_Kim.png)
Trans-eQTLs Can Be Used to Identify Tissue-Specific Gene Regulatory Networks


Abstract

1. Introduction
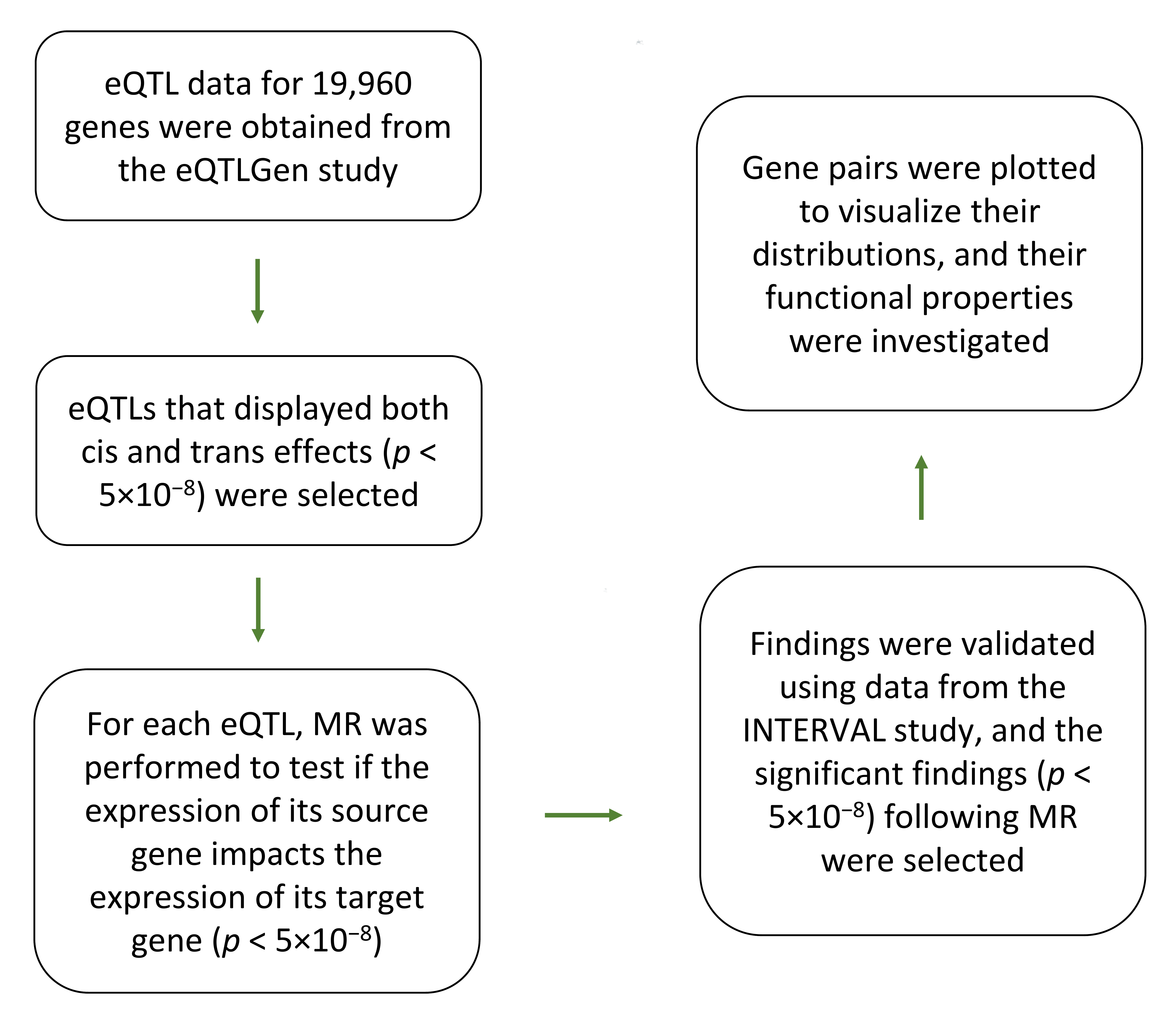
2. Methods
2.1. Data for the Discovery Step
2.2. Data for the Validation Step
2.3. Mendelian Randomization
2.4. Phenom-Wide Association Study
2.5. eQTL Assesment
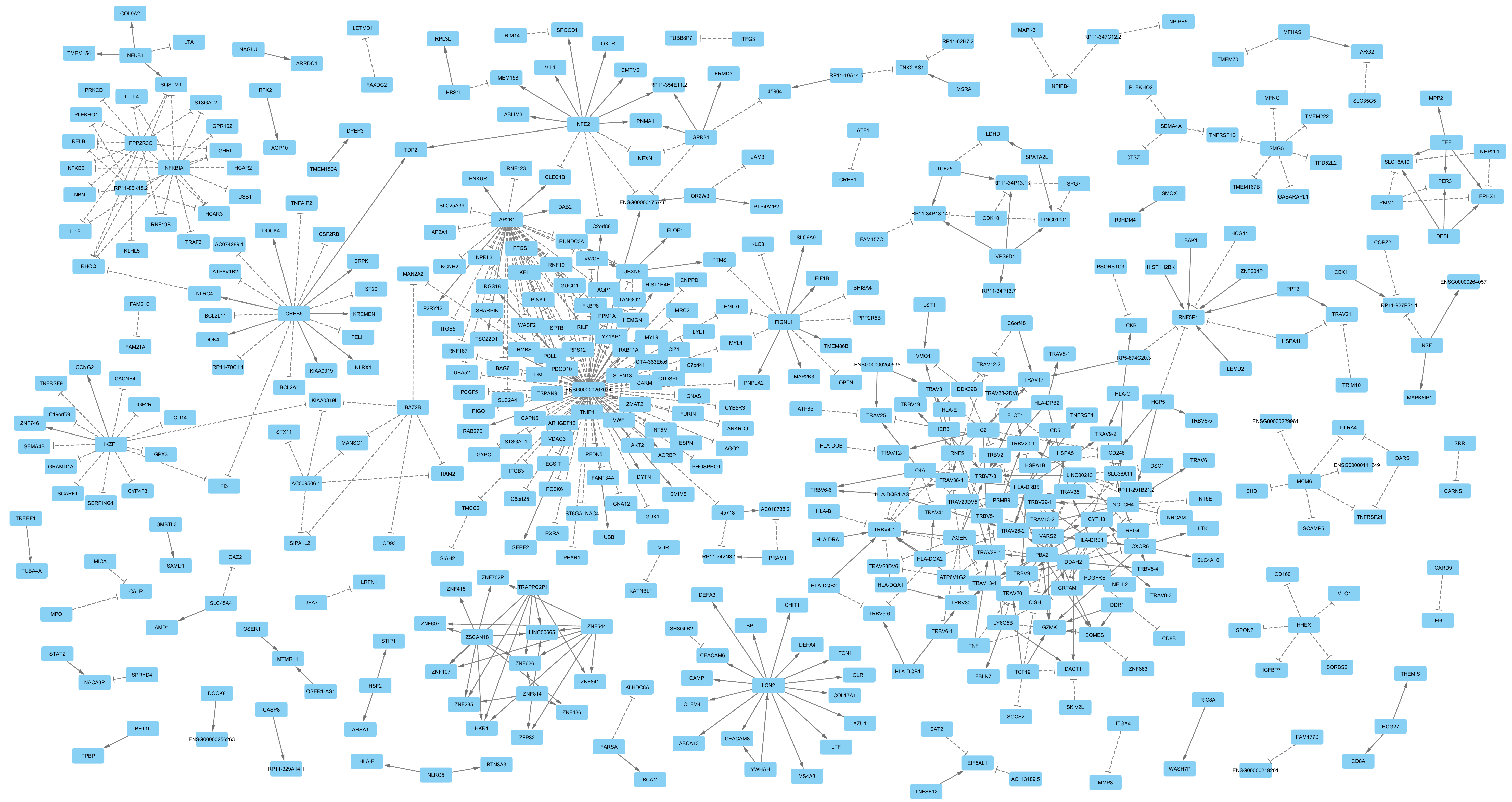
3. Results
4. Discussion
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Badia-i-Mompel, P.; Wessels, L.; Müller-Dott, S.; Trimbour, R.; Ramirez Flores, R.O.; Argelaguet, R.; Saez-Rodriguez, J. Gene regulatory network inference in the era of single-cell multi-omics. Nat. Rev. Genet. 2023, 24, 739–754. [Google Scholar] [CrossRef]
- McCalla, S.G.; Fotuhi Siahpirani, A.; Li, J.; Pyne, S.; Stone, M.; Periyasamy, V.; Shin, J.; Roy, S. Identifying strengths and weaknesses of methods for computational network inference from single-cell RNA-seq data. G3 Genes Genomes Genet. 2023, 13, jkad004. [Google Scholar] [CrossRef]
- Mercatelli, D.; Scalambra, L.; Triboli, L.; Ray, F.; Giorgi, F.M. Gene regulatory network inference resources: A practical overview. Biochim. Biophys. Acta Gene Regul. Mech. 2020, 1863, 194430. [Google Scholar] [CrossRef] [PubMed]
- GTEx Consortium. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 2020, 369, 1318–1330. [Google Scholar] [CrossRef] [PubMed]
- Price, A.L.; Helgason, A.; Thorleifsson, G.; McCarroll, S.A.; Kong, A.; Stefansson, K. Single-tissue and cross-tissue heritability of gene expression via identity-by-descent in related or unrelated individuals. PLoS Genet. 2011, 7, e1001317. [Google Scholar] [CrossRef] [PubMed]
- Nica, A.C.; Dermitzakis, E.T. Expression quantitative trait loci: Present and future. Philos. Trans. R. Soc. B Biol. Sci. 2013, 368, 20120362. [Google Scholar] [CrossRef]
