
Redox Imbalance in Cystic Fibrosis: The Multifaceted Role of Oxidative Stress



Abstract

1. Introduction
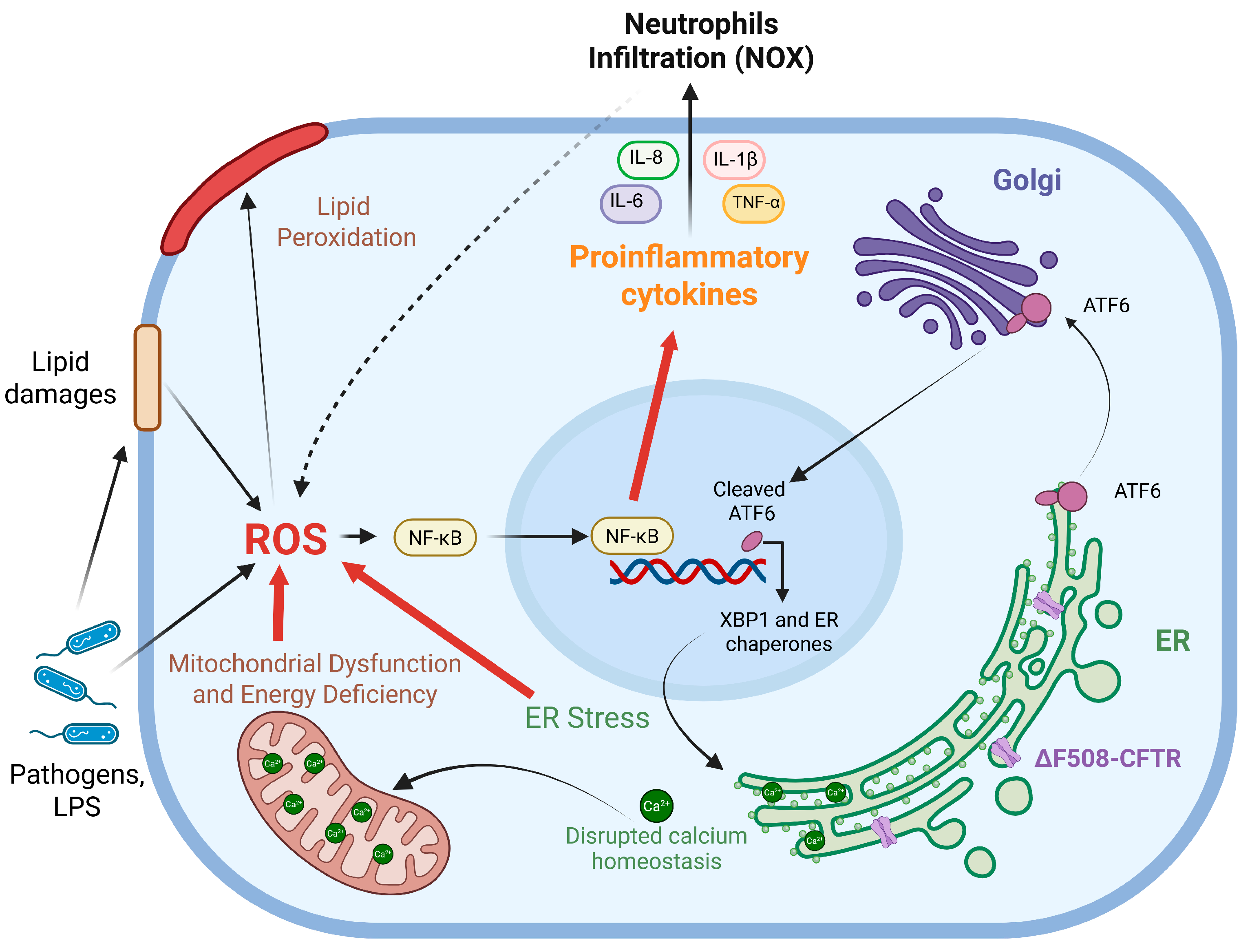
2. Oxidative Stress in Cystic Fibrosis
3. The Connection Between Inflammation and Oxidative Stress
4. Sources of Oxidative Stress in CF
4.1. ER Stress
4.2. Mitochondrial Dysfunction
4.3. Bacterial Infections
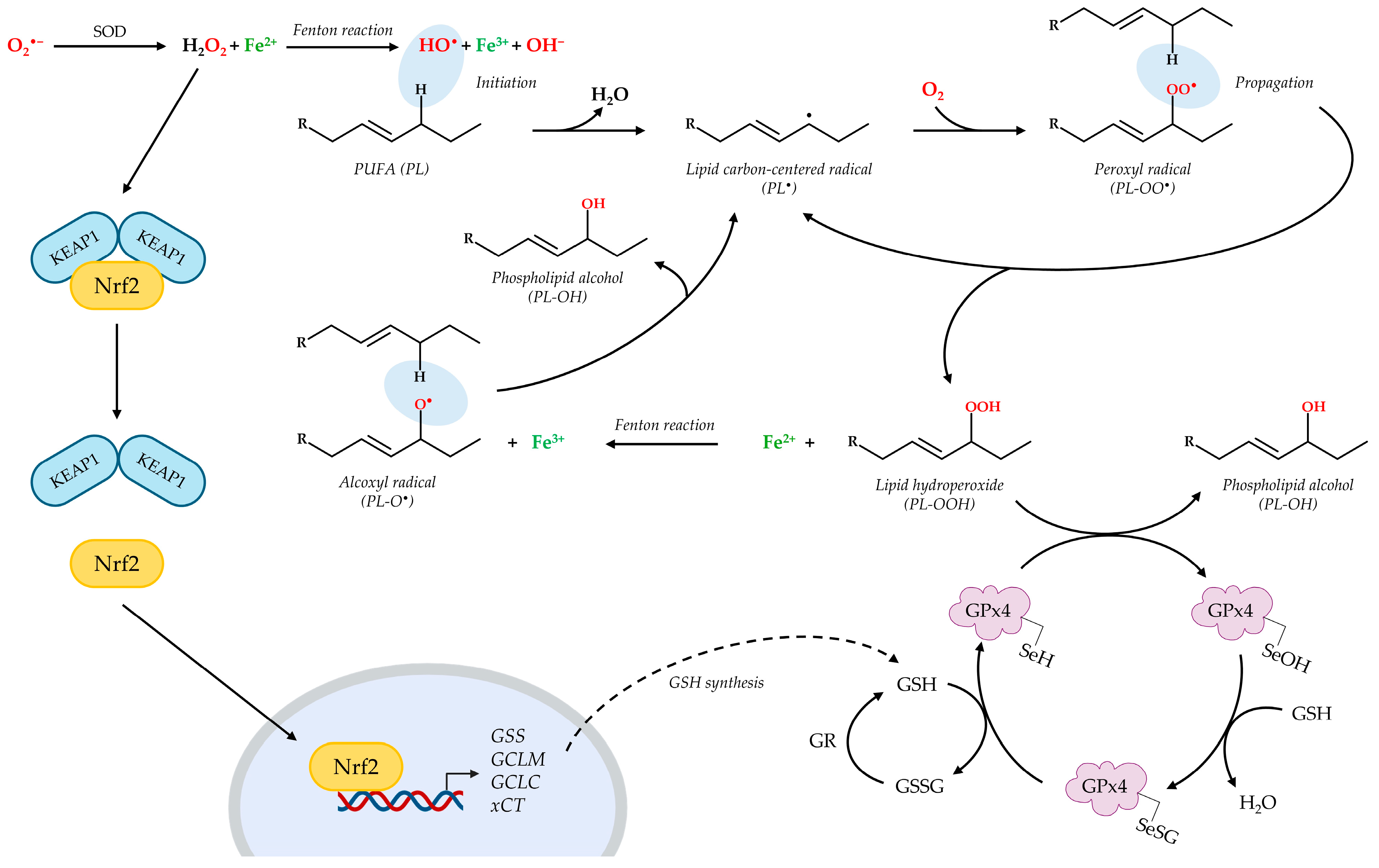
5. Oxidative Stress Can Lead to Lipid Peroxidation
6. Altered Lipid Metabolism and Lipid Imbalance in CF
7. Therapeutic Approaches Against Oxidative Stress in CF
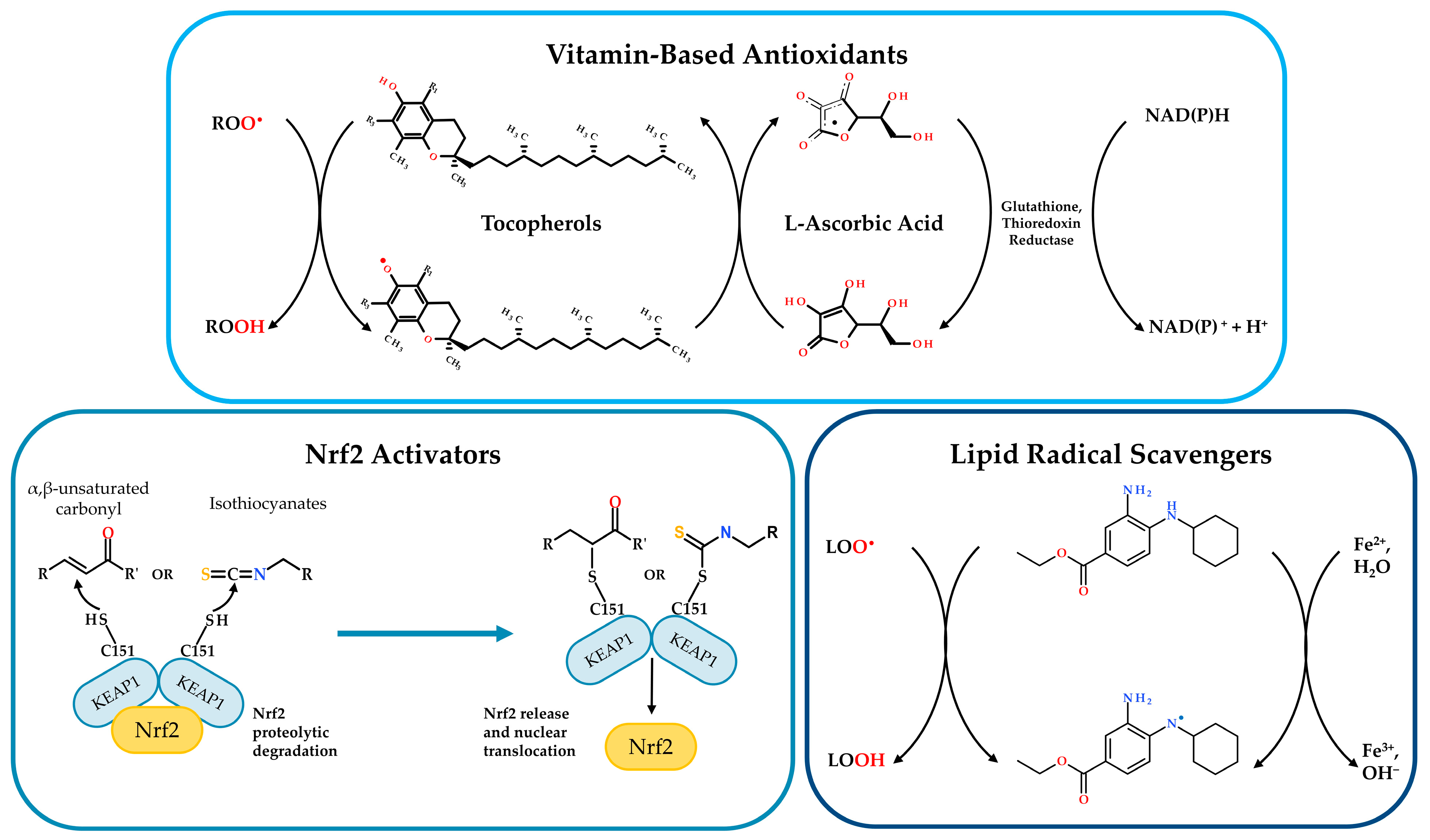
7.1. Activators of Nrf2/GSH Signaling Pathway
7.2. Vitamin-Based Antioxidants
7.3. Molecules Addressing LPO or Lipid Imbalance

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Therapeutic Approach | Mechanism of Action | Model System(s) Tested In | Effects of Treatment | Prospects for Improvement |
|---|---|---|---|---|
| Activators of Nrf2/GSH signaling pathway | ||||
| General Nrf2 Activators (Michael Acceptors) [95,96,97,98,99] | Interaction with KEAP1 cysteine residues (Michael reaction), Nrf2 translocation to nucleus and induction of ARE target genes (incl. GSH synthesis enzymes). | General cellular mechanism description. | Stimulation of the antioxidant defense system. | Nrf2 activators are considered potentially promising. No effect on CFTR function in single treatment. |
| Curcumin [100,101,102] | Nrf2 activation (Michael reaction against KEAP1). Investigated for addressing ΔF508 mutation. | In vitro: Baby hamster kidney cells (ΔF508-CFTR transfected). In vivo: ΔF508 CF mice. CFTR-depleted zebrafish. | Initial studies: Improved ΔF508-CFTR processing (in vitro); corrected ΔF508-CFTR trafficking (mice). Later studies: failed to replicate initial findings. Recent studies: Restored Nrf2 activity (zebrafish), normalized inflammation. | Inconsistent results highlight challenges; potential benefits in specific contexts like inflammation and tissue damage. |
| Resveratrol [103,104,105,106] | Nrf2 activation (Michael reaction against KEAP1). | In vitro: Primary sinonasal epithelial cells from humans, mice, and pigs: CFBE (wt, ΔF508-CFTR) and HEK293T-CFTR expressing either wild type or F508del CFTR In vivo: wt and ΔF508-CFTR mice | Initial promise: Improved ΔF508-CFTR maturation/trafficking; increased airway fluid secretion and mucociliary clearance. Conflicting results: Beneficial effects not confirmed in primary human airway cells from patients. | Conflicting results cast doubt on its consistent efficacy as a therapeutic agent for CF. |
| Dimethyl fumarate (DMF) [107] | Nrf2 activation (Michael reaction against KEAP1); anti-inflammatory and antioxidant effects. Modulation inflammatory response to LPS. | CF (ΔF508: CFBE, HNE cells) and non-CF bronchial epithelial cells. | Reinstated pro-inflammatory cytokine expression and ROS levels in ΔF508 CF cells; restored Trikafta™ efficacy reduced by LPS. Association with CFTR modulators. | Promising as an adjunctive therapy with CFTR modulators, particularly in managing inflammation. |
| GSH Supplementation [109,110,111,112,113,114] | Direct increment of GSH levels; reduction of oxidant burden; inactivation of P. aeruginosa metabolite pyocyanin. | P. aeruginosa biofilms (CF isolates); Clinical trials (human inhalation); Pediatric patients (oral adm.); Children with pancreatic insufficiency (oral adm.). | Biofilms: Disrupted P. aeruginosa biofilms. Inhalation: Well tolerated, but no improvement in lung function, oxidative stress, or inflammation; reduced BALF PGE2, higher lymphocytes (immune modulation). Oral: Controversial; some benefit in pediatric patients with moderate disease, not in those with pancreatic insufficiency. | Inhalation limited by degradation and lack of lung function improvement. Oral route shows highly specific and limited benefits. Overall efficacy remains a challenge. |
| N-acetylcysteine (NAC) [115] | GSH precursor; antioxidant; modulation of airway inflammation; prevention of biofilm formation (P. aeruginosa). | CF patients, clinical investigation/use. | Inhalation observed to provide great efficacy. | Considered promising, especially via inhalation. |
