
Design, Synthesis, and Anti-Proliferative Action of Purine/Pteridine-Based Derivatives as Dual Inhibitors of EGFR and BRAFV600E

 , , , ,
, , , ,
Abstract
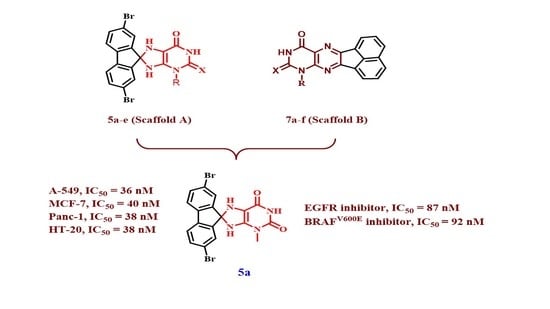
1. Introduction
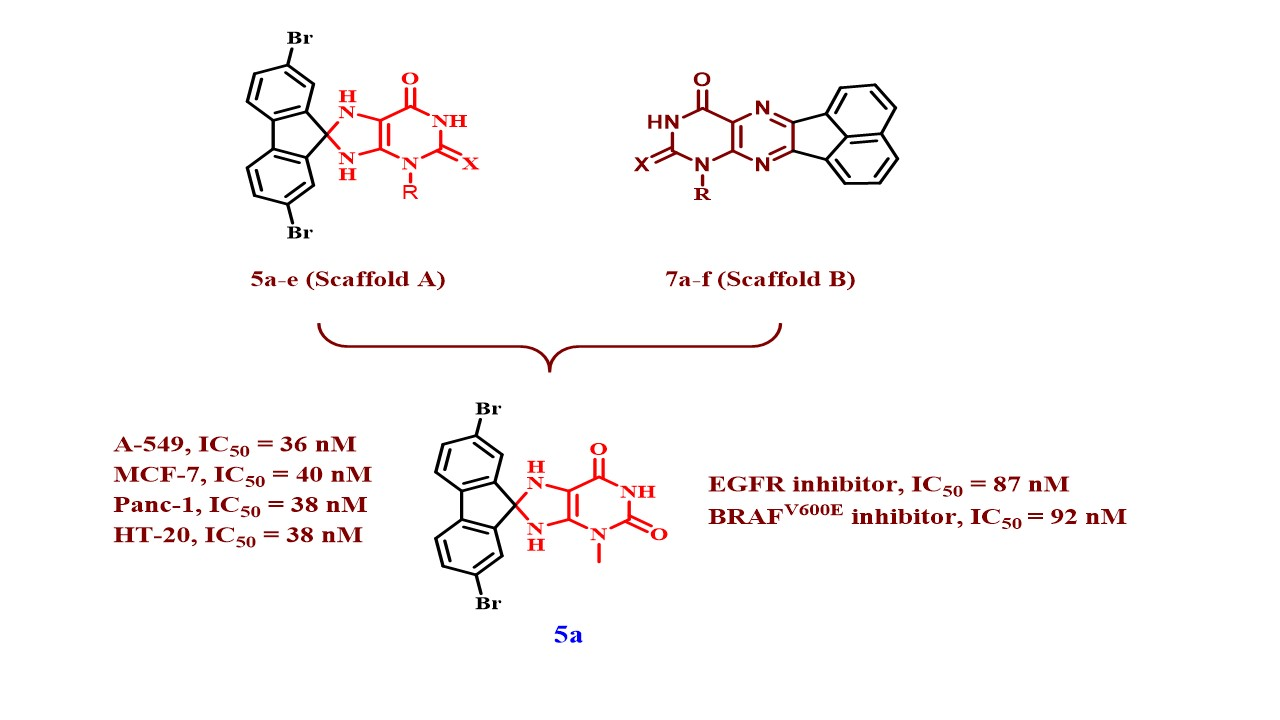