- Pasaniuc, B.; Price, A.L. Dissecting the genetics of complex traits using summary association statistics. Nat. Rev. Genet. 2017, 18, 117–127. [Google Scholar] [CrossRef]
- Zhu, Z.; Zhang, F.; Hu, H.; Bakshi, A.; Robinson, M.R.; Powell, J.E.; Montgomery, G.W.; Goddard, M.E.; Wray, N.R.; Visscher, P.M.; et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 2016, 48, 481–487. [Google Scholar] [CrossRef]
- Davey Smith, G.; Hemani, G. Mendelian randomization: Genetic anchors for causal inference in epidemiological studies. Hum. Mol. Genet. 2014, 23, R89–R98. [Google Scholar] [CrossRef]
- Zhu, Z.; Zheng, Z.; Zhang, F.; Wu, Y.; Trzaskowski, M.; Maier, R.; Robinson, M.R.; McGrath, J.J.; Visscher, P.M.; Wray, N.R.; et al. Causal associations between risk factors and common diseases inferred from GWAS summary data. Nat. Commun. 2018, 9, 224. [Google Scholar] [CrossRef]
- Võsa, U.; Claringbould, A.; Westra, H.-J.; Bonder, M.J.; Deelen, P.; Zeng, B.; Kirsten, H.; Saha, A.; Kreuzhuber, R.; Yazar, S.; et al. Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat. Genet. 2021, 53, 1300–1310. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Ritchie, S.C.; Liang, Y.; Timmers, P.R.H.J.; Pietzner, M.; Lannelongue, L.; Lambert, S.A.; Tahir, U.A.; May-Wilson, S.; Foguet, C.; et al. An atlas of genetic scores to predict multi-omic traits. Nature 2023, 616, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience 2015, 4, 7. [Google Scholar] [CrossRef]
- Park, J.-H.; Wacholder, S.; Gail, M.H.; Peters, U.; Jacobs, K.B.; Chanock, S.J.; Chatterjee, N. Estimation of effect size distribution from genome-wide association studies and implications for future discoveries. Nat. Genet. 2010, 42, 570–575. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef]
