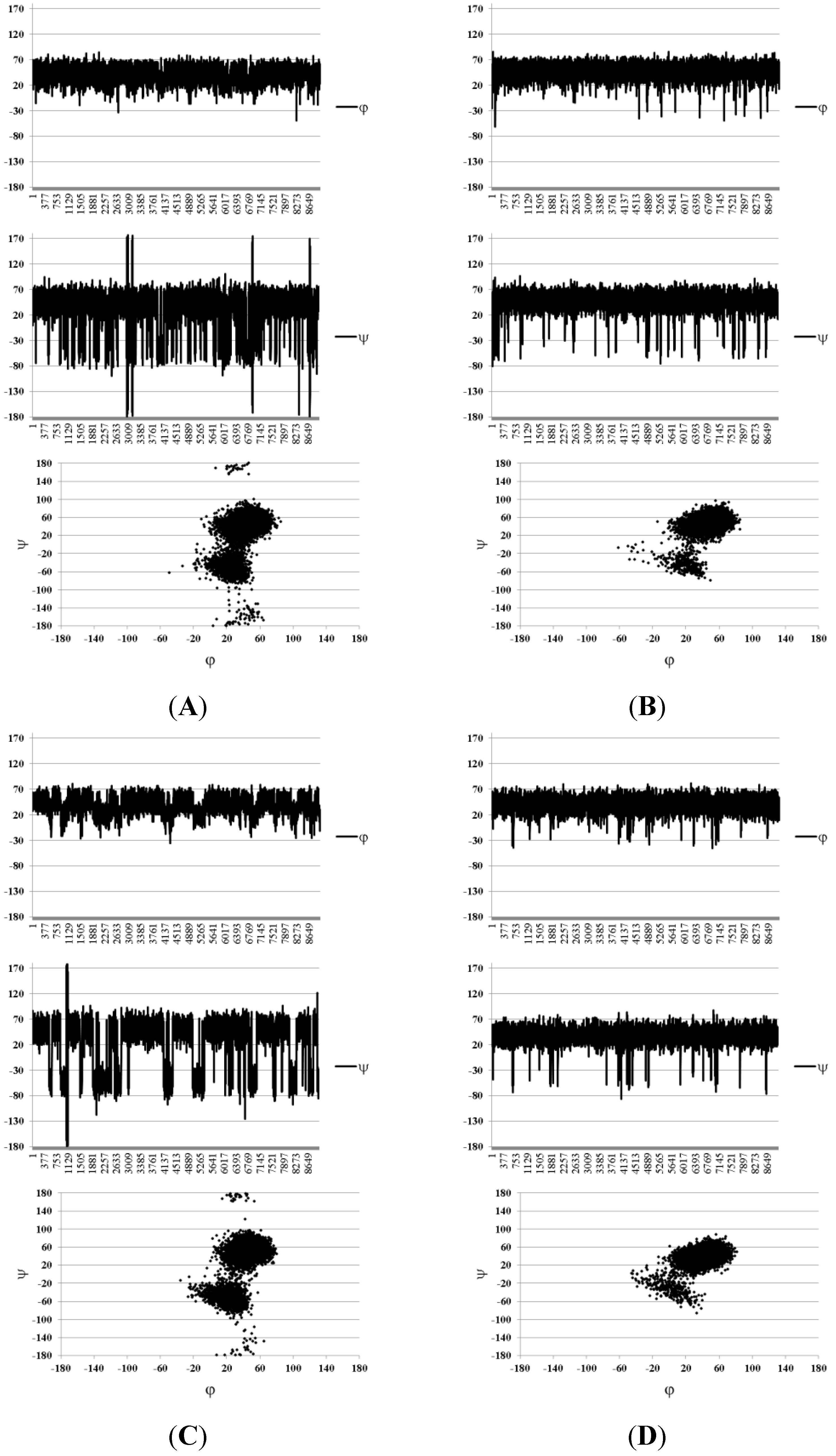
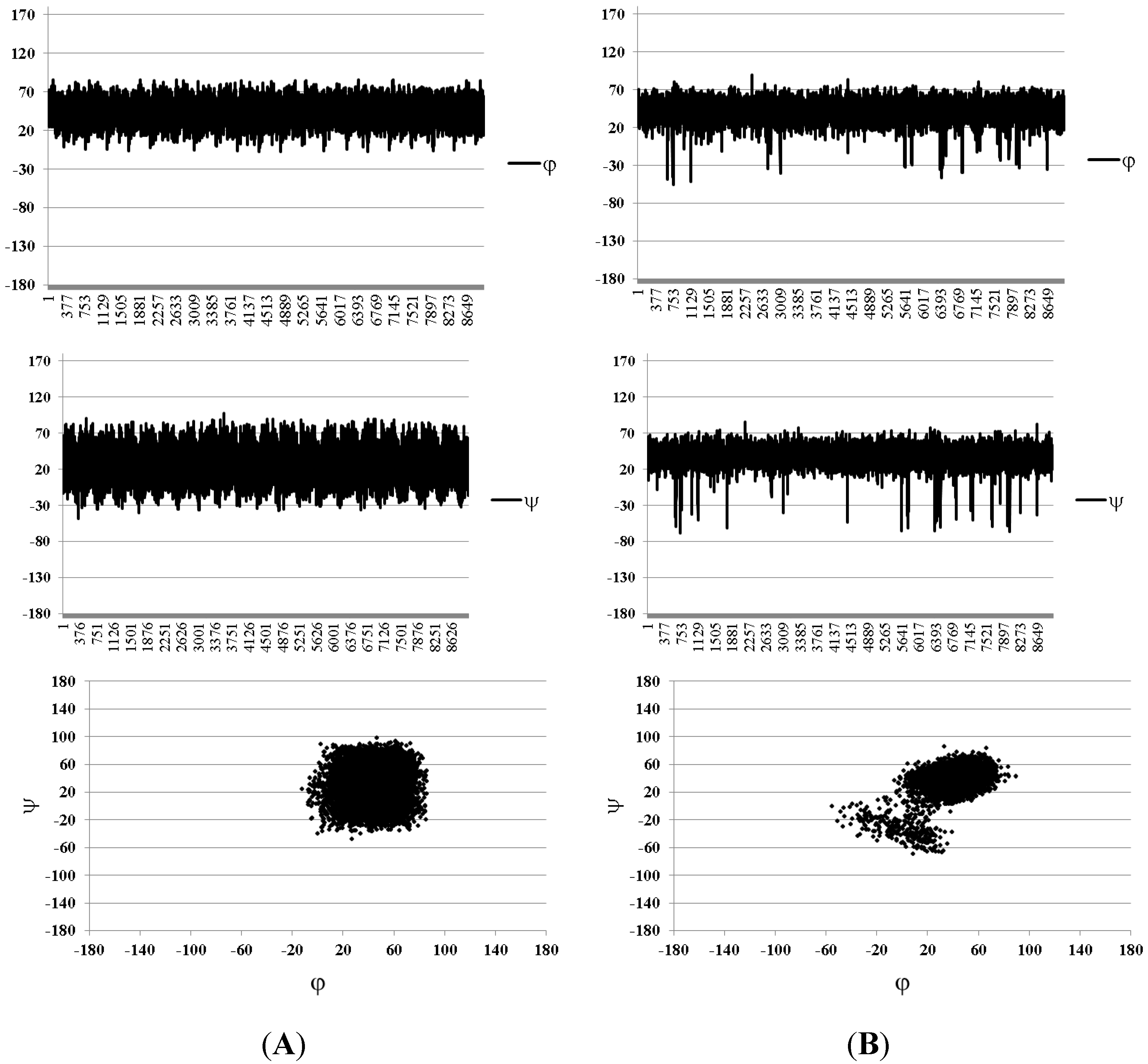
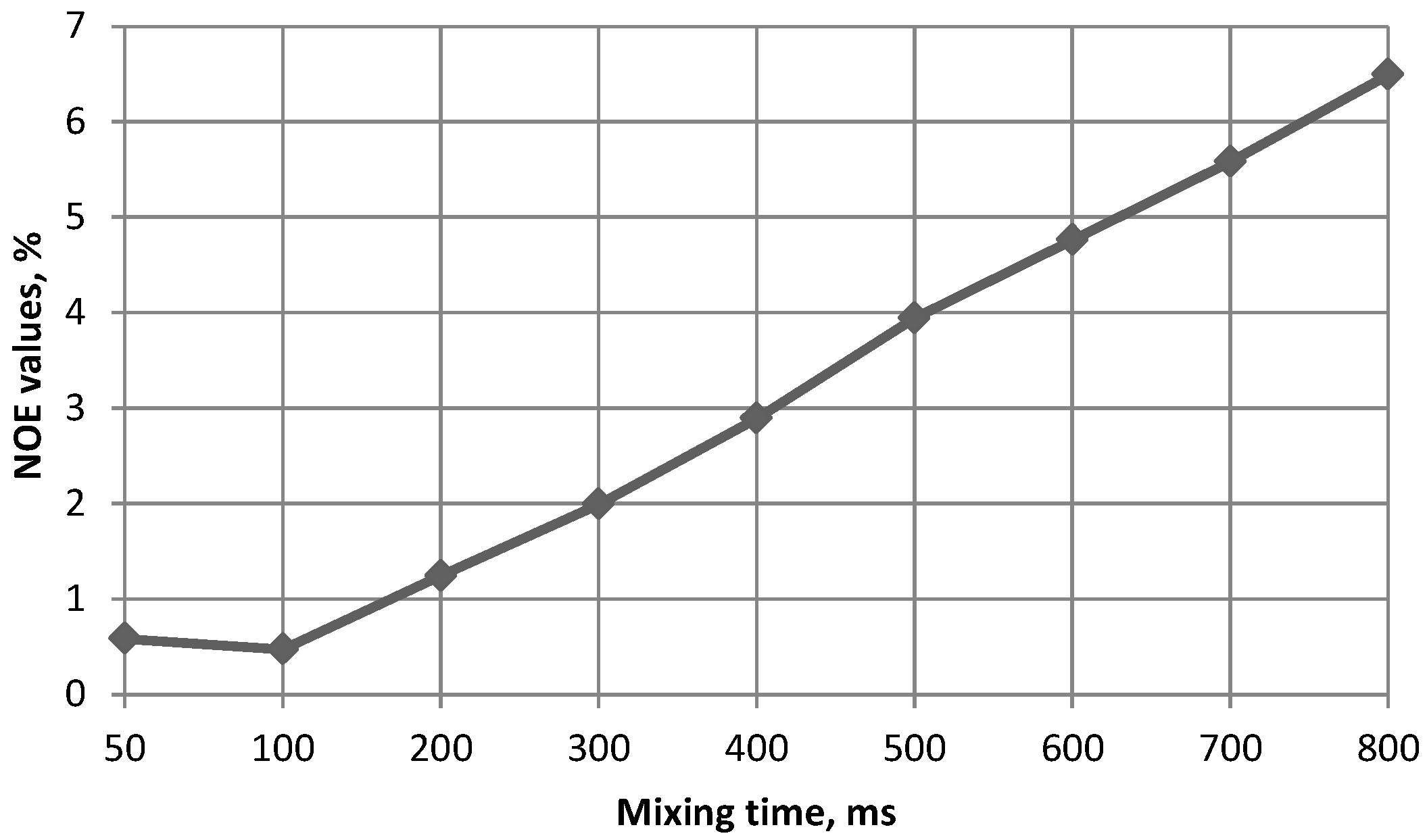
| Compound Number | H1(Fuc)–H3(GlA) | H1(Fuc)–H4(GlA) | H1(Fuc)–H2(GlA) | H1(Fuc`)–H3(Fuc) | H1(Fuc`)–H4(Fuc) |
|---|---|---|---|---|---|
| 1 | 1 (exp) | 0 (exp) | 0 (exp) | n.a.* | n.a.* |
| 1 (SASA) | 0.26 (SASA) | 0.12 (SASA) | |||
| 1 (water) | 0.05 (water) | 0.01 (water) | |||
| 4 | 1 (exp) | 0 (exp) | 0 (exp) | 0.62 (exp) | 0.7 (exp) |
| 1 (SASA) | 0.33 (SASA) | 0.09 (SASA) | 0.81 (SASA) | 3.0 (SASA) | |
| 1 (water) | 0 (water) | 0.01 (water) | 0.7 (water) | 1 (water) |
3. Experimental Section
3.1. Materials
3.2. Methods
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pomin, V.H. Fucanomics and galactanomics: Current status in drug discovery, mechanisms of action and role of the well defined structures. Biochim. Biophys. Acta 2012, 1820, 1971–1979. [Google Scholar] [CrossRef] [PubMed]
- Jiao, G.; Yu, G.; Zhang, J.; Ewart, S. Chemical structures and bioactivities of sulfated polysaccharides from marine algae. Mar. Drugs 2011, 9, 196–223. [Google Scholar] [CrossRef] [PubMed]
- Pomin, V.H. Holothurian fucosylated chondroitin sulfate. Mar. Drugs 2014, 12, 232–254. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, J.L.; Varki, A.; Borsig, L. Heparin attenuates metastasis mainly due to inhibition of P- and L-selectin, but non-anticoagulant heparins can have additional effects. Thromb. Res. 2007, 120, S107–S111. [Google Scholar] [CrossRef]