| NACA (N-acetylcysteine amide) and diNACA [116] | Formulations to enhance NAC bioavailability. diNACA also for reducing mucus viscosity/elasticity. | Implied for CF, COPD, bronchitis, radiation-induced pneumonia. | Being evaluated. | Potential to enhance NAC’s therapeutic benefits by improving bioavailability and mucolytic effects (diNACA). |
| Vitamin-based antioxidants | ||||
| Antioxidants, β-carotene, Antioxidant-enriched multivitamins [119,120] | As supplement to counteract malabsorption of fat-soluble vitamins and antioxidants. | CF patients (pancreatic-insufficient for multivitamins). | Generally, no lung function improvement (oral adm.). Increased blood antioxidant levels. Multivitamins: modest reduction in some inflammation markers; no lung improvement; lower risk of first pulmonary exacerbation and reduced antibiotic need. | No impact on lung function, but potential improvement of systemic antioxidant status and reduced exacerbation frequency. |
| Ascorbic acid (vitamin C) [121] | Inhibits bacterial quorum sensing, affecting chemotaxis and biofilm formation. | In vitro: B. subtilis, E. coli, P. aeruginosa, M. abscessus biofilms. | Impaired biofilm formation and promoted destruction of existing biofilms. | Interesting in vitro antibiofilm activity; clinical relevance and efficacy in CF patients require further investigation. |
| Vitamin A and E status [123] | Observation of vitamin levels during CFTR modulator treatment. | CF Patients under lumacaftor/ivacaftor treatments. | Plasma vitamin A significantly increased; Vitamin E and Vit E/cholesterol ratio slightly decreased (alongside improved lung function from modulators). | Need to monitor and adjust vitamins supplementation during CFTR modulator therapies due to altered vitamins levels. |
| Molecules addressing LPO or lipid imbalance | ||||
| Deferoxamine [86] | Iron chelator; inhibition of Lipid Peroxidation (LPO). | CF and non-CF primary cells from human epithelium; primary cell from CF mice (Cftrtm1eur F508del) and pigs (CFTR-KO) | Partially correction of lipid profile. | Potential use if iron-mediated LPO significantly contributes to CF pathology; further research needed. |
| Ferrostatin-1 [23,60] | Scavenger of lipid radicals | Mice lacking CFTR expression (with P. aeruginosa infection). | Blocked peroxidative damage and ferroptotic cell death induced by P. aeruginosa | Not a current clinical drug candidate, but its mechanism is promising against CF inflammation and infection. |
| Fenretinide [124] | Modulation of fatty acid imbalance and MUC5AC and MUC5B expression. | CF patients; Mice lacking CFTR expression (with P. aeruginosa infection). | Exhibited beneficial effects on lipid homeostasis and redox balance. | Potential positive effects on lipid and redox equilibrium; it warrants further investigation. |
| Myriocin [125] | Sphingolipid synthesis inhibitor. | CF cell models (IB3-1 cells, primary cells ΔF508/W1282X) | Significantly decreased dyslipidemia; positive effect on modulating immunity and inflammatory response. | Promising for addressing CF-associated dyslipidemia and its impact on inflammation and immunity. |
7.4. MicroRNAs as Potential Targets to Regulate Oxidative Stress
| Pathway | miRNAs | Outcome | Reference |
|---|---|---|---|
| Inflammation | ↑ miR-146a | IL-6 overproduction | [135] |
| ↓ miR-17 and mi-R93 | IL-8 overproduction | [136,137] | |
| ↑ miR-199a-3p | IKKβ increased expression and NF-κB hyperactivation | [133] | |
| ↑ miR-181b | Alteration of ALX/FPR2-dependent pathway | [138] | |
| Oxidative stress | ↑ miR-34a | SIRT1/-6 deacetylases decreased expression | [141] |
| ↑ miR-570-3p | SIRT1 deacetylase decreased expression | [142] | |
| ↑ miR-551b | Downregulation of catalase expression | [143] | |
| ↑ miR-21 | Downregulation of SOD3 and TNFα | [144] | |
| ↑ miR-200c | Inhibition of Nrf2 expression | [145] | |
| ER stress | ↑ miR-221 | Downregulation of ATF6 levels | [146] |
8. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| 1O2 | Singlet oxygen |
| AA | Arachidonic acid |
| ARE | Antioxidant response element |
| ASL | Airway surface liquid |
| ATF6 | Activating transcription factor 6 |
| BALF | Bronchoalveolar lavage fluid |
| CF | Cystic fibrosis |
| CFBE | Cystic fibrosis bronchial epithelial |
| CFTR | Cystic Fibrosis Transmembrane Conductance Regulator |
| COPD | Chronic obstructive pulmonary disease |
| CoQ10 | Coenzyme Q10 |
| DHA | Docosahexaenoic acids |
| ER | Endoplasmic reticulum |
| ETI | Elexacaftor/tezacaftor/ivacaftor |
| Fe2+ | Ferrous iron |
| Fe3+ | Ferric iron |
| GCL | Glutamate-cysteine ligase |
| GCLC | Glutamate-cysteine ligase catalytic subunit |
| GCLM | Glutamate-cysteine ligase modifier subunit |
| GPx4 | Glutathione peroxidase 4 |
| GR | Glutathione reductase |
| GSH | Glutathione |
| GSS | Glutathione synthetase |
| GSSG | Glutathione disulfide |
| H2O2 | Hydrogen peroxide |
| HNE | Human nasal epithelial |
| IL | Interleukin |
| IRE1α | Inositol-requiring enzyme 1 α |
| KEAP1 | Kelch-like ECH-associated protein 1 |
| LOXs | Lipoxygenases |
| LPO | Lipid peroxidation |
| miRNAs | MicroRNAs |
| MRPs | Multidrug resistant proteins |
| NAC | N-acetylcysteine |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NOX | NADPH oxidase |
| Nrf2 | Nuclear factor (erythroid-derived 2)-like 2 |
| O2•− | Superoxide anions |
| HO• | Hydroxyl radicals |
| PERK | Protein kinase RNA-like ER Kinase |
| PL-O• | Alkoxyl radical |
| PL-OH | Phospholipid alcohols |
| PL-OO• | Peroxyl radical |
| PL-OOH | Lipid hydroperoxide |
| PLs | Phospholipids |
| PM | Plasma membrane |
| PUFAs | Polyunsaturated fatty acids |
| ROS | Reactive oxygen species |
| SOD | Superoxide dismutase |
| UPR | Unfolded protein response |
| XBP1 | X-box binding protein 1 |
| xCT | System Xc− light chain |
References
- Farinha, C.M.; Callebaut, I. Molecular mechanisms of cystic fibrosis—How mutations lead to misfunction and guide therapy. Biosci. Rep. 2022, 42, BSR20212006. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, B.P.; Freedman, S.D. Cystic fibrosis. Lancet 2009, 373, 1891–1904. [Google Scholar] [CrossRef] [PubMed]
- Rommens, J.M.; Iannuzzi, M.C.; Kerem, B.; Drumm, M.L.; Melmer, G.; Dean, M.; Rozmahel, R.; Cole, J.L.; Kennedy, D.; Hidaka, N.; et al. Identification of the cystic fibrosis gene: Chromosome walking and jumping. Science 1989, 245, 1059–1065. [Google Scholar] [CrossRef] [PubMed]
- Shteinberg, M.; Haq, I.J.; Polineni, D.; Davies, J.C. Cystic fibrosis. Lancet 2021, 397, 2195–2211. [Google Scholar] [CrossRef]
- Moliteo, E.; Sciacca, M.; Palmeri, A.; Papale, M.; Manti, S.; Parisi, G.F.; Leonardi, S. Cystic Fibrosis and Oxidative Stress: The Role of CFTR. Molecules 2022, 27, 5324. [Google Scholar] [CrossRef]
- Gadsby, D.C.; Vergani, P.; Csanady, L. The ABC protein turned chloride channel whose failure causes cystic fibrosis. Nature 2006, 440, 477–483. [Google Scholar] [CrossRef]
- Henderson, A.G.; Ehre, C.; Button, B.; Abdullah, L.H.; Cai, L.H.; Leigh, M.W.; DeMaria, G.C.; Matsui, H.; Donaldson, S.H.; Davis, C.W.; et al. Cystic fibrosis airway secretions exhibit mucin hyperconcentration and increased osmotic pressure. J. Clin. Investig. 2014, 124, 3047–3060. [Google Scholar] [CrossRef]
- Sellers, Z.M.; Illek, B.; Figueira, M.F.; Hari, G.; Joo, N.S.; Sibley, E.; Souza-Menezes, J.; Morales, M.M.; Fischer, H.; Wine, J.J. Impaired PGE2-stimulated Cl- and HCO3- secretion contributes to cystic fibrosis airway disease. PLoS ONE 2017, 12, e0189894. [Google Scholar] [CrossRef]
- Hill, D.B.; Long, R.F.; Kissner, W.J.; Atieh, E.; Garbarine, I.C.; Markovetz, M.R.; Fontana, N.C.; Christy, M.; Habibpour, M.; Tarran, R.; et al. Pathological mucus and impaired mucus clearance in cystic fibrosis patients result from increased concentration, not altered pH. Eur. Respir. J. 2018, 52, 1801297. [Google Scholar] [CrossRef]
- Cystic Fibrosis Mutation Database (CFTR1). Available online: http://www.genet.sickkids.on.ca/StatisticsPage.html (accessed on 16 April 2025).
