
PNU-74654 Suppresses TNFR1/IKB Alpha/p65 Signaling and Induces Cell Death in Testicular Cancer

 ,
,  ,
,  ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. MTT Assay
2.3. Flow Cytometry Analysis
2.4. Hoechst 33,342 Staining
2.5. Human Apoptosis Array for Proteome Profiling
2.6. Protein Extraction from Cells and Western Blotting
2.7. Statistical Analysis
3. Results
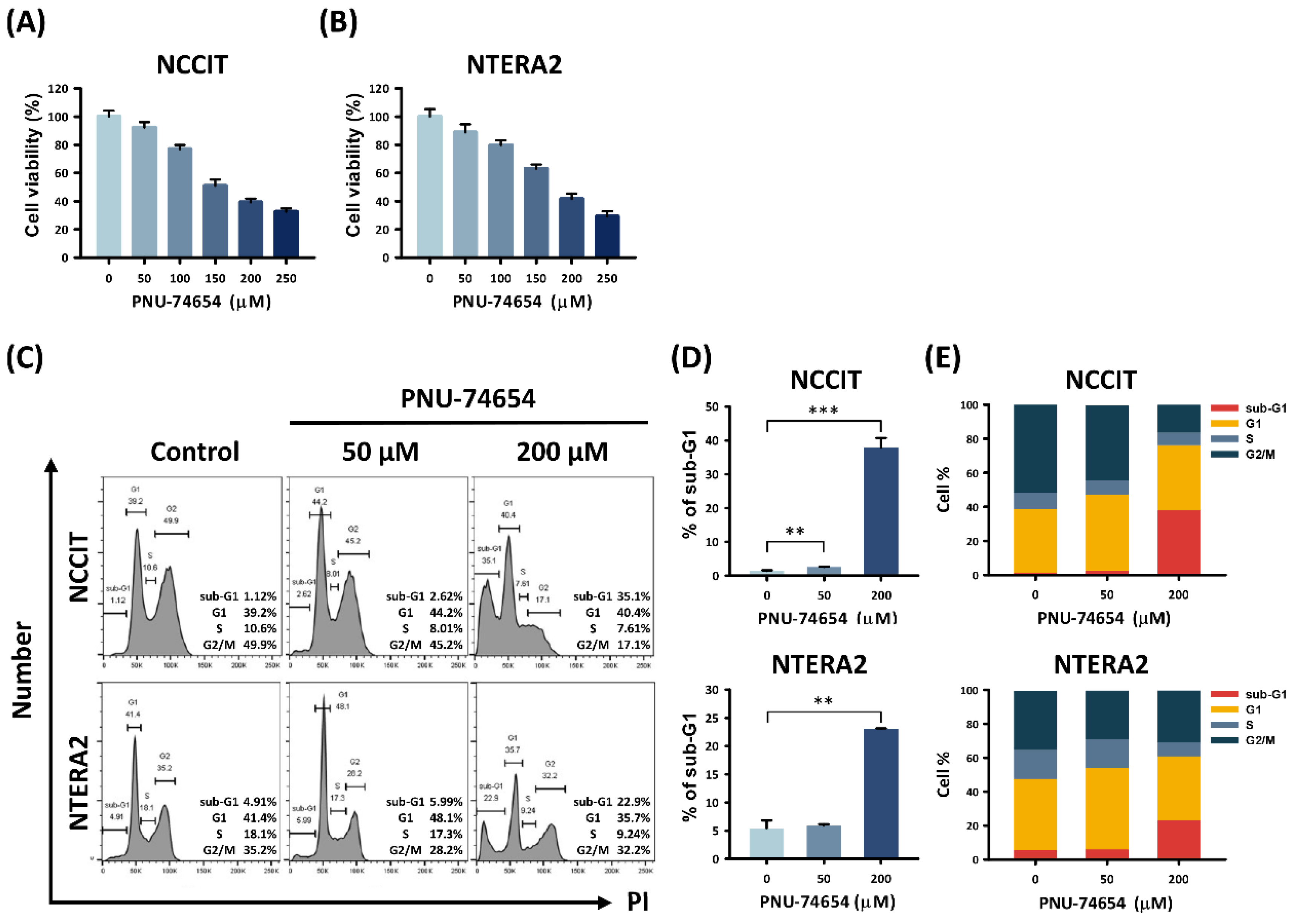
3.1. PNU-74654 Had a Cytotoxic Effect on TC Cells and Elicited Cell Death
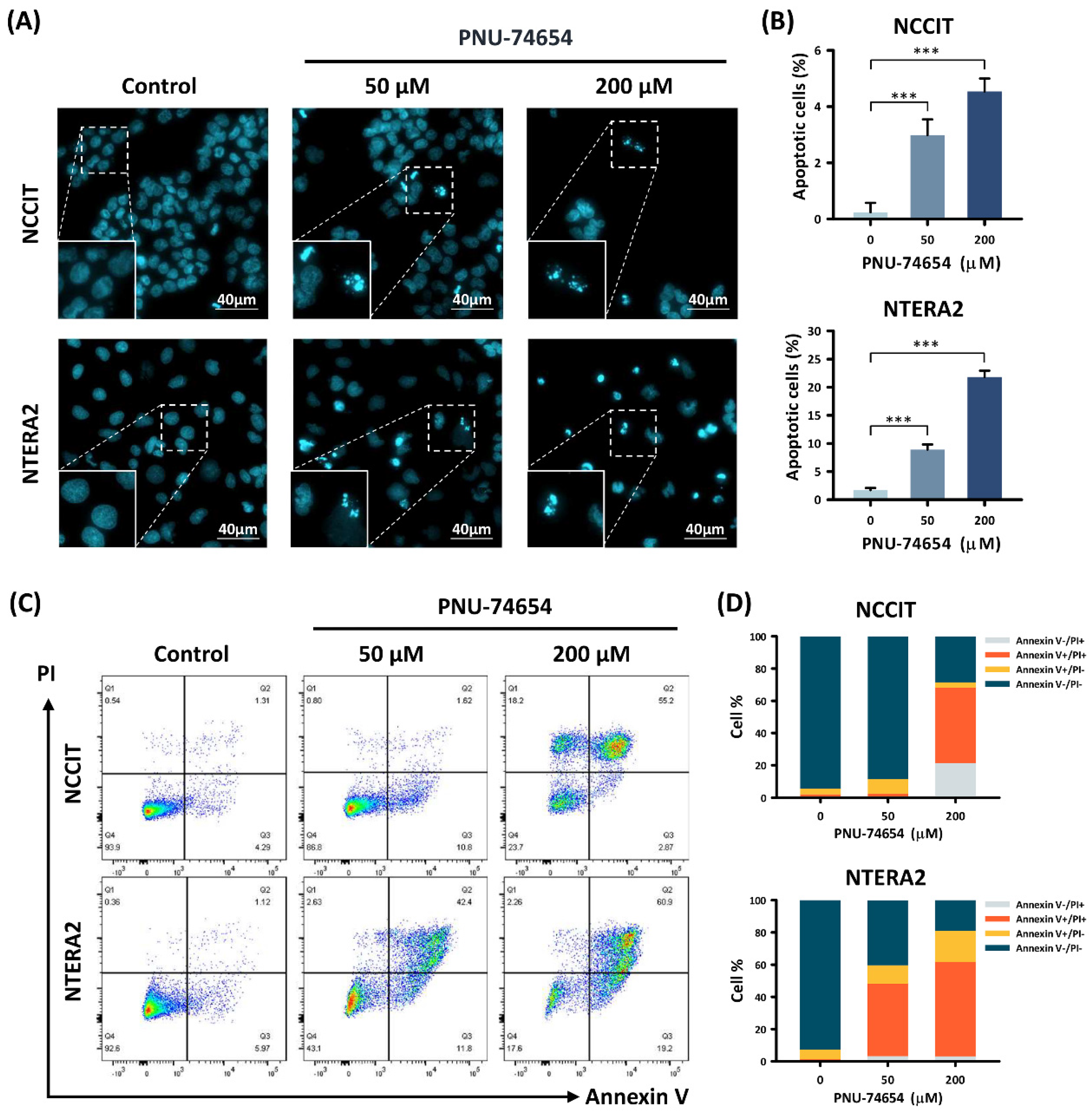
3.2. Apoptosis Induced by PNU-74654 in TC Cells
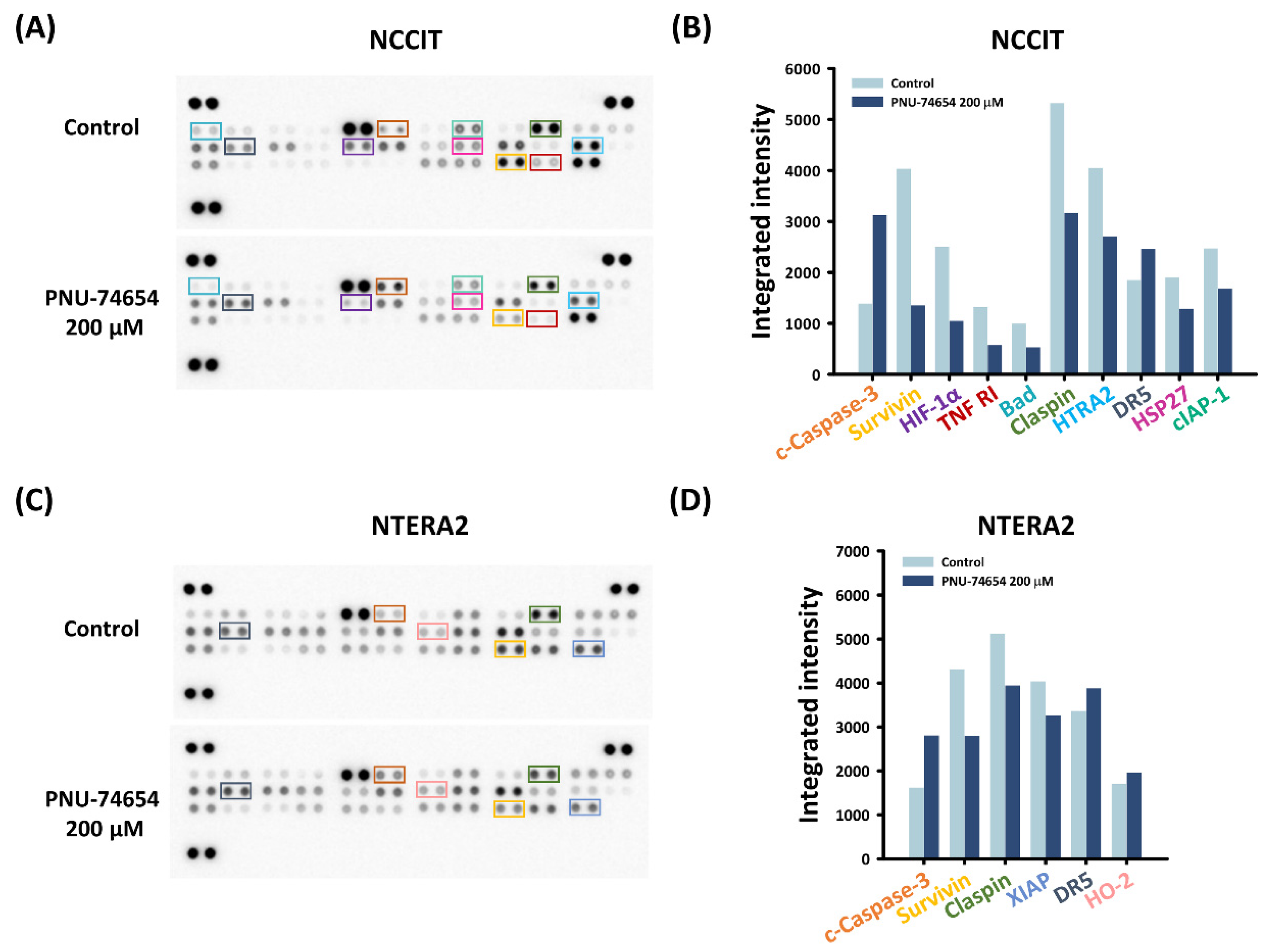
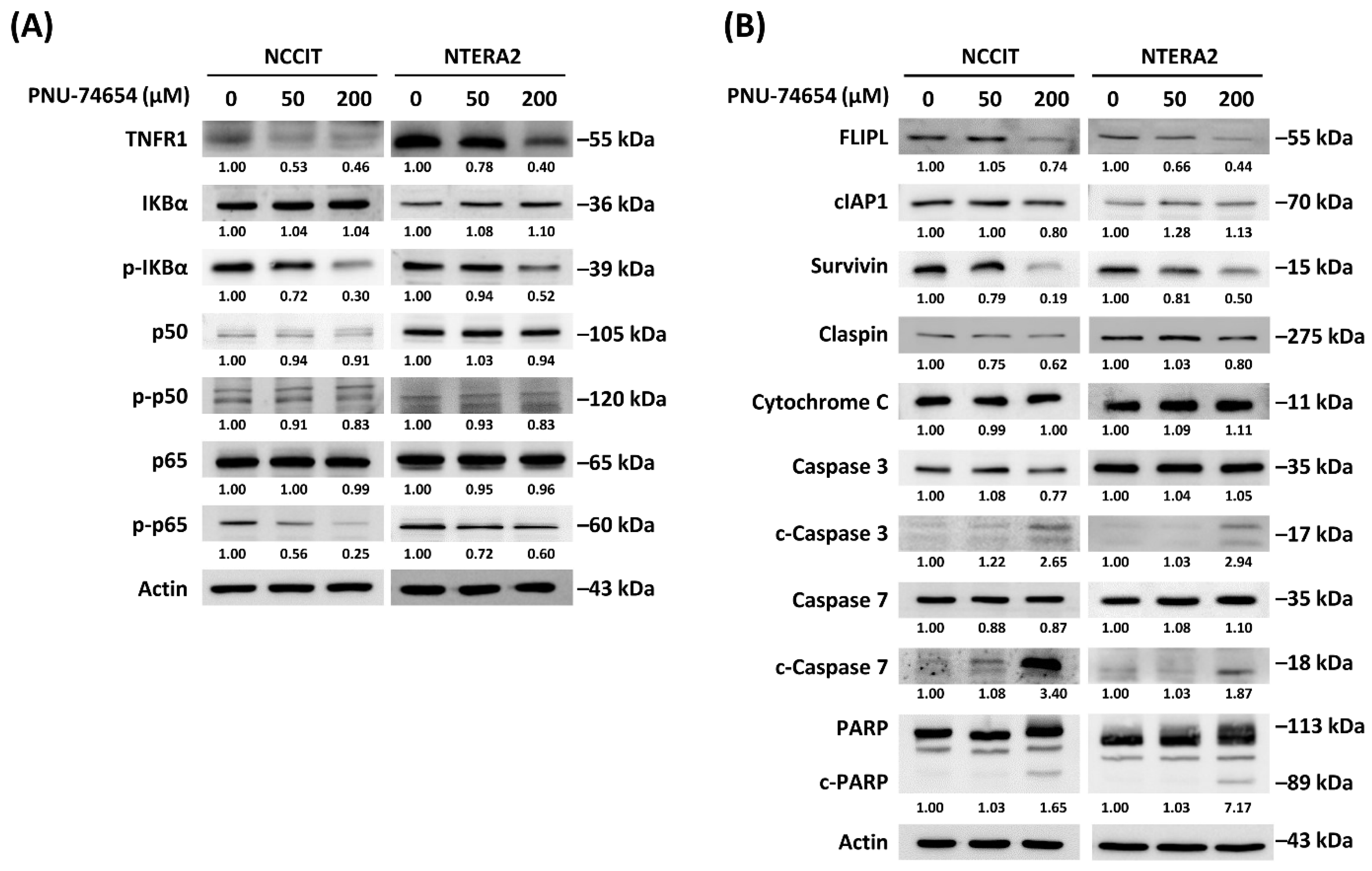
3.3. Inhibition of TNF R1 Triggered NF-κB Anti-Apoptotic Pathway and Execution Phase of Apoptosis Contributed to PNU-74654-Induced Apoptosis in TC Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Chen, W.J.; Huang, C.Y.; Huang, Y.H.; Wang, S.C.; Hsieh, T.Y.; Chen, S.L.; Sung, W.W.; Lee, T.H. Correlations between Mortality-to-Incidence Ratios and Health Care Disparities in Testicular Cancer. Int. J. Env. Res. Public Health 2019, 17, 130. [Google Scholar] [CrossRef]
- Znaor, A.; Lortet-Tieulent, J.; Jemal, A.; Bray, F. International variations and trends in testicular cancer incidence and mortality. Eur. Urol. 2014, 65, 1095–1106. [Google Scholar] [CrossRef]
- McGlynn, K.A.; Trabert, B. Adolescent and adult risk factors for testicular cancer. Nat. Rev. Urol. 2012, 9, 339–349. [Google Scholar] [CrossRef]