- Wang, L.; Brown, J.R.; Varki, A.; Esko, J.D. Heparin’s anti-inflammatory effects require glucosamine 6-O-sulfation and are mediated by blockade of L- and P-selectins. J. Clin. Invest. 2002, 110, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Petitou, M.; van Boeckel, C.A. A synthetic antithrombin III binding pentasaccharide is now a drug! What comes next? Angew. Chem. Int. Ed. Engl. 2004, 43, 3118–3133. [Google Scholar] [CrossRef] [PubMed]
- Gray, E.; Hogwood, J.; Mulloy, B. The anticoagulant and antithrombotic mechanisms of heparin. Handb. Exp. Pharmacol. 2012, 207, 43–61. [Google Scholar] [PubMed]
- Borsig, L.; Wang, L.; Cavalcante, M.C.; Cardilo-Reis, L.; Ferreira, P.L.; Mourão, P.A.; Esko, J.D.; Pavão, M.S. Selectin blocking activity of a fucosylated chondroitin sulfate glycosaminoglycan from sea cucumber. Effect on tumor metastasis and neutrophil recruitment. J. Biol. Chem. 2007, 282, 14984–14991. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, R.J.; Oliveira, S.N.; Pomin, V.H.; Mecawi, A.S.; Araujo, I.G.; Mourão, P.A. Effects of oversulfated and fucosylated chondroitin sulfates on coagulation. Challenges for the study of anticoagulant polysaccharides. Thromb. Haemost. 2010, 103, 994–1004. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhang, D.; Wang, S.; Tao, L.; Wang, A.; Chen, W.; Zhu, Z.; Zheng, S.; Gao, X.; Lu, Y. Holothurian glycosaminoglycan inhibits metastasis and thrombosis via targeting of nuclear factor-κB/tissue factor/Factor Xa pathway in melanoma B16F10 cells. PLoS One 2013, 8, e56557. [Google Scholar] [CrossRef] [PubMed]