- De Boeck, K. Cystic fibrosis in the year 2020: A disease with a new face. Acta Paediatr. 2020, 109, 893–899. [Google Scholar] [CrossRef]
- Allen, L.; Carr, S.B.; Davies, G.; Downey, D.; Egan, M.; Forton, J.T.; Gray, R.; Haworth, C.; Horsley, A.; Smyth, A.R.; et al. Future therapies for cystic fibrosis. Nat. Commun. 2023, 14, 693. [Google Scholar] [CrossRef] [PubMed]
- Castaldo, A.; Gelzo, M.; Iacotucci, P.; Longobardi, A.; Taccetti, G.; Terlizzi, V.; Carnovale, V. One year of treatment with elexacaftor/tezacaftor/ivacaftor in patients with cystic fibrosis homozygous for the F508del mutation causes a significant increase in liver biochemical indexes. Front. Mol. Biosci. 2023, 10, 1327958. [Google Scholar] [CrossRef] [PubMed]
- Vanzacaftor, tezacaftor, and deutivacaftor (Alyftrek) for cystic fibrosis. Med. Lett. Drugs Ther. 2025, 67, 41–43. [CrossRef] [PubMed]
- Hoppe, J.E.; Kasi, A.S.; Pittman, J.E.; Jensen, R.; Thia, L.P.; Robinson, P.; Tirakitsoontorn, P.; Ramsey, B.; Mall, M.A.; Taylor-Cousar, J.L.; et al. Vanzacaftor-tezacaftor-deutivacaftor for children aged 6-11 years with cystic fibrosis (RIDGELINE Trial VX21-121-105): An analysis from a single-arm, phase 3 trial. Lancet Respir. Med. 2025, 13, 244–255. [Google Scholar] [CrossRef]
- Keating, C.; Yonker, L.M.; Vermeulen, F.; Prais, D.; Linnemann, R.W.; Trimble, A.; Kotsimbos, T.; Mermis, J.; Braun, A.T.; O’Carroll, M.; et al. Vanzacaftor-tezacaftor-deutivacaftor versus elexacaftor-tezacaftor-ivacaftor in individuals with cystic fibrosis aged 12 years and older (SKYLINE Trials VX20-121-102 and VX20-121-103): Results from two randomised, active-controlled, phase 3 trials. Lancet Respir. Med. 2025, 13, 256–271. [Google Scholar] [CrossRef]
- Galli, F.; Battistoni, A.; Gambari, R.; Pompella, A.; Bragonzi, A.; Pilolli, F.; Iuliano, L.; Piroddi, M.; Dechecchi, M.C.; Cabrini, G. Oxidative stress and antioxidant therapy in cystic fibrosis. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2012, 1822, 690–713. [Google Scholar] [CrossRef]
- Ziady, A.G.; Hansen, J. Redox balance in cystic fibrosis. Int. J. Biochem. Cell Biol. 2014, 52, 113–123. [Google Scholar] [CrossRef]
- Recchiuti, A.; Mattoscio, D.; Isopi, E. Roles, Actions, and Therapeutic Potential of Specialized Pro-resolving Lipid Mediators for the Treatment of Inflammation in Cystic Fibrosis. Front. Pharmacol. 2019, 10, 252. [Google Scholar] [CrossRef]
- Benabdeslam, H.; Abidi, H.; Garcia, I.; Bellon, G.; Gilly, R.; Revol, A. Lipid peroxidation and antioxidant defenses in cystic fibrosis patients. Clin. Chem. Lab. Med. 1999, 37, 511–516. [Google Scholar] [CrossRef]
- Ribeiro, C.M.; Boucher, R.C. Role of endoplasmic reticulum stress in cystic fibrosis-related airway inflammatory responses. Proc. Am. Thorac. Soc. 2010, 7, 387–394. [Google Scholar] [CrossRef]
- Atlante, A.; Favia, M.; Bobba, A.; Guerra, L.; Casavola, V.; Reshkin, S.J. Characterization of mitochondrial function in cells with impaired cystic fibrosis transmembrane conductance regulator (CFTR) function. J. Bioenerg. Biomembr. 2016, 48, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Ousingsawat, J.; Schreiber, R.; Gulbins, E.; Kamler, M.; Kunzelmann, K.P. aeruginosa Induced Lipid Peroxidation Causes Ferroptotic Cell Death in Airways. Cell Physiol. Biochem. 2021, 55, 590–604. [Google Scholar] [CrossRef]
- Alfadda, A.A.; Sallam, R.M. Reactive oxygen species in health and disease. J. Biomed. Biotechnol. 2012, 2012, 936486. [Google Scholar] [CrossRef]
- Riley, P.A. Free radicals in biology: Oxidative stress and the effects of ionizing radiation. Int. J. Radiat. Biol. 1994, 65, 27–33. [Google Scholar] [CrossRef]
- Fang, F.C. Antimicrobial reactive oxygen and nitrogen species: Concepts and controversies. Nat. Rev. Microbiol. 2004, 2, 820–832. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef] [PubMed]
- Averill-Bates, D. Reactive oxygen species and cell signaling. Review. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2024, 1871, 119573. [Google Scholar] [CrossRef]
- Sies, H. Oxidative Stress: Eustress and Distress in Redox Homeostasis. In Stress: Physiology, Biochemistry, and Pathology; Handbook of Stress Series, Volume 3; Academic Press: New York, NY, USA, 2019; pp. 153–163. [Google Scholar] [CrossRef]
- Cohen, T.S.; Prince, A. Cystic fibrosis: A mucosal immunodeficiency syndrome. Nat. Med. 2012, 18, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Kariya, C.; Leitner, H.; Min, E.; van Heeckeren, C.; van Heeckeren, A.; Day, B.J. A role for CFTR in the elevation of glutathione levels in the lung by oral glutathione administration. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, L1590–L1597. [Google Scholar] [CrossRef]
- Velsor, L.W.; Kariya, C.; Kachadourian, R.; Day, B.J. Mitochondrial oxidative stress in the lungs of cystic fibrosis transmembrane conductance regulator protein mutant mice. Am. J. Respir. Cell Mol. Biol. 2006, 35, 579–586. [Google Scholar] [CrossRef]
- Rogers, C.S.; Stoltz, D.A.; Meyerholz, D.K.; Ostedgaard, L.S.; Rokhlina, T.; Taft, P.J.; Rogan, M.P.; Pezzulo, A.A.; Karp, P.H.; Itani, O.A.; et al. Disruption of the CFTR gene produces a model of cystic fibrosis in newborn pigs. Science 2008, 321, 1837–1841. [Google Scholar] [CrossRef] [PubMed]
- Roesch, E.A.; Nichols, D.P.; Chmiel, J.F. Inflammation in cystic fibrosis: An update. Pediatr. Pulmonol. 2018, 53, S30–S50. [Google Scholar] [CrossRef]
- Hubeau, C.; Lorenzato, M.; Couetil, J.P.; Hubert, D.; Dusser, D.; Puchelle, E.; Gaillard, D. Quantitative analysis of inflammatory cells infiltrating the cystic fibrosis airway mucosa. Clin. Exp. Immunol. 2001, 124, 69–76. [Google Scholar] [CrossRef]
- Sorio, C.; Montresor, A.; Bolomini-Vittori, M.; Caldrer, S.; Rossi, B.; Dusi, S.; Angiari, S.; Johansson, J.E.; Vezzalini, M.; Leal, T.; et al. Mutations of Cystic Fibrosis Transmembrane Conductance Regulator Gene Cause a Monocyte-Selective Adhesion Deficiency. Am. J. Respir. Crit. Care Med. 2016, 193, 1123–1133. [Google Scholar] [CrossRef]
- Kettle, A.J.; Chan, T.; Osberg, I.; Senthilmohan, R.; Chapman, A.L.; Mocatta, T.J.; Wagener, J.S. Myeloperoxidase and protein oxidation in the airways of young children with cystic fibrosis. Am. J. Respir. Crit. Care Med. 2004, 170, 1317–1323. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef]