- Baird, D.C.; Meyers, G.J.; Hu, J.S. Testicular Cancer: Diagnosis and Treatment. Am. Fam. Physician 2018, 97, 261–268. [Google Scholar]
- Pai, S.G.; Carneiro, B.A.; Mota, J.M.; Costa, R.; Leite, C.A.; Barroso-Sousa, R.; Kaplan, J.B.; Chae, Y.K.; Giles, F.J. Wnt/beta-catenin pathway: Modulating anticancer immune response. J. Hematol. Oncol. 2017, 10, 101. [Google Scholar] [CrossRef]
- van Ooyen, A.; Kwee, V.; Nusse, R. The nucleotide sequence of the human int-1 mammary oncogene; evolutionary conservation of coding and non-coding sequences. EMBO J. 1985, 4, 2905–2909. [Google Scholar] [CrossRef]
- Niehrs, C. The complex world of WNT receptor signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 767–779. [Google Scholar] [CrossRef]
- Kemler, R. From cadherins to catenins: Cytoplasmic protein interactions and regulation of cell adhesion. Trends Genet. 1993, 9, 317–321. [Google Scholar] [CrossRef]
- Stadeli, R.; Hoffmans, R.; Basler, K. Transcription under the control of nuclear Arm/beta-catenin. Curr. Biol. 2006, 16, R378–R385. [Google Scholar] [CrossRef]
- Molenaar, M.; van de Wetering, M.; Oosterwegel, M.; Peterson-Maduro, J.; Godsave, S.; Korinek, V.; Roose, J.; Destree, O.; Clevers, H. XTcf-3 transcription factor mediates beta-catenin-induced axis formation in Xenopus embryos. Cell 1996, 86, 391–399. [Google Scholar] [CrossRef]
- Huber, O.; Korn, R.; McLaughlin, J.; Ohsugi, M.; Herrmann, B.G.; Kemler, R. Nuclear localization of beta-catenin by interaction with transcription factor LEF-1. Mech. Dev. 1996, 59, 3–10. [Google Scholar] [CrossRef]