- Panagos, C.G.; Thomson, D.S.; Moss, C.; Hughes, A.D.; Kelly, M.S.; Liu, Y.; Chai, W.; Venkatasamy, R.; Spina, D.; Page, C.P.; et al. Fucosylated chondroitin sulfates from the body wall of the sea cucumber Holothuria forskali: Conformation, selectin binding, and biological activity. J. Biol. Chem. 2014, 289, 28284–28298. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Wu, M.; Xu, L.; Lian, W.; Xiang, J.; Lu, F.; Gao, N.; Xiao, C.; Wang, S.; Zhao, J. Comparison of physicochemical characteristics and anticoagulant activities of polysaccharides from three sea cucumbers. Mar. Drugs 2013, 11, 399–417. [Google Scholar] [CrossRef] [PubMed]
- Myrona, P.; Siddiqueeb, S.; Al Azad, S. Fucosylated chondroitin sulfate diversity in sea cucumbers: A review. Carbohydr. Polym. 2014, 112, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Mourão, P.A.; Giumarães, B.; Mulloy, B.; Thomas, S.; Gray, E. Antithrombotic activity of a fucosylated chondroitin sulphate from echinoderm: Sulphated fucose branches on the polysaccharide account for its antithrombotic action. Br. J. Haematol. 1998, 101, 647–652. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Xue, C.; Yin, L.; Tang, Q.; Yu, G.; Chai, W. Comparison of structures and anticoagulant activities of fucosylated chondroitin sulfates from different sea cucumbers. Carbohydr. Polym. 2011, 83, 688–695. [Google Scholar] [CrossRef]
