Abstract
Retinitis pigmentosa (RP) encompasses a diverse range of hereditary, degenerative retinal ailments, presenting notable obstacles to molecular genetic diagnoses due to the intricate array of variants in different genes involved. This study enrolled 21 probands and their families who have been diagnosed with nonsyndromic RP but without a previous molecular diagnosis. We employed whole-exome sequencing (WES) to detect possible harmful gene variations in individuals with unknown-cause RP at the molecular level. WES allowed the identification of ten potential disease-causing variants in eight different genes. In 8 out of the total 21 patients, this method successfully identified the underlying molecular causes, such as putative pathogenic variants in genes including CRB1, KLHL7, PDE6B, RDH12, RP1, RPE65, USH2A, and RHO. A novel variant was identified in one of these genes, specifically PDE6B, providing valuable information on prospective targets for future enhanced gene therapeutic approaches.
1. Introduction
Approximately one in every four thousand individuals worldwide is afflicted with RP, the most common form of inherited retinal dystrophies (IRDs). RP (MIM: 268,000) is defined by the primary degeneration of rods, often presenting as progressive night blindness and subsequent visual field constriction. The condition begins with cones malfunctioning, leading to decreased visual acuity and central vision loss [1,2]. Moreover, in most patients, a symptom known as “bone spicule” pigmentation is observed in the retina. It is marked by the gradual degeneration of rod and cone photoreceptors, resulting in significant vision impairment in both eyes [3]. Cmmon symptoms encompass advancing nyctalopia, visual field constriction, and visual acuity decrease.
Usually, RP patients share similar genetic backgrounds, with phenotypic heterogeneity among people. Furthermore, RP is a genetically diverse illness with autosomal-dominant, autosomal-recessive, or X-linked inheritance. Genetic, allelic, and phenotypic variation make the diagnosis of RP patients a difficult process. About 93 genes are linked to nonsyndromic RP variants, and the RetNet database (https://web.sph.uth.edu/RetNet/ (accessed on 20 May 2023)) provides up-to-date information [4,5].
This research aimed to discover the putative pathogenic gene associated with Korean families exhibiting retinitis pigmentosa via whole-exome sequencing. Furthermore, we aimed to assess the diagnostic efficacy of this approach and to determine the relationship between candidate genes and clinical characteristics.
2. Materials and Methods
2.1. Clinical Examination
A group of 21 families who were diagnosed with RP but did not have any previous molecular test results, and who had a well-documented family history, were chosen from an elite eye clinic in Seoul, Korea, between January 2021 and October 2022.
All patients visiting the elite eye clinic received comprehensive ophthalmic examinations, including best-corrected visual acuity (BCVA) and intraocular pressure (IOP) measurements, slit lamp biomicroscopy, color fundus photography (Topcon Inc., Tokyo, Japan), ocular biometry applying optical low-coherence reflectometry (Lenstar 900 Optical Biometer, Haag-Streit, Koeniz, Switzerland), OCT and OCT angiography (Heidelberg Engineering, Max-Jarecki-Straße 8, 69115 Heidelberg, Germany), and stationary perimetry tests (Humphery field analyzer; Carl Zeiss Meditec, Inc., Dublin, CA, USA).
Written informed consent was obtained from all participants or their guardians, and the study received approval from a Bioethics Committee authorized by the Ministry of Health and Welfare (MOHW) of and Hanyang University and OneOmics. All study protocols followed the principles of the Declaration of Helsinki.
2.2. Whole-Exome Sequencing
Using Exgene Blood SV mini (GeneAll, Seoul, Republic of Korea), DNA was extracted from the blood or saliva of patients and their families according to the manufacturer’s protocol [6]. Exomes were captured and amplified via PCR by using a xGen Exome Research Panel V2 (Integrated DNA Technologies, Coralville, IA, USA) exome kit [7]. Paired-end sequencing was performed using Novaseq 6000 (Illumina, San Diego, CA, USA) [8]. Variant discoveries were performed using TAK’s best practice. Discovered variants were functionally annotated by utilizing the software tool snpEff (version 5.0) in conjunction with the genome annotation release GRCh38.102 [9]. To assess in silico variant effects, dbNSFP v4.5 (Gencode release 29/Ensembl version 94) was annotated using SnpSift v5.0 [10,11]. dbSNP build 155 and the CLINVAR database were annotated using bcftools v1.3 [12,13,14]. Variants have a frequency of more than 1% in GnomAD and the KRGDB [15,16,17].