- Ong, G.; Ragetli, R.; Mnich, K.; Doble, B.W.; Kammouni, W.; Logue, S.E. IRE1 signaling increases PERK expression during chronic ER stress. Cell Death Dis. 2024, 15, 276. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Blohmke, C.J.; Mayer, M.L.; Tang, A.C.; Hirschfeld, A.F.; Fjell, C.D.; Sze, M.A.; Falsafi, R.; Wang, S.; Hsu, K.; Chilvers, M.A.; et al. Atypical activation of the unfolded protein response in cystic fibrosis airway cells contributes to p38 MAPK-mediated innate immune responses. J. Immunol. 2012, 189, 5467–5475. [Google Scholar] [CrossRef]
- Trouve, P.; Ferec, C.; Genin, E. The Interplay between the Unfolded Protein Response, Inflammation and Infection in Cystic Fibrosis. Cells 2021, 10, 2980. [Google Scholar] [CrossRef]
- Zeeshan, H.M.; Lee, G.H.; Kim, H.R.; Chae, H.J. Endoplasmic Reticulum Stress and Associated ROS. Int. J. Mol. Sci. 2016, 17, 327. [Google Scholar] [CrossRef] [PubMed]
- Ashby, M.C.; Tepikin, A.V. ER calcium and the functions of intracellular organelles. Semin. Cell Dev. Biol. 2001, 12, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Bartoszewski, R.; Rab, A.; Jurkuvenaite, A.; Mazur, M.; Wakefield, J.; Collawn, J.F.; Bebok, Z. Activation of the unfolded protein response by ΔF508 CFTR. Am. J. Respir. Cell Mol. Biol. 2008, 39, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Kerbiriou, M.; Le Drevo, M.A.; Ferec, C.; Trouve, P. Coupling cystic fibrosis to endoplasmic reticulum stress: Differential role of Grp78 and ATF6. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2007, 1772, 1236–1249. [Google Scholar] [CrossRef]
- Nichols, D.P.; Chmiel, J.F. Inflammation and its genesis in cystic fibrosis. Pediatr. Pulmonol. 2015, 50 (Suppl. S40), S39–S56. [Google Scholar] [CrossRef]
- Rimessi, A.; Previati, M.; Nigro, F.; Wieckowski, M.R.; Pinton, P. Mitochondrial reactive oxygen species and inflammation: Molecular mechanisms, diseases and promising therapies. Int. J. Biochem. Cell Biol. 2016, 81, 281–293. [Google Scholar] [CrossRef]
- Patergnani, S.; Vitto, V.A.M.; Pinton, P.; Rimessi, A. Mitochondrial Stress Responses and “Mito-Inflammation” in Cystic Fibrosis. Front. Pharmacol. 2020, 11, 581114. [Google Scholar] [CrossRef]
- Favia, M.; de Bari, L.; Bobba, A.; Atlante, A. An Intriguing Involvement of Mitochondria in Cystic Fibrosis. J. Clin. Med. 2019, 8, 1890. [Google Scholar] [CrossRef]
- Li, X.; Zhang, W.; Cao, Q.; Wang, Z.; Zhao, M.; Xu, L.; Zhuang, Q. Mitochondrial dysfunction in fibrotic diseases. Cell Death Discov. 2020, 6, 80. [Google Scholar] [CrossRef]
- Cui, X.; Zhou, Z.; Tu, H.; Wu, J.; Zhou, J.; Yi, Q.; Liu, O.; Dai, X. Mitophagy in fibrotic diseases: Molecular mechanisms and therapeutic applications. Front. Physiol. 2024, 15, 1430230. [Google Scholar] [CrossRef]
- Kleme, M.L.; Sane, A.; Garofalo, C.; Seidman, E.; Brochiero, E.; Berthiaume, Y.; Levy, E. CFTR Deletion Confers Mitochondrial Dysfunction and Disrupts Lipid Homeostasis in Intestinal Epithelial Cells. Nutrients 2018, 10, 836. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Maity, S. ER Stress-Sensor Proteins and ER-Mitochondrial Crosstalk-Signaling Beyond (ER) Stress Response. Biomolecules 2021, 11, 173. [Google Scholar] [CrossRef] [PubMed]
- Rimessi, A.; Vitto, V.A.M.; Patergnani, S.; Pinton, P. Update on Calcium Signaling in Cystic Fibrosis Lung Disease. Front. Pharmacol. 2021, 12, 581645. [Google Scholar] [CrossRef] [PubMed]
- Rimessi, A.; Bezzerri, V.; Patergnani, S.; Marchi, S.; Cabrini, G.; Pinton, P. Mitochondrial Ca2+-dependent NLRP3 activation exacerbates the Pseudomonas aeruginosa-driven inflammatory response in cystic fibrosis. Nat. Commun. 2015, 6, 6201. [Google Scholar] [CrossRef]
- Riquelme, S.A.; Lozano, C.; Moustafa, A.M.; Liimatta, K.; Tomlinson, K.L.; Britto, C.; Khanal, S.; Gill, S.K.; Narechania, A.; Azcona-Gutierrez, J.M.; et al. CFTR-PTEN-dependent mitochondrial metabolic dysfunction promotes Pseudomonas aeruginosa airway infection. Sci. Transl. Med. 2019, 11, eaav4634. [Google Scholar] [CrossRef]
- Cavinato, L.; Genise, E.; Luly, F.R.; Di Domenico, E.G.; Del Porto, P.; Ascenzioni, F. Escaping the Phagocytic Oxidative Burst: The Role of SODB in the Survival of Pseudomonas aeruginosa Within Macrophages. Front. Microbiol. 2020, 11, 326. [Google Scholar] [CrossRef]
- Valgimigli, L. Lipid Peroxidation and Antioxidant Protection. Biomolecules 2023, 13, 1291. [Google Scholar] [CrossRef]
- Miotto, G.; Rossetto, M.; Di Paolo, M.L.; Orian, L.; Venerando, R.; Roveri, A.; Vuckovic, A.M.; Bosello Travain, V.; Zaccarin, M.; Zennaro, L.; et al. Insight into the mechanism of ferroptosis inhibition by ferrostatin-1. Redox Biol. 2020, 28, 101328. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef]
- Hadian, K.; Stockwell, B.R. The therapeutic potential of targeting regulated non-apoptotic cell death. Nat. Rev. Drug Discov. 2023, 22, 723–742. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.J.; Frazer, D.M. Current understanding of iron homeostasis. Am. J. Clin. Nutr. 2017, 106, 1559S–1566S. [Google Scholar] [CrossRef] [PubMed]
- Ghio, A.J.; Roggli, V.L.; Soukup, J.M.; Richards, J.H.; Randell, S.H.; Muhlebach, M.S. Iron accumulates in the lavage and explanted lungs of cystic fibrosis patients. J. Cyst. Fibros. 2013, 12, 390–398. [Google Scholar] [CrossRef]
- D’Orazio, S.; Mattoscio, D. Dysregulation of the Arachidonic Acid Pathway in Cystic Fibrosis: Implications for Chronic Inflammation and Disease Progression. Pharmaceuticals 2024, 17, 1185. [Google Scholar] [CrossRef] [PubMed]
- Ursini, F.; Bosello Travain, V.; Cozza, G.; Miotto, G.; Roveri, A.; Toppo, S.; Maiorino, M. A white paper on Phospholipid Hydroperoxide Glutathione Peroxidase (GPx4) forty years later. Free Radic. Biol. Med. 2022, 188, 117–133. [Google Scholar] [CrossRef]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef]
- Maiorino, M.; Conrad, M.; Ursini, F. GPx4, Lipid Peroxidation, and Cell Death: Discoveries, Rediscoveries, and Open Issues. Antioxid. Redox Signal. 2018, 29, 61–74. [Google Scholar] [CrossRef]
- Weaver, K.; Skouta, R. The Selenoprotein Glutathione Peroxidase 4: From Molecular Mechanisms to Novel Therapeutic Opportunities. Biomedicines 2022, 10, 891. [Google Scholar] [CrossRef]
- Brigelius-Flohe, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta (BBA) Gen. Subj. 2013, 1830, 3289–3303. [Google Scholar] [CrossRef]
- Ursini, F.; Maiorino, M.; Gregolin, C. The selenoenzyme phospholipid hydroperoxide glutathione peroxidase. Biochim. Biophys. Acta (BBA) Gen. Subj. 1985, 839, 62–70. [Google Scholar] [CrossRef]
- Roum, J.H.; Buhl, R.; McElvaney, N.G.; Borok, Z.; Crystal, R.G. Systemic deficiency of glutathione in cystic fibrosis. J. Appl. Physiol. 1993, 75, 2419–2424. [Google Scholar] [CrossRef]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef]