- Ustyuzhanina, N.E.; Fomitskaya, P.A; Gerbst, A.G.; Dmitrenok, A.S.; Nifantiev, N.E. Synthesis of the oligosaccharides related to side chains of fucosylated chondroitin sulfates from sea cucumbers. Mar. Drugs 2015, 13, 770–787. [Google Scholar] [CrossRef]
- Mulloy, B. Long-range carbon-proton coupling constants: Application to conformational studies of oligosaccharides. Carbohydr. Res. 1988, 184, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Ooi, T.; Oobatake, M.; Némethy, G.; Scheraga, H.A. Accessible surface areas as a measure of the thermodynamic parameters of hydration of peptides. Proc. Natl. Acad. Sci. USA 1987, 84, 3086–3090. [Google Scholar] [CrossRef] [PubMed]
- Gerbst, A.G.; Grachev, A.A.; Yashunsky, D.V.; Tsvetkov, Y.E.; Shashkov, A.S.; Nifantiev, N.E. Theoretical and experimental conformational studies of oligoglucosides structurally related to fragments of fungal cell wall β-(1→3)-d-Glucan. J. Carbohydr. Chem. 2013, 32, 205–221. [Google Scholar] [CrossRef]
- Grachev, A.A.; Gerbst, A.G.; Ustuzhanina, N.E.; Shashkov, A.S.; Usov, A.I.; Nifantiev, N.E. NMR Investigation of the influence of sulfate groups at C-2 and C-4 on the conformational behavior of fucoidan fragments with homo-(1→3)-linked backbone. J. Carbohydr. Chem. 2006, 25, 315–330. [Google Scholar] [CrossRef]
- TINKER Molecular Modeling. Available online: http://dasher.wustl.edu/tinker (accessed on 6 February 2015).