2. Results and Discussion
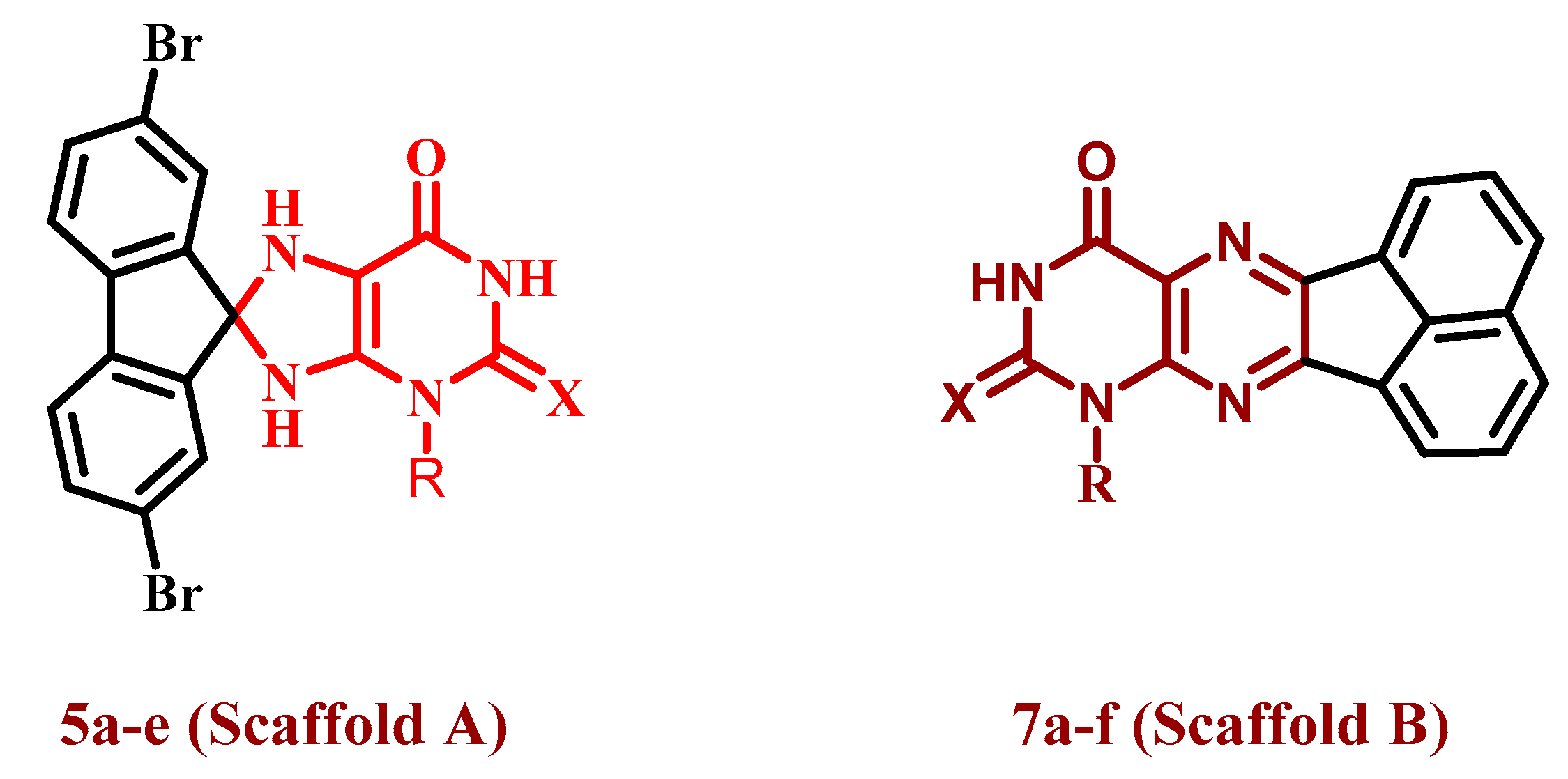
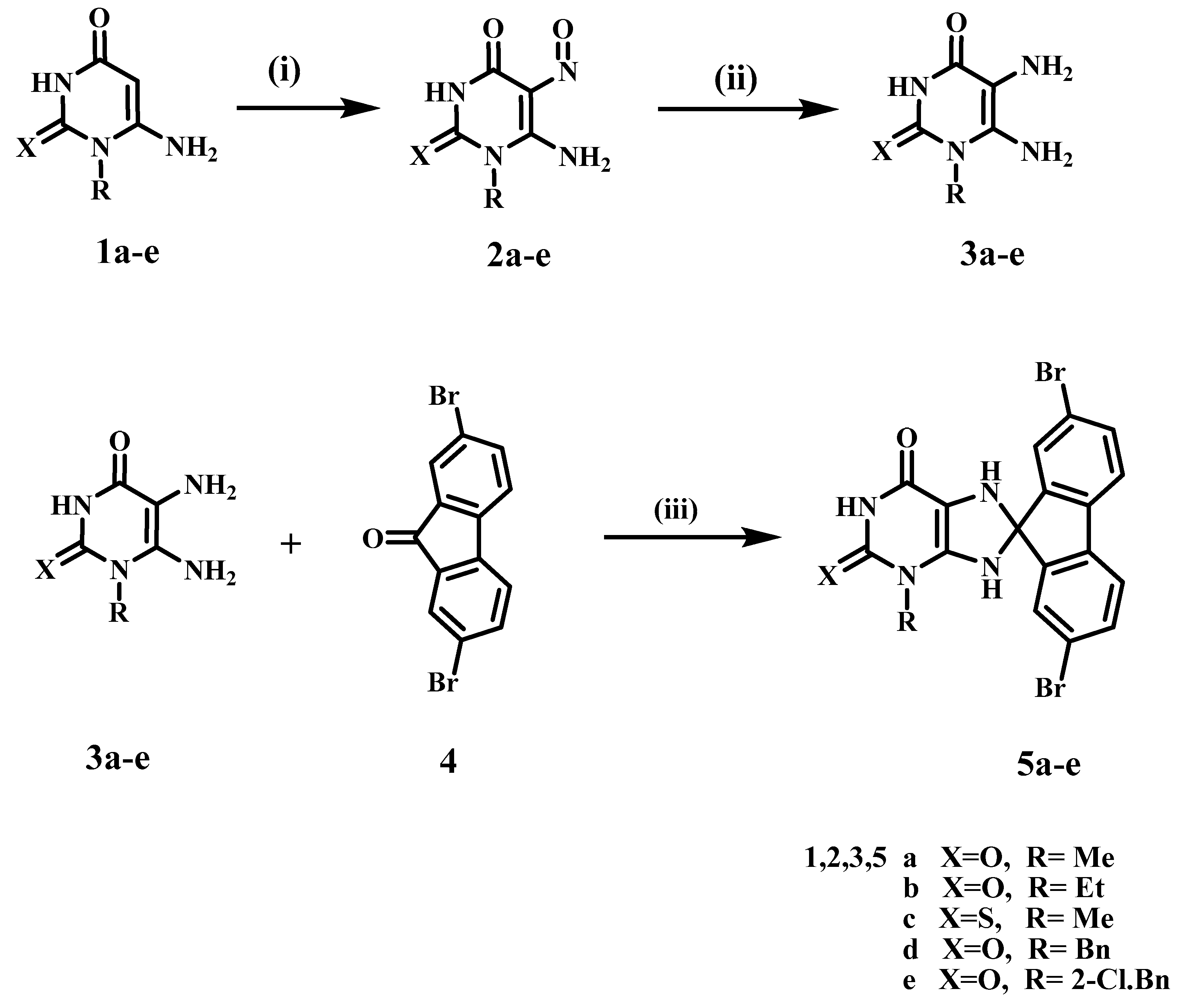