- Adinolfi, S.; Patinen, T.; Jawahar Deen, A.; Pitkanen, S.; Harkonen, J.; Kansanen, E.; Kublbeck, J.; Levonen, A.L. The KEAP1-NRF2 pathway: Targets for therapy and role in cancer. Redox Biol. 2023, 63, 102726. [Google Scholar] [CrossRef]
- He, F.; Ru, X.; Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 2020, 21, 4777. [Google Scholar] [CrossRef]
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta (BBA) Gen. Subj. 2013, 1830, 3143–3153. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Sato, H.; Kuriyama-Matsumura, K.; Sato, K.; Maebara, K.; Wang, H.; Tamba, M.; Itoh, K.; Yamamoto, M.; Bannai, S. Electrophile response element-mediated induction of the cystine/glutamate exchange transporter gene expression. J. Biol. Chem. 2002, 277, 44765–44771. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Kinter, M.; Shank, S.; Cotton, C.; Kelley, T.J.; Ziady, A.G. Dysfunction of Nrf-2 in CF epithelia leads to excess intracellular H2O2 and inflammatory cytokine production. PLoS ONE 2008, 3, e3367. [Google Scholar] [CrossRef]
- de Bari, L.; Favia, M.; Bobba, A.; Lassandro, R.; Guerra, L.; Atlante, A. Aberrant GSH reductase and NOX activities concur with defective CFTR to pro-oxidative imbalance in cystic fibrosis airways. J. Bioenerg. Biomembr. 2018, 50, 117–129. [Google Scholar] [CrossRef]
- Yoshida, M.; Minagawa, S.; Araya, J.; Sakamoto, T.; Hara, H.; Tsubouchi, K.; Hosaka, Y.; Ichikawa, A.; Saito, N.; Kadota, T.; et al. Involvement of cigarette smoke-induced epithelial cell ferroptosis in COPD pathogenesis. Nat. Commun. 2019, 10, 3145. [Google Scholar] [CrossRef]
- Nagasaki, T.; Schuyler, A.J.; Zhao, J.; Samovich, S.N.; Yamada, K.; Deng, Y.; Ginebaugh, S.P.; Christenson, S.A.; Woodruff, P.G.; Fahy, J.V.; et al. 15LO1 dictates glutathione redox changes in asthmatic airway epithelium to worsen type 2 inflammation. J. Clin. Investig. 2022, 132, e151685. [Google Scholar] [CrossRef]
- Dar, H.H.; Tyurina, Y.Y.; Mikulska-Ruminska, K.; Shrivastava, I.; Ting, H.C.; Tyurin, V.A.; Krieger, J.; St Croix, C.M.; Watkins, S.; Bayir, E.; et al. Pseudomonas aeruginosa utilizes host polyunsaturated phosphatidylethanolamines to trigger theft-ferroptosis in bronchial epithelium. J. Clin. Investig. 2018, 128, 4639–4653. [Google Scholar] [CrossRef]
- Maniam, P.; Essilfie, A.T.; Kalimutho, M.; Ling, D.; Frazer, D.M.; Phipps, S.; Anderson, G.J.; Reid, D.W. Increased susceptibility of cystic fibrosis airway epithelial cells to ferroptosis. Biol. Res. 2021, 54, 38. [Google Scholar] [CrossRef] [PubMed]
- Veltman, M.; De Sanctis, J.B.; Stolarczyk, M.; Klymiuk, N.; Bahr, A.; Brouwer, R.W.; Oole, E.; Shah, J.; Ozdian, T.; Liao, J.; et al. CFTR Correctors and Antioxidants Partially Normalize Lipid Imbalance but not Abnormal Basal Inflammatory Cytokine Profile in CF Bronchial Epithelial Cells. Front. Physiol. 2021, 12, 619442. [Google Scholar] [CrossRef] [PubMed]
- Dei Cas, M.; Zulueta, A.; Mingione, A.; Caretti, A.; Ghidoni, R.; Signorelli, P.; Paroni, R. An Innovative Lipidomic Workflow to Investigate the Lipid Profile in a Cystic Fibrosis Cell Line. Cells 2020, 9, 1197. [Google Scholar] [CrossRef]
- Loberto, N.; Mancini, G.; Bassi, R.; Carsana, E.V.; Tamanini, A.; Pedemonte, N.; Dechecchi, M.C.; Sonnino, S.; Aureli, M. Sphingolipids and plasma membrane hydrolases in human primary bronchial cells during differentiation and their altered patterns in cystic fibrosis. Glycoconj. J. 2020, 37, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Dobi, D.; Loberto, N.; Bassi, R.; Pistocchi, A.; Lunghi, G.; Tamanini, A.; Aureli, M. Cross-talk between CFTR and sphingolipids in cystic fibrosis. FEBS Open Bio 2023, 13, 1601–1614. [Google Scholar] [CrossRef]
- Ghidoni, R.; Caretti, A.; Signorelli, P. Role of Sphingolipids in the Pathobiology of Lung Inflammation. Mediat. Inflamm. 2015, 2015, 487508. [Google Scholar] [CrossRef]
- Balasubramanian, A.; Sundrud, M.S. ATP-dependent transporters: Emerging players at the crossroads of immunity and metabolism. Front. Immunol. 2023, 14, 1286696. [Google Scholar] [CrossRef]
- He, P.; Gelissen, I.C.; Ammit, A.J. Regulation of ATP binding cassette transporter A1 (ABCA1) expression: Cholesterol-dependent and—Independent signaling pathways with relevance to inflammatory lung disease. Respir. Res. 2020, 21, 250. [Google Scholar] [CrossRef]
- Strandvik, B. Fatty acid metabolism in cystic fibrosis. Prostaglandins Leukot. Essent. Fatty Acids 2010, 83, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Njoroge, S.W.; Laposata, M.; Katrangi, W.; Seegmiller, A.C. DHA and EPA reverse cystic fibrosis-related FA abnormalities by suppressing FA desaturase expression and activity. J. Lipid Res. 2012, 53, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Keum, Y.S.; Choi, B.Y. Molecular and chemical regulation of the Keap1-Nrf2 signaling pathway. Molecules 2014, 19, 10074–10089. [Google Scholar] [CrossRef] [PubMed]
- Egbujor, M.C.; Buttari, B.; Profumo, E.; Telkoparan-Akillilar, P.; Saso, L. An Overview of NRF2-Activating Compounds Bearing α,β-Unsaturated Moiety and Their Antioxidant Effects. Int. J. Mol. Sci. 2022, 23, 8466. [Google Scholar] [CrossRef]
- Hoch, C.C.; Shoykhet, M.; Weiser, T.; Griesbaum, L.; Petry, J.; Hachani, K.; Multhoff, G.; Bashiri Dezfouli, A.; Wollenberg, B. Isothiocyanates in medicine: A comprehensive review on phenylethyl-, allyl-, and benzyl-isothiocyanates. Pharmacol. Res. 2024, 201, 107107. [Google Scholar] [CrossRef]
- Taguchi, K.; Motohashi, H.; Yamamoto, M. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells 2011, 16, 123–140. [Google Scholar] [CrossRef]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef]
- Ashrafizadeh, M.; Ahmadi, Z.; Mohammadinejad, R.; Farkhondeh, T.; Samarghandian, S. Curcumin Activates the Nrf2 Pathway and Induces Cellular Protection Against Oxidative Injury. Curr. Mol. Med. 2020, 20, 116–133. [Google Scholar] [CrossRef]
- Egan, M.E.; Pearson, M.; Weiner, S.A.; Rajendran, V.; Rubin, D.; Glockner-Pagel, J.; Canny, S.; Du, K.; Lukacs, G.L.; Caplan, M.J. Curcumin, a major constituent of turmeric, corrects cystic fibrosis defects. Science 2004, 304, 600–602. [Google Scholar] [CrossRef]
- Song, Y.; Sonawane, N.D.; Salinas, D.; Qian, L.; Pedemonte, N.; Galietta, L.J.; Verkman, A.S. Evidence against the rescue of defective ΔF508-CFTR cellular processing by curcumin in cell culture and mouse models. J. Biol. Chem. 2004, 279, 40629–40633. [Google Scholar] [CrossRef]