- He, T.C.; Sparks, A.B.; Rago, C.; Hermeking, H.; Zawel, L.; da Costa, L.T.; Morin, P.J.; Vogelstein, B.; Kinzler, K.W. Identification of c-MYC as a target of the APC pathway. Science 1998, 281, 1509–1512. [Google Scholar] [CrossRef]
- Saegusa, M.; Hashimura, M.; Kuwata, T.; Hamano, M.; Okayasu, I. Beta-catenin simultaneously induces activation of the p53-p21WAF1 pathway and overexpression of cyclin D1 during squamous differentiation of endometrial carcinoma cells. Am. J. Pathol. 2004, 164, 1739–1749. [Google Scholar] [CrossRef]
- Lapham, A.; Adams, J.E.; Paterson, A.; Lee, M.; Brimmell, M.; Packham, G. The Bcl-w promoter is activated by beta-catenin/TCF4 in human colorectal carcinoma cells. Gene 2009, 432, 112–117. [Google Scholar] [CrossRef]
- Rao, T.P.; Kuhl, M. An updated overview on Wnt signaling pathways: A prelude for more. Circ. Res. 2010, 106, 1798–1806. [Google Scholar] [CrossRef]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/beta-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef]
- de Sousa, E.M.F.; Vermeulen, A.L. Wnt Signaling in Cancer Stem Cell Biology. Cancers 2016, 8, 60. [Google Scholar] [CrossRef]
- Fearon, E.R. Molecular genetics of colorectal cancer. Ann. Rev. Pathol. 2011, 6, 479–507. [Google Scholar] [CrossRef]
- Seshagiri, S.; Stawiski, E.W.; Durinck, S.; Modrusan, Z.; Storm, E.E.; Conboy, C.B.; Chaudhuri, S.; Guan, Y.; Janakiraman, V.; Jaiswal, B.S.; et al. Recurrent R-spondin fusions in colon cancer. Nature 2012, 488, 660–664. [Google Scholar] [CrossRef]