- Kolberg, L.; Raudvere, U.; Kuzmin, I.; Adler, P.; Vilo, J.; Peterson, H. g:Profiler—Interoperable web service for functional enrichment analysis and gene identifier mapping (2023 update). Nucleic Acids Res. 2023, 51, W207–W212. [Google Scholar] [CrossRef]
- Barabási, A.-L.; Albert, R. Emergence of scaling in random networks. Science 1999, 286, 509–512. [Google Scholar] [CrossRef]
- Li, E.; Liu, H.; Huang, L.; Zhang, X.; Dong, X.; Song, W.; Zhao, H.; Lai, J. Long-range interactions between proximal and distal regulatory regions in maize. Nat. Commun. 2019, 10, 2633. [Google Scholar] [CrossRef]
- Krzyzanowska, K.; Mittermayer, F.; Wolzt, M.; Schernthaner, G. ADMA, cardiovascular disease and diabetes. Diabetes Res. Clin. Pract. 2008, 82, S122–S126. [Google Scholar] [CrossRef]
- Hasegawa, K.; Wakino, S.; Kimoto, M.; Minakuchi, H.; Fujimura, K.; Hosoya, K.; Komatsu, M.; Kaneko, Y.; Kanda, T.; Tokuyama, H. The Hydrolase DDAH2 Enhances Pancreatic Insulin Secretion by Transcriptional Regulation of Secretagogin Through a Sirt1-Dependent Mechanism in Mice. FASEB J. 2013, 27, 2301–2315. [Google Scholar] [CrossRef]
- Chen, C.-H.; Zhao, J.-F.; Hsu, C.-P.; Kou, Y.R.; Lu, T.-M.; Lee, T.-S. The detrimental effect of asymmetric dimethylarginine on cholesterol efflux of macrophage foam cells: Role of the NOX/ROS signaling. Free Radic. Biol. Med. 2019, 143, 354–365. [Google Scholar] [CrossRef]
- Asymmetric Dimethylarginine (ADMA): A Novel Risk Factor for Endothelial Dysfunction | Circulation. Available online: https://www-ahajournals-org.proxy.bib.uottawa.ca/doi/10.1161/01.CIR.98.18.1842 (accessed on 17 July 2025).