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Leimkuhler, B.; Skeel, R. Symplectic numerical integrators in constrained Hamiltonian systems. J. Comp. Phys. 1994, 112, 117–125. [Google Scholar] [CrossRef]
- Meissner, A.; Sørensen, O.W. Measurement of J(H,H) and long-range J(X,H) coupling constants in small molecules. Broadband XLOC and J-HMBC. Magn. Reson. Chem. 2001, 39, 49–52. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gerbst, A.G.; Dmitrenok, A.S.; Ustyuzhanina, N.E.; Nifantiev, N.E. Conformational Analysis of the Oligosaccharides Related to Side Chains of Holothurian Fucosylated Chondroitin Sulfates. Mar. Drugs 2015, 13, 936-947. https://doi.org/10.3390/md13020936
Gerbst AG, Dmitrenok AS, Ustyuzhanina NE, Nifantiev NE. Conformational Analysis of the Oligosaccharides Related to Side Chains of Holothurian Fucosylated Chondroitin Sulfates. Marine Drugs. 2015; 13(2):936-947. https://doi.org/10.3390/md13020936
Chicago/Turabian StyleGerbst, Alexey G., Andrey S. Dmitrenok, Nadezhda E. Ustyuzhanina, and Nikolay E. Nifantiev. 2015. "Conformational Analysis of the Oligosaccharides Related to Side Chains of Holothurian Fucosylated Chondroitin Sulfates" Marine Drugs 13, no. 2: 936-947. https://doi.org/10.3390/md13020936
APA StyleGerbst, A. G., Dmitrenok, A. S., Ustyuzhanina, N. E., & Nifantiev, N. E. (2015). Conformational Analysis of the Oligosaccharides Related to Side Chains of Holothurian Fucosylated Chondroitin Sulfates. Marine Drugs, 13(2), 936-947. https://doi.org/10.3390/md13020936