- Storm, E.E.; Durinck, S.; de Sousa e Melo, F.; Tremayne, J.; Kljavin, N.; Tan, C.; Ye, X.; Chiu, C.; Pham, T.; Hongo, J.A.; et al. Targeting PTPRK-RSPO3 colon tumours promotes differentiation and loss of stem-cell function. Nature 2016, 529, 97–100. [Google Scholar] [CrossRef]
- Satoh, S.; Daigo, Y.; Furukawa, Y.; Kato, T.; Miwa, N.; Nishiwaki, T.; Kawasoe, T.; Ishiguro, H.; Fujita, M.; Tokino, T.; et al. AXIN1 mutations in hepatocellular carcinomas, and growth suppression in cancer cells by virus-mediated transfer of AXIN1. Nat. Genet. 2000, 24, 245–250. [Google Scholar] [CrossRef]
- Bafico, A.; Liu, G.; Goldin, L.; Harris, V.; Aaronson, S.A. An autocrine mechanism for constitutive Wnt pathway activation in human cancer cells. Cancer Cell 2004, 6, 497–506. [Google Scholar] [CrossRef]
- He, B.; You, L.; Uematsu, K.; Xu, Z.; Lee, A.Y.; Matsangou, M.; McCormick, F.; Jablons, D.M. A monoclonal antibody against Wnt-1 induces apoptosis in human cancer cells. Neoplasia 2004, 6, 7–14. [Google Scholar] [CrossRef]
- You, L.; He, B.; Xu, Z.; Uematsu, K.; Mazieres, J.; Mikami, I.; Reguart, N.; Moody, T.W.; Kitajewski, J.; McCormick, F.; et al. Inhibition of Wnt-2-mediated signaling induces programmed cell death in non-small-cell lung cancer cells. Oncogene 2004, 23, 6170–6174. [Google Scholar] [CrossRef]