- Kode, A.; Rajendrasozhan, S.; Caito, S.; Yang, S.R.; Megson, I.L.; Rahman, I. Resveratrol induces glutathione synthesis by activation of Nrf2 and protects against cigarette smoke-mediated oxidative stress in human lung epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L478–L488. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Blount, A.C.; McNicholas, C.M.; Skinner, D.F.; Chestnut, M.; Kappes, J.C.; Sorscher, E.J.; Woodworth, B.A. Resveratrol enhances airway surface liquid depth in sinonasal epithelium by increasing cystic fibrosis transmembrane conductance regulator open probability. PLoS ONE 2013, 8, e81589. [Google Scholar] [CrossRef] [PubMed]
- Dhooghe, B.; Bouckaert, C.; Capron, A.; Wallemacq, P.; Leal, T.; Noel, S. Resveratrol increases F508del-CFTR dependent salivary secretion in cystic fibrosis mice. Biol. Open 2015, 4, 929–936. [Google Scholar] [CrossRef]
- Jai, Y.; Shah, K.; Bridges, R.J.; Bradbury, N.A. Evidence against resveratrol as a viable therapy for the rescue of defective ΔF508 CFTR. Biochim. Biophys. Acta (BBA) Gen. Subj. 2015, 1850, 2377–2384. [Google Scholar] [CrossRef]
- Laselva, O.; Allegretta, C.; Di Gioia, S.; Avolio, C.; Conese, M. Anti-Inflammatory and Anti-Oxidant Effect of Dimethyl Fumarate in Cystic Fibrosis Bronchial Epithelial Cells. Cells 2021, 10, 2132. [Google Scholar] [CrossRef]
- Borcherding, D.C.; Siefert, M.E.; Lin, S.; Brewington, J.; Sadek, H.; Clancy, J.P.; Plafker, S.M.; Ziady, A.G. Clinically-approved CFTR modulators rescue Nrf2 dysfunction in cystic fibrosis airway epithelia. J. Clin. Investig. 2019, 129, 3448–3463. [Google Scholar] [CrossRef] [PubMed]
- Das, T.; Simone, M.; Ibugo, A.I.; Witting, P.K.; Manefield, M.; Manos, J. Glutathione Enhances Antibiotic Efficiency and Effectiveness of DNase I in Disrupting Pseudomonas aeruginosa Biofilms While Also Inhibiting Pyocyanin Activity, Thus Facilitating Restoration of Cell Enzymatic Activity, Confluence and Viability. Front. Microbiol. 2017, 8, 2429. [Google Scholar] [CrossRef]
- Griese, M.; Kappler, M.; Eismann, C.; Ballmann, M.; Junge, S.; Rietschel, E.; van Koningsbruggen-Rietschel, S.; Staab, D.; Rolinck-Werninghaus, C.; Mellies, U.; et al. Inhalation treatment with glutathione in patients with cystic fibrosis. A randomized clinical trial. Am. J. Respir. Crit. Care Med. 2013, 188, 83–89. [Google Scholar] [CrossRef]
- Calabrese, C.; Tosco, A.; Abete, P.; Carnovale, V.; Basile, C.; Magliocca, A.; Quattrucci, S.; De Sanctis, S.; Alatri, F.; Mazzarella, G.; et al. Randomized, single blind, controlled trial of inhaled glutathione vs placebo in patients with cystic fibrosis. J. Cyst. Fibros. 2015, 14, 203–210. [Google Scholar] [CrossRef]
- Hartl, D.; Starosta, V.; Maier, K.; Beck-Speier, I.; Rebhan, C.; Becker, B.F.; Latzin, P.; Fischer, R.; Ratjen, F.; Huber, R.M.; et al. Inhaled glutathione decreases PGE2 and increases lymphocytes in cystic fibrosis lungs. Free Radic. Biol. Med. 2005, 39, 463–472. [Google Scholar] [CrossRef]
- Visca, A.; Bishop, C.T.; Hilton, S.; Hudson, V.M. Oral reduced L-glutathione improves growth in pediatric cystic fibrosis patients. J. Pediatr. Gastroenterol. Nutr. 2015, 60, 802–810. [Google Scholar] [CrossRef] [PubMed]
- Bozic, M.; Goss, C.H.; Tirouvanziam, R.M.; Baines, A.; Kloster, M.; Antoine, L.; Borowitz, D.; Schwarzenberg, S.J. Oral Glutathione and Growth in Cystic Fibrosis: A Multicenter, Randomized, Placebo-controlled, Double-blind Trial. J. Pediatr. Gastroenterol. Nutr. 2020, 71, 771–777. [Google Scholar] [CrossRef]
- Guerini, M.; Condro, G.; Friuli, V.; Maggi, L.; Perugini, P. N-acetylcysteine (NAC) and Its Role in Clinical Practice Management of Cystic Fibrosis (CF): A Review. Pharmaceuticals 2022, 15, 217. [Google Scholar] [CrossRef]
- Wall, G.M. N-Acetylcysteine Amide (NACA) and (2R,2′R′)-3,3′-Disulfanediylbis(2-acetamidopropanamide) (DINACA) for the Prevention and Treatment of Radiation Pneumonitis and the Treatment of Pulmonary Function in Cystic. Fibrosis. Patent WO2020146660, 16 July 2020. [Google Scholar]
- Ciofu, O.; Smith, S.; Lykkesfeldt, J. Antioxidant supplementation for lung disease in cystic fibrosis. Cochrane Database Syst. Rev. 2019, 10, CD007020. [Google Scholar] [CrossRef]
- Wood, L.G.; Fitzgerald, D.A.; Lee, A.K.; Garg, M.L. Improved antioxidant and fatty acid status of patients with cystic fibrosis after antioxidant supplementation is linked to improved lung function. Am. J. Clin. Nutr. 2003, 77, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Renner, S.; Rath, R.; Rust, P.; Lehr, S.; Frischer, T.; Elmadfa, I.; Eichler, I. Effects of β-carotene supplementation for six months on clinical and laboratory parameters in patients with cystic fibrosis. Thorax 2001, 56, 48–52. [Google Scholar] [CrossRef] [PubMed]
- Sagel, S.D.; Khan, U.; Jain, R.; Graff, G.; Daines, C.L.; Dunitz, J.M.; Borowitz, D.; Orenstein, D.M.; Abdulhamid, I.; Noe, J.; et al. Effects of an Antioxidant-enriched Multivitamin in Cystic Fibrosis. A Randomized, Controlled, Multicenter Clinical Trial. Am. J. Respir. Crit. Care Med. 2018, 198, 639–647. [Google Scholar] [CrossRef]
- Pandit, S.; Ravikumar, V.; Abdel-Haleem, A.M.; Derouiche, A.; Mokkapati, V.; Sihlbom, C.; Mineta, K.; Gojobori, T.; Gao, X.; Westerlund, F.; et al. Low Concentrations of Vitamin C Reduce the Synthesis of Extracellular Polymers and Destabilize Bacterial Biofilms. Front. Microbiol. 2017, 8, 2599. [Google Scholar] [CrossRef]
- Dalton, J.P.; Uy, B.; Phummarin, N.; Copp, B.R.; Denny, W.A.; Swift, S.; Wiles, S. Effect of common and experimental anti-tuberculosis treatments on Mycobacterium tuberculosis growing as biofilms. PeerJ 2016, 4, e2717. [Google Scholar] [CrossRef]
- Sommerburg, O.; Hammerling, S.; Schneider, S.P.; Okun, J.; Langhans, C.D.; Leutz-Schmidt, P.; Wielputz, M.O.; Siems, W.; Graber, S.Y.; Mall, M.A.; et al. CFTR Modulator Therapy with Lumacaftor/Ivacaftor Alters Plasma Concentrations of Lipid-Soluble Vitamins A and E in Patients with Cystic Fibrosis. Antioxidants 2021, 10, 483. [Google Scholar] [CrossRef]
- Guilbault, C.; De Sanctis, J.B.; Wojewodka, G.; Saeed, Z.; Lachance, C.; Skinner, T.A.; Vilela, R.M.; Kubow, S.; Lands, L.C.; Hajduch, M.; et al. Fenretinide corrects newly found ceramide deficiency in cystic fibrosis. Am. J. Respir. Cell Mol. Biol. 2008, 38, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Signorelli, P.; Pivari, F.; Barcella, M.; Merelli, I.; Zulueta, A.; Dei Cas, M.; Rosso, L.; Ghidoni, R.; Caretti, A.; Paroni, R.; et al. Myriocin modulates the altered lipid metabolism and storage in cystic fibrosis. Cell. Signal. 2021, 81, 109928. [Google Scholar] [CrossRef] [PubMed]