- Lundman, P.; Eriksson, M.J.; Stühlinger, M.; Cooke, J.P.; Hamsten, A.; Tornvall, P. Mild-to-moderate hypertriglyceridemia in young men is associated with endothelial dysfunction and increased plasma concentrations of asymmetric dimethylarginine. J. Am. Coll. Cardiol. 2001, 38, 111–116. [Google Scholar] [CrossRef]
- Wink, D.A.; Hines, H.B.; Cheng, R.Y.S.; Switzer, C.H.; Flores-Santana, W.; Vitek, M.P.; Ridnour, L.A.; Colton, C.A. Nitric oxide and redox mechanisms in the immune response. J. Leukoc. Biol. 2011, 89, 873–891. [Google Scholar] [CrossRef]
- Huang, S.; Li, Z.; Wu, Z.; Liu, C.; Yu, M.; Wen, M.; Zhang, L.; Wang, X. DDAH2 suppresses RLR-MAVS-mediated innate antiviral immunity by stimulating nitric oxide-activated, Drp1-induced mitochondrial fission. Sci. Signal. 2021, 14, eabc7931. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| GO-BP ID | Description | p | Corrected p 1 |
|---|---|---|---|
| 0002250 | Adaptive immune response | 1.0 × 10−25 | 2.3 × 10−22 |
| 0002504 | Antigen processing and presentation of peptide or polysaccharide antigen via MHC class II | 3.2 × 10−9 | 7.2 × 10−6 |
| 0002376 | Immune system process | 1.5 × 10−8 | 3.4 × 10−5 |
| 0002503 | Peptide antigen assembly with MHC class II protein complex | 1.5 × 10−8 | 3.4 × 10−5 |
| 0009617 | Response to bacterium | 1.7 × 10−8 | 3.9 × 10−5 |
| 0019882 | Antigen processing and presentation | 2.2 × 10−8 | 4.9 × 10−5 |
| 0007166 | Cell surface receptor signaling pathway | 2.8 × 10−7 | 6.4 × 10−4 |
| 0045087 | Innate immune response | 3.6 × 10−7 | 8.1 × 10−4 |
| 0019886 | Antigen processing and presentation of exogenous peptide antigen via MHC class II | 2.6 × 10−6 | 5.9 × 10−3 |
| 0006955 | Immune response | 2.8 × 10−6 | 6.2 × 10−3 |
| Trait | PMID | Panel 2 | B | SE | p | NSNP |
|---|---|---|---|---|---|---|
| Multiple sclerosis | 31604244 | D | 1.49 | 0.06 | 2.7 × 10−117 | 9 |
| V | 1.64 | 0.07 | 5.3 × 10−134 | 28 | ||