To avoid missing disease-related variations with an allele frequency of 1% or higher, the detected variants were filtered based on an allele frequency of below 1% in the KRGDB [18]. Each variant that successfully passed the screening process was examined against 93 genes associated with RP as listed in the RetNet database [19].
The pathogenicity of the chosen variants was assessed based on the norms and recommendations set out by the American College of Medical Genetics and Genomics (ACMG) guidelines using Intervar [20,21]. GATK-gCNV analysis allows for a germline copy number variation (CNV) pipeline [22].
2.3. Sanger Confirmation
The novel candidate variations were confirmed via capillary sequencing. Sanger sequencing was conducted on the relevant gene segments to conduct a segregation analysis and determine the inheritance pattern of the modified alleles in specific families using the DNA samples of available relatives.
3. Results
The present study includes 21 patients who have been diagnosed with nonsyndromic RP. The average age at which RP was diagnosed was 31.72 ± 12.76 years (mean ± SD), whereas the mean age of examination was assessed as being 37.78 ± 12.39 years (Table 1). All participants had typical characteristics of RP, such as the first manifestation of night blindness being the primary symptom, followed by a gradual reduction in the visual field over subsequent years.

Table 1.
Clinical symptoms and signs in RP patients. BCVA: best-corrected visual acuity; IOP: intraocular pressure; OD: right eye; OS: left eye; SPH: sphere (the correction for nearsightedness or farsightedness is spherical, equal in all meridians of the eye); LP: light perception; HM: hand motion; f/c: finger count; PL: plano (no nearsightedness or farsightedness correction); RNFLD: retinal nerve fiber layer defect; and ERM: epiretinal membrane.
Cataracts were detected in patients FRP_0196, FRP_0170, FRP_0043, FRP_0207, FRP_0355, FRP_0157, FRP_0221, and FRP_0243. Detailed ophthalmological observations of the patients are presented in Table 1, and the clinical characteristics of family with novel variants of PDE6B gene are documented in Figure 1.
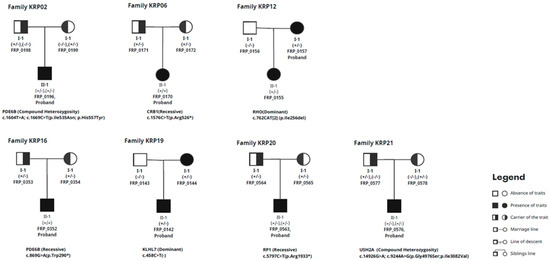
Figure 1.
Full pedigree chart of trio families.
Thirteen families had cases that were not resolved even after WES was performed. WES identified potentially causal variations in genes associated with RP in 8 out of the 21 families (Table 2). These variants include one new variant and eight variants that have been previously described. The genes involved include PDE6B, CRB1, PRE65, RHO, KLHL7, RP1, and USH2A. The in silico investigation of CNVs utilizing GATK’s gCNV did not detect any noteworthy structural changes. In addition, variants from the remaining 13 families did not meet the criteria outlined by the ACMG standard.
Table 2.
Pathogenic DNA variants in RP patients. Variants of the *PED6B gene (KRP16 family) have not been reported previously.
A novel variant was identified from gene PDE6B (c.869G>A, p.Trp290*). On PDE6B, c.869G>A(p.Trp290*) is a putative pathogenic novel variant in the KRP16 family. The proband was diagnosed with retinitis pigmentosa (Figure 2). The patient did not exhibit any indications or symptoms that were present in his mother and brother. The variant tester agreed on the potential impact of stop-gain (automatically causing disease).
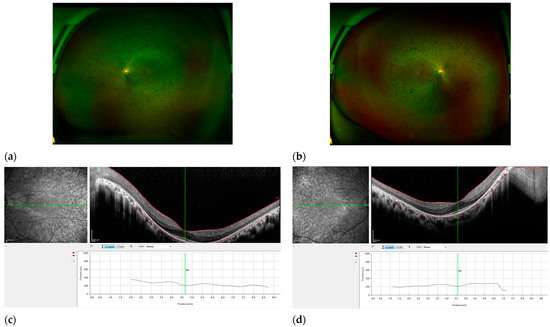
Figure 2.