- Lavergne, E.; Hendaoui, I.; Coulouarn, C.; Ribault, C.; Leseur, J.; Eliat, P.A.; Mebarki, S.; Corlu, A.; Clément, B.; Musso, O. Blocking Wnt signaling by SFRP-like molecules inhibits in vivo cell proliferation and tumor growth in cells carrying active beta-catenin. Oncogene 2011, 30, 423–433. [Google Scholar] [CrossRef]
- Takahashi-Yanaga, F.; Kahn, M. Targeting Wnt signaling: Can we safely eradicate cancer stem cells? Clin Cancer Res 2010, 16, 3153–3162. [Google Scholar] [CrossRef]
- Trosset, J.Y. Inhibition of protein-protein interactions: The discovery of druglike beta-catenin inhibitors by combining virtual and biophysical screening. Proteins 2006, 64, 60–67. [Google Scholar] [CrossRef]
- Lévy, L.; Neuveut, C.; Renard, C.A.; Charneau, P.; Branchereau, S.; Gauthier, F.; Van Nhieu, J.T.; Cherqui, D.; Petit-Bertron, A.F.; Mathieu, D.; et al. Transcriptional activation of interleukin-8 by beta-catenin-Tcf4. J. Biol. Chem. 2002, 277, 42386–42393. [Google Scholar] [CrossRef]
- Yamada, T.; Takaoka, A.S.; Naishiro, Y.; Hayashi, R.; Maruyama, K.; Maesawa, C.; Ochiai, A.; Hirohashi, S. Transactivation of the multidrug resistance 1 gene by T-cell factor 4/beta-catenin complex in early colorectal carcinogenesis. Cancer Res. 2000, 60, 4761–4766. [Google Scholar]
- Sanchez-Tillo, E.; de Barrios, O.; Siles, L.; Cuatrecasas, M.; Castells, A.; Postigo, A. beta-catenin/TCF4 complex induces the epithelial-to-mesenchymal transition (EMT)-activator ZEB1 to regulate tumor invasiveness. Proc. Natl. Acad. Sci. USA 2011, 108, 19204–19209. [Google Scholar] [CrossRef]