| LDL | 32493714 | D | −0.08 | 0.01 | 6.7 × 10−39 | 12 |
| V | −0.10 | 0.01 | 1.0 × 10−39 | 20 | ||
| Type 1 diabetes | 34127860 | D | −1.02 | 0.05 | 6.8 × 10−88 | 11 |
| V | −0.25 | 0.04 | 8.2 × 10−9 | 30 | ||
| Rheumatoid arthritis | 24390342 | D | 0.76 | 0.08 | 4.6 × 10−24 | 10 |
| V | 0.60 | 0.07 | 3.7 × 10−20 | 27 | ||
| Psoriasis | UKBB 1 | D | −0.01 | 0.001 | 3.9 × 10−22 | 12 |
| V | −0.01 | 0.001 | 3.3 × 10−19 | 29 | ||
| Primary biliary cholangitis | 22961000 | D | −0.81 | 0.10 | 8.2 × 10−15 | 7 |
| V | −0.96 | 0.11 | 1.1 × 10−19 | 24 | ||
| Crohn’s disease | 26192919 | D | −0.25 | 0.04 | 4.8 × 10−12 | 8 |
| V | −0.36 | 0.04 | 1.3 × 10−17 | 23 | ||
| High cholesterol | UKBB | D | −0.02 | 0.002 | 8.5 × 10−12 | 12 |
| V | −0.02 | 0.002 | 3.9 × 10−17 | 29 | ||
| Type 2 diabetes | 30297969 | D | −0.11 | 0.02 | 1.6 × 10−9 | 11 |
| V | −0.12 | 0.02 | 1.0 × 10−8 | 29 | ||
| Triglycerides | 24097068 | D | −0.07 | 0.01 | 3.1 × 10−9 | 8 |
| V | −0.07 | 0.01 | 4.9 × 10−8 | 25 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nikpay, M. Trans-eQTLs Can Be Used to Identify Tissue-Specific Gene Regulatory Networks. Curr. Issues Mol. Biol. 2025, 47, 594. https://doi.org/10.3390/cimb47080594
Nikpay M. Trans-eQTLs Can Be Used to Identify Tissue-Specific Gene Regulatory Networks. Current Issues in Molecular Biology. 2025; 47(8):594. https://doi.org/10.3390/cimb47080594
Chicago/Turabian StyleNikpay, Majid. 2025. "Trans-eQTLs Can Be Used to Identify Tissue-Specific Gene Regulatory Networks" Current Issues in Molecular Biology 47, no. 8: 594. https://doi.org/10.3390/cimb47080594
APA StyleNikpay, M. (2025). Trans-eQTLs Can Be Used to Identify Tissue-Specific Gene Regulatory Networks. Current Issues in Molecular Biology, 47(8), 594. https://doi.org/10.3390/cimb47080594