Features of RP patient KRP16. Fundus photography of the right eye (a) and left eye (b), exhibiting characteristic traits of RP, such as optic disc pallor, narrowed retinal arteries, and pigmentation resembling bone spicules in the mid-peripheral area. Scale bar, 200 μm. (c,d) OCT revealed a reduction in the thickness of the retina and the absence of photoreceptor cells outside the fovea. The right eye has observable solitary cystic alterations in the foveola, as well as a delicate epiretinal membrane in the temporal region of the macula (scale bar, 200 μm).
4. Discussion
Inherited RP impacts people of all age groups and covers a wide range of diseases with significant genetic and phenotypic diversity. The pathogenesis of these disorders involves pathogenic variations that may vary from single-nucleotide changes to chromosomal rearrangements [23].
These variants impact genes that encode various signaling and structural components. Several genetic investigations have been carried out to comprehend the hereditary foundations of RP, encompassing molecular genetic research and studies on the aggregation of families. These investigations have revealed a genetic predisposition causing RP, revealing many pathogenic genes or susceptibility regions that are significantly linked to its development in Korean population specifically.
Trio-based WES is a more comprehensive method compared to WES focused on a single individual (proband-centered WES). This approach allows for the discovery of gene variations and a thorough evaluation of their pathogenicity. Additionally, it permits the investigation of gene variants using the principles of cogenetic segregation. This methodology can create an improved strong network that maps the association between genes and phenotypes, enabling a more comprehensive exploration of the inheritance patterns of mutant genes. In addition, because of the scarcity of genetic studies focusing on IRDs and the urgent need for RP research, specifically within the unique population of Korea, we recruited a group of 21 families who have been diagnosed with RP. Using a trio-based WES technique, our goal is to obtain a more comprehensive understanding of the genetic factors involved in RP. Specifically, we seek to uncover how genetic variations contribute to the development of this disorder within families.
The variant p.Trp290* is believed to cause the denaturation of PDE6B by interfering with the structure of the GAF2 region of the protein [24]. Phosphodiesterase 6B (PDE6B) variants often cause autosomal recessive retinitis pigmentosa, also known as rod–cone dystrophy. The PDE6B protein plays a crucial role in phototransduction. It is crucial to comprehend the pathogenicity and functional significance of identified PDE6B polymorphisms to provide genetic information to families and facilitate participation in therapeutic trials for autosomal-recessive PDE6B-related retinitis pigmentosa. PDE6A and PDE6B combine to create a heterodimer that is blocked by a homodimer consisting of two γ subunits, which are produced by PDE6G. PDE6A and PDE6B each contain two potential noncatalytic domains (GAF1 and GAF2) in the N-terminus, which interact with cGMP and the polycationic region of two γ subunits in the inhibitory state of the complex [25].
Given that the one cause of RP materializes through genetic influence, our study includes an examination of documented variants in RP risk genes: PDE6B, CRB1, RPE65, RHO, KLHL7, RP1, and USH2A. These genes hold pivotal roles in bone spicule formation, modulating scleral thickness, influencing choroidal blood flow, and orchestrating other factors germane to the onset of RP. The identification of pathogenic variants in these genes promises to enrich our understanding of the pathogenesis of RP.
The objective of this work was to conduct a multimodal study that could characterize a unique form of IRD in Korean population, broadening understanding of how a novel pathogenic variant affects the phenotype differently from a similar one [26].
To gain a more comprehensive understanding of the genetic components that contribute to RP, it is essential to have a broader sequencing coverage that encompasses noncoding areas. This may be achieved by utilizing advanced technologies like WGS, along with a larger collection of well-organized clinical data. Moreover, it is crucial to carry out additional functional investigations and ex vivo experimental validations to verify the several risk genes for RP that have been found in our research.
To summarize, our study’s thorough investigation of RP families from Korea tackles a gap in genetic research in this area. Through the consolidation and integration of data, our goal is to create a helpful resource for genetic counselling and personalized treatment strategies.