- Leal, L.F.; Bueno, A.C.; Gomes, D.C.; Abduch, R.; de Castro, M.; Antonini, S.R. Inhibition of the Tcf/beta-catenin complex increases apoptosis and impairs adrenocortical tumor cell proliferation and adrenal steroidogenesis. Oncotarget 2015, 6, 43016–43032. [Google Scholar] [CrossRef]
- Rahmani, F.; Amerizadeh, F.; Hassanian, S.M.; Hashemzehi, M.; Nasiri, S.N.; Fiuji, H.; Ferns, G.A.; Khazaei, M.; Avan, A. PNU-74654 enhances the antiproliferative effects of 5-FU in breast cancer and antagonizes thrombin-induced cell growth via the Wnt pathway. J. Cell Physiol. 2019, 234, 14123–14132. [Google Scholar] [CrossRef]
- Wang, S.C.; Chang, Y.C.; Wu, M.Y.; Yu, C.Y.; Chen, S.L.; Sung, W.W. Intravesical Instillation of Azacitidine Suppresses Tumor Formation through TNF-R1 and TRAIL-R2 Signaling in Genotoxic Carcinogen-Induced Bladder Cancer. Cancers 2021, 13, 3933. [Google Scholar] [CrossRef]
- Bagrodia, A.; Lee, B.H.; Lee, W.; Cha, E.K.; Sfakianos, J.P.; Iyer, G.; Pietzak, E.J.; Gao, S.P.; Zabor, E.C.; Ostrovnaya, I.; et al. Genetic Determinants of Cisplatin Resistance in Patients with Advanced Germ Cell Tumors. J. Clin. Oncol. 2016, 34, 4000–4007. [Google Scholar] [CrossRef]
- Okpanyi, V.; Schneider, D.T.; Zahn, S.; Sievers, S.; Calaminus, G.; Nicholson, J.C.; Palmer, R.D.; Leuschner, I.; Borkhardt, A.; Schönberger, S. Analysis of the adenomatous polyposis coli (APC) gene in childhood and adolescent germ cell tumors. Pediatr. Blood Cancer 2011, 56, 384–391. [Google Scholar] [CrossRef]
- Baichwal, V.R.; Baeuerle, P.A. Activate NF-kappa B or die? Curr. Biol. 1997, 7, R94–R96. [Google Scholar] [CrossRef]