- Liessi, N.; Tomati, V.; Capurro, V.; Loberto, N.; Garcia-Aloy, M.; Franceschi, P.; Aureli, M.; Pedemonte, N.; Armirotti, A. The combination elexacaftor/tezacaftor/ivacaftor (ETI) modulates the de novo synthethic pathway of ceramides in a genotype-independent manner. J. Cyst. Fibros. 2023, 22, 680–682. [Google Scholar] [CrossRef]
- Ciobanu, D.Z.; Liessi, N.; Tomati, V.; Capurro, V.; Bertozzi, S.M.; Summa, M.; Bertorelli, R.; Loberto, N.; Dobi, D.; Aureli, M.; et al. Tezacaftor is a direct inhibitor of sphingolipid delta-4 desaturase enzyme (DEGS). J. Cyst. Fibros. 2024, 23, 1167–1172. [Google Scholar] [CrossRef] [PubMed]
- Jonas, S.; Izaurralde, E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat. Rev. Genet. 2015, 16, 421–433. [Google Scholar] [CrossRef]
- Matoulkova, E.; Michalova, E.; Vojtesek, B.; Hrstka, R. The role of the 3′ untranslated region in post-transcriptional regulation of protein expression in mammalian cells. RNA Biol. 2012, 9, 563–576. [Google Scholar] [CrossRef]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef]
- Lima, J.F.; Cerqueira, L.; Figueiredo, C.; Oliveira, C.; Azevedo, N.F. Anti-miRNA oligonucleotides: A comprehensive guide for design. RNA Biol. 2018, 15, 338–352. [Google Scholar] [CrossRef]
- Gillen, A.E.; Gosalia, N.; Leir, S.H.; Harris, A. MicroRNA regulation of expression of the cystic fibrosis transmembrane conductance regulator gene. Biochem. J. 2011, 438, 25–32. [Google Scholar] [CrossRef]
- Bardin, P.; Sonneville, F.; Corvol, H.; Tabary, O. Emerging microRNA Therapeutic Approaches for Cystic Fibrosis. Front. Pharmacol. 2018, 9, 1113. [Google Scholar] [CrossRef]
- De Palma, F.D.E.; Raia, V.; Kroemer, G.; Maiuri, M.C. The Multifaceted Roles of MicroRNAs in Cystic Fibrosis. Diagnostics 2020, 10, 1102. [Google Scholar] [CrossRef] [PubMed]
- Luly, F.R.; Leveque, M.; Licursi, V.; Cimino, G.; Martin-Chouly, C.; Theret, N.; Negri, R.; Cavinato, L.; Ascenzioni, F.; Del Porto, P. MiR-146a is over-expressed and controls IL-6 production in cystic fibrosis macrophages. Sci. Rep. 2019, 9, 16259. [Google Scholar] [CrossRef] [PubMed]
- Oglesby, I.K.; Vencken, S.F.; Agrawal, R.; Gaughan, K.; Molloy, K.; Higgins, G.; McNally, P.; McElvaney, N.G.; Mall, M.A.; Greene, C.M. miR-17 overexpression in cystic fibrosis airway epithelial cells decreases interleukin-8 production. Eur. Respir. J. 2015, 46, 1350–1360. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, E.; Borgatti, M.; Montagner, G.; Bianchi, N.; Finotti, A.; Lampronti, I.; Bezzerri, V.; Dechecchi, M.C.; Cabrini, G.; Gambari, R. Expression of microRNA-93 and Interleukin-8 during Pseudomonas aeruginosa-mediated induction of proinflammatory responses. Am. J. Respir. Cell Mol. Biol. 2014, 50, 1144–1155. [Google Scholar] [CrossRef]
- Pierdomenico, A.M.; Patruno, S.; Codagnone, M.; Simiele, F.; Mari, V.C.; Plebani, R.; Recchiuti, A.; Romano, M. microRNA-181b is increased in cystic fibrosis cells and impairs lipoxin A(4) receptor-dependent mechanisms of inflammation resolution and antimicrobial defense. Sci. Rep. 2017, 7, 13519. [Google Scholar] [CrossRef]
- Lettieri-Barbato, D.; Aquilano, K.; Punziano, C.; Minopoli, G.; Faraonio, R. MicroRNAs, Long Non-Coding RNAs, and Circular RNAs in the Redox Control of Cell Senescence. Antioxidants 2022, 11, 480. [Google Scholar] [CrossRef]
- Carbonell, T.; Gomes, A.V. MicroRNAs in the regulation of cellular redox status and its implications in myocardial ischemia-reperfusion injury. Redox Biol. 2020, 36, 101607. [Google Scholar] [CrossRef]
- Baker, J.R.; Vuppusetty, C.; Colley, T.; Papaioannou, A.I.; Fenwick, P.; Donnelly, L.; Ito, K.; Barnes, P.J. Oxidative stress dependent microRNA-34a activation via PI3Kα reduces the expression of sirtuin-1 and sirtuin-6 in epithelial cells. Sci. Rep. 2016, 6, 35871. [Google Scholar] [CrossRef]
- Baker, J.R.; Vuppusetty, C.; Colley, T.; Hassibi, S.; Fenwick, P.S.; Donnelly, L.E.; Ito, K.; Barnes, P.J. MicroRNA-570 is a novel regulator of cellular senescence and inflammaging. FASEB J. 2019, 33, 1605–1616. [Google Scholar] [CrossRef]
- Xu, X.; Wells, A.; Padilla, M.T.; Kato, K.; Kim, K.C.; Lin, Y. A signaling pathway consisting of miR-551b, catalase and MUC1 contributes to acquired apoptosis resistance and chemoresistance. Carcinogenesis 2014, 35, 2457–2466. [Google Scholar] [CrossRef]
- Zhang, X.; Ng, W.L.; Wang, P.; Tian, L.; Werner, E.; Wang, H.; Doetsch, P.; Wang, Y. MicroRNA-21 modulates the levels of reactive oxygen species by targeting SOD3 and TNFα. Cancer Res. 2012, 72, 4707–4713. [Google Scholar] [CrossRef] [PubMed]
- Cortez, M.A.; Valdecanas, D.; Zhang, X.; Zhan, Y.; Bhardwaj, V.; Calin, G.A.; Komaki, R.; Giri, D.K.; Quini, C.C.; Wolfe, T.; et al. Therapeutic delivery of miR-200c enhances radiosensitivity in lung cancer. Mol. Ther. 2014, 22, 1494–1503. [Google Scholar] [CrossRef]
- Oglesby, I.K.; Agrawal, R.; Mall, M.A.; McElvaney, N.G.; Greene, C.M. miRNA-221 is elevated in cystic fibrosis airway epithelial cells and regulates expression of ATF6. Mol. Cell. Pediatr. 2015, 2, 1. [Google Scholar] [CrossRef] [PubMed]
- Seyhan, A.A. Trials and Tribulations of MicroRNA Therapeutics. Int. J. Mol. Sci. 2024, 25, 1469. [Google Scholar] [CrossRef] [PubMed]
- De Santi, C.; Greene, C.M. Challenges facing microRNA therapeutics for cystic fibrosis lung disease. Epigenomics 2020, 12, 179–181. [Google Scholar] [CrossRef]
- Finotti, A.; Gambari, R. Perspectives in MicroRNA Therapeutics for Cystic Fibrosis. Non-Coding RNA 2025, 11, 3. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Artusi, I.; Rubin, M.; Cozza, G. Redox Imbalance in Cystic Fibrosis: The Multifaceted Role of Oxidative Stress. Pharmaceuticals 2025, 18, 784. https://doi.org/10.3390/ph18060784
Artusi I, Rubin M, Cozza G. Redox Imbalance in Cystic Fibrosis: The Multifaceted Role of Oxidative Stress. Pharmaceuticals. 2025; 18(6):784. https://doi.org/10.3390/ph18060784
Chicago/Turabian StyleArtusi, Ilaria, Michela Rubin, and Giorgio Cozza. 2025. "Redox Imbalance in Cystic Fibrosis: The Multifaceted Role of Oxidative Stress" Pharmaceuticals 18, no. 6: 784. https://doi.org/10.3390/ph18060784
APA StyleArtusi, I., Rubin, M., & Cozza, G. (2025). Redox Imbalance in Cystic Fibrosis: The Multifaceted Role of Oxidative Stress. Pharmaceuticals, 18(6), 784. https://doi.org/10.3390/ph18060784