Author Contributions
Conceptualization, I.K.; methodology, J.-Y.L.; software, Y.P.; validation, I.K.; formal analysis, Y.K.; resources, J.-Y.L.; data curation, Y.K.; writing—original draft preparation, Y.P.; writing—review and editing, Y.P.; visualization, Y.P.; supervision, I.K; project administration, I.K. and J.-Y.L.; funding acquisition, J.-Y.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by a Biotechnology and Healthcare for Korea–India International Joint R&D Partnering of the National Research Foundation grant funded by the Korean government (NRF-2020K1A3A1A57080204, funded by the NRF).
Institutional Review Board Statement
This study was conducted according to the guidelines of the Declaration of Helsinki and approved by the Public Institutional Bioethics Committee designated by the MOHW of One Omics (P01-202107-31-012, 20 July 2021).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The data used in this study are available from the corresponding author upon request.
Conflicts of Interest
Author Jong-Young Lee was employed by the company “OneOmics Co., Ltd.”. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Becherucci, V.; Bacci, G.M.; Marziali, E.; Sodi, A.; Bambi, F.; Caputo, R. The New Era of Therapeutic Strategies for the Treatment of Retinitis Pigmentosa: A Narrative Review of Pathomolecular Mechanisms for the Development of Cell-Based Therapies. Biomedicines 2023, 11, 2656. [Google Scholar] [CrossRef] [PubMed]
- Smirnov, V.M.; Nassisi, M.; Solis Hernandez, C.; Méjécase, C.; El Shamieh, S.; Condroyer, C.; Antonio, A.; Meunier, I.; Andrieu, C.; Defoort-Dhellemmes, S.; et al. Retinal Phenotype of Patients with Isolated Retinal Degeneration Due to CLN3 Pathogenic Variants in a French Retinitis Pigmentosa Cohort. JAMA Ophthalmol. 2021, 139, 278–291. [Google Scholar] [CrossRef]
- Hamel, C. Retinitis pigmentosa. Orphanet J. Rare Dis. 2006, 1, 40. [Google Scholar] [CrossRef]
- Martin-Merida, I.; Avila-Fernandez, A.; Del Pozo-Valero, M.; Blanco-Kelly, F.; Zurita, O.; Perez-Carro, R.; Aguilera-Garcia, D.; Riveiro-Alvarez, R.; Arteche, A.; Trujillo-Tiebas, M.J.; et al. Genomic Landscape of Sporadic Retinitis Pigmentosa: Findings from 877 Spanish Cases. Ophthalmology 2019, 126, 1181–1188. [Google Scholar] [CrossRef]
- Bravo-Gil, N.; González-Del Pozo, M.; Martín-Sánchez, M.; Méndez-Vidal, C.; Rodríguez-de la Rúa, E.; Borrego, S.; Antiñolo, G. Unravelling the genetic basis of simplex Retinitis Pigmentosa cases. Sci. Rep. 2017, 7, 41937. [Google Scholar] [CrossRef]
- Choi, B.G.; Hong, J.Y.; Hong, J.R.; Hur, M.S.; Kim, S.M.; Lee, Y.W.; Choe, Y.B.; Ahn, K.J. The IL17F His161Arg polymorphism, a potential risk locus for psoriasis, increases serum levels of interleukin-17F in an Asian population. Sci. Rep. 2019, 9, 18921. [Google Scholar] [CrossRef]
- Tilleman, L.; Heindryckx, B.; Deforce, D.; Van Nieuwerburgh, F. Pan-cancer pharmacogenetics: Targeted sequencing panels or exome sequencing? Pharmacogenomics 2020, 21, 1073–1084. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Guo, R.; Guo, J.; Qi, Z.; Li, W.; Hao, C. Parallel Tests of Whole Exome Sequencing and Copy Number Variant Sequencing Increase the Diagnosis Yields of Rare Pediatric Disorders. Front. Genet. 2020, 11, 473. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Liu, X.; Jian, X.; Boerwinkle, E. dbNSFP: A lightweight database of human nonsynonymous SNPs and their functional predictions. Hum. Mutat. 2011, 32, 894–899. [Google Scholar] [CrossRef]