- Strasser, A.; Cory, S.; Adams, J.M. Deciphering the rules of programmed cell death to improve therapy of cancer and other diseases. EMBO J. 2011, 30, 3667–3683. [Google Scholar] [CrossRef]
- Smits, V.A.J.; Cabrera, E.; Freire, R.; Gillespie, D.A. Claspin—Checkpoint adaptor and DNA replication factor. FEBS J. 2019, 286, 441–455. [Google Scholar] [CrossRef]
- Semple, J.I.; Smits, V.A.; Fernaud, J.R.; Mamely, I.; Freire, R. Cleavage and degradation of Claspin during apoptosis by caspases and the proteasome. Cell Death Differ. 2007, 14, 1433–1442. [Google Scholar] [CrossRef][Green Version]
- Sah, N.K.; Khan, Z.; Khan, G.J.; Bisen, P.S. Structural, functional and therapeutic biology of survivin. Cancer Lett. 2006, 244, 164–171. [Google Scholar] [CrossRef]
- Nusse, R.; Clevers, H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef]
- Takam Kamga, P.; Dal Collo, G.; Cassaro, A.; Bazzoni, R.; Delfino, P.; Adamo, A.; Bonato, A.; Carbone, C.; Tanasi, I.; Bonifacio, M.; et al. Small Molecule Inhibitors of Microenvironmental Wnt/beta-Catenin Signaling Enhance the Chemosensitivity of Acute Myeloid Leukemia. Cancers 2020, 12, 2696. [Google Scholar] [CrossRef]
- Zhang, F.; Li, P.; Liu, S.; Yang, M.; Zeng, S.; Deng, J.; Chen, D.; Yi, Y.; Liu, H.J.O. β-Catenin-CCL2 feedback loop mediates crosstalk between cancer cells and macrophages that regulates breast cancer stem cells. Oncogene 2021, 40, 5854–5865. [Google Scholar] [CrossRef]
- Maria, A.G.; Silva Borges, K.; Lira, R.; Hassib Thomé, C.; Berthon, A.; Drougat, L.; Kiseljak-Vassiliades, K.; Wierman, M.E.; Faucz, F.R.; Faça, V.M.; et al. Inhibition of Aurora kinase A activity enhances the antitumor response of beta-catenin blockade in human adrenocortical cancer cells. Mol. Cell. Endocrinol. 2021, 528, 111243. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, W.-J.; Sung, W.-W.; Yu, C.-Y.; Luan, Y.-Z.; Chang, Y.-C.; Chen, S.-L.; Lee, T.-H. PNU-74654 Suppresses TNFR1/IKB Alpha/p65 Signaling and Induces Cell Death in Testicular Cancer. Curr. Issues Mol. Biol. 2022, 44, 222-232. https://doi.org/10.3390/cimb44010016
Chen W-J, Sung W-W, Yu C-Y, Luan Y-Z, Chang Y-C, Chen S-L, Lee T-H. PNU-74654 Suppresses TNFR1/IKB Alpha/p65 Signaling and Induces Cell Death in Testicular Cancer. Current Issues in Molecular Biology. 2022; 44(1):222-232. https://doi.org/10.3390/cimb44010016
Chicago/Turabian StyleChen, Wen-Jung, Wen-Wei Sung, Chia-Ying Yu, Yu-Ze Luan, Ya-Chuan Chang, Sung-Lang Chen, and Tsung-Hsien Lee. 2022. "PNU-74654 Suppresses TNFR1/IKB Alpha/p65 Signaling and Induces Cell Death in Testicular Cancer" Current Issues in Molecular Biology 44, no. 1: 222-232. https://doi.org/10.3390/cimb44010016
APA StyleChen, W.-J., Sung, W.-W., Yu, C.-Y., Luan, Y.-Z., Chang, Y.-C., Chen, S.-L., & Lee, T.-H. (2022). PNU-74654 Suppresses TNFR1/IKB Alpha/p65 Signaling and Induces Cell Death in Testicular Cancer. Current Issues in Molecular Biology, 44(1), 222-232. https://doi.org/10.3390/cimb44010016