- Liu, X.; Li, C.; Mou, C.; Dong, Y.; Tu, Y. dbNSFP v4: A comprehensive database of transcript-specific functional predictions and annotations for human nonsynonymous and splice-site SNVs. Genome Med. 2020, 12, 103. [Google Scholar] [CrossRef] [PubMed]
- Sherry, S.T.; Ward, M.-H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [PubMed]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef]
- Landrum, M.J.; Chitipiralla, S.; Brown, G.R.; Chen, C.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; Kaur, K.; Liu, C. ClinVar: Improvements to accessing data. Nucleic Acids Res. 2020, 48, D835–D844. [Google Scholar] [CrossRef]
- Jung, K.S.; Hong, K.-W.; Jo, H.Y.; Choi, J.; Ban, H.-J.; Cho, S.B.; Chung, M. KRGDB: The large-scale variant database of 1722 Koreans based on whole genome sequencing. Database 2020, 2020, baz146. [Google Scholar] [CrossRef]
- Gudmundsson, S.; Singer-Berk, M.; Watts, N.A.; Phu, W.; Goodrich, J.K.; Solomonson, M.; Consortium, G.A.D.; Rehm, H.L.; MacArthur, D.G.; O’Donnell-Luria, A. Variant interpretation using population databases: Lessons from gnomAD. Hum. Mutat. 2022, 43, 1012–1030. [Google Scholar] [CrossRef]
- Jeon, S.; Bhak, Y.; Choi, Y.; Jeon, Y.; Kim, S.; Jang, J.; Jang, J.; Blazyte, A.; Kim, C.; Kim, Y. Korean Genome Project: 1094 Korean personal genomes with clinical information. Sci. Adv. 2020, 6, eaaz7835. [Google Scholar] [CrossRef]
- Saint Pierre, A.; Génin, E. How important are rare variants in common disease? Brief. Funct. Genom. 2014, 13, 353–361. [Google Scholar] [CrossRef]
- Daiger, S.P. Identifying retinal disease genes: How far have we come, how far do we have to go? Novartis Found. Symp. 2004, 255, 17–27; discussion 27–36, 177–178. [Google Scholar] [CrossRef]
- Li, Q.; Wang, K. InterVar: Clinical interpretation of genetic variants by the 2015 ACMG-AMP guidelines. Am. J. Hum. Genet. 2017, 100, 267–280. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Babadi, M.; Fu, J.M.; Lee, S.K.; Smirnov, A.N.; Gauthier, L.D.; Walker, M.; Benjamin, D.I.; Zhao, X.; Karczewski, K.J.; Wong, I.; et al. GATK-gCNV enables the discovery of rare copy number variants from exome sequencing data. Nat. Genet. 2023, 55, 1589–1597. [Google Scholar] [CrossRef] [PubMed]
- Perez-Carro, R.; Corton, M.; Sánchez-Navarro, I.; Zurita, O.; Sanchez-Bolivar, N.; Sánchez-Alcudia, R.; Lelieveld, S.H.; Aller, E.; Lopez-Martinez, M.A.; López-Molina, M.I.; et al. Panel-based NGS Reveals Novel Pathogenic Mutations in Autosomal Recessive Retinitis Pigmentosa. Sci. Rep. 2016, 6, 19531. [Google Scholar] [CrossRef]
- Watson, C.J.G.; Nash, B.M.; Loi, T.H.; Grigg, J.R.; Jamieson, R.V. Genetic Variants and Impact in PDE6B Rod-Cone Dystrophy. In Advances in Vision Research, Volume III: Genetic Eye Research around the Globe; Prakash, G., Iwata, T., Eds.; Springer: Singapore, 2021; pp. 197–206. [Google Scholar]
- Khateb, S.; Nassisi, M.; Bujakowska, K.M.; Méjécase, C.; Condroyer, C.; Antonio, A.; Foussard, M.; Démontant, V.; Mohand-Saïd, S.; Sahel, J.A.; et al. Longitudinal Clinical Follow-up and Genetic Spectrum of Patients with Rod-Cone Dystrophy Associated with Mutations in PDE6A and PDE6B. JAMA Ophthalmol. 2019, 137, 669–679. [Google Scholar] [CrossRef]
- D’Esposito, F.; Cennamo, G.; de Crecchio, G.; Maltese, P.E.; Cecchin, S.; Bertelli, M.; Ziccardi, L.; Esposito Veneruso, P.; Magli, A.; Cennamo, G.; et al. Multimodal Imaging in Autosomal Dominant Cone-Rod Dystrophy Caused by Novel CRX Variant. Ophthalmic Res. 2018, 60, 169–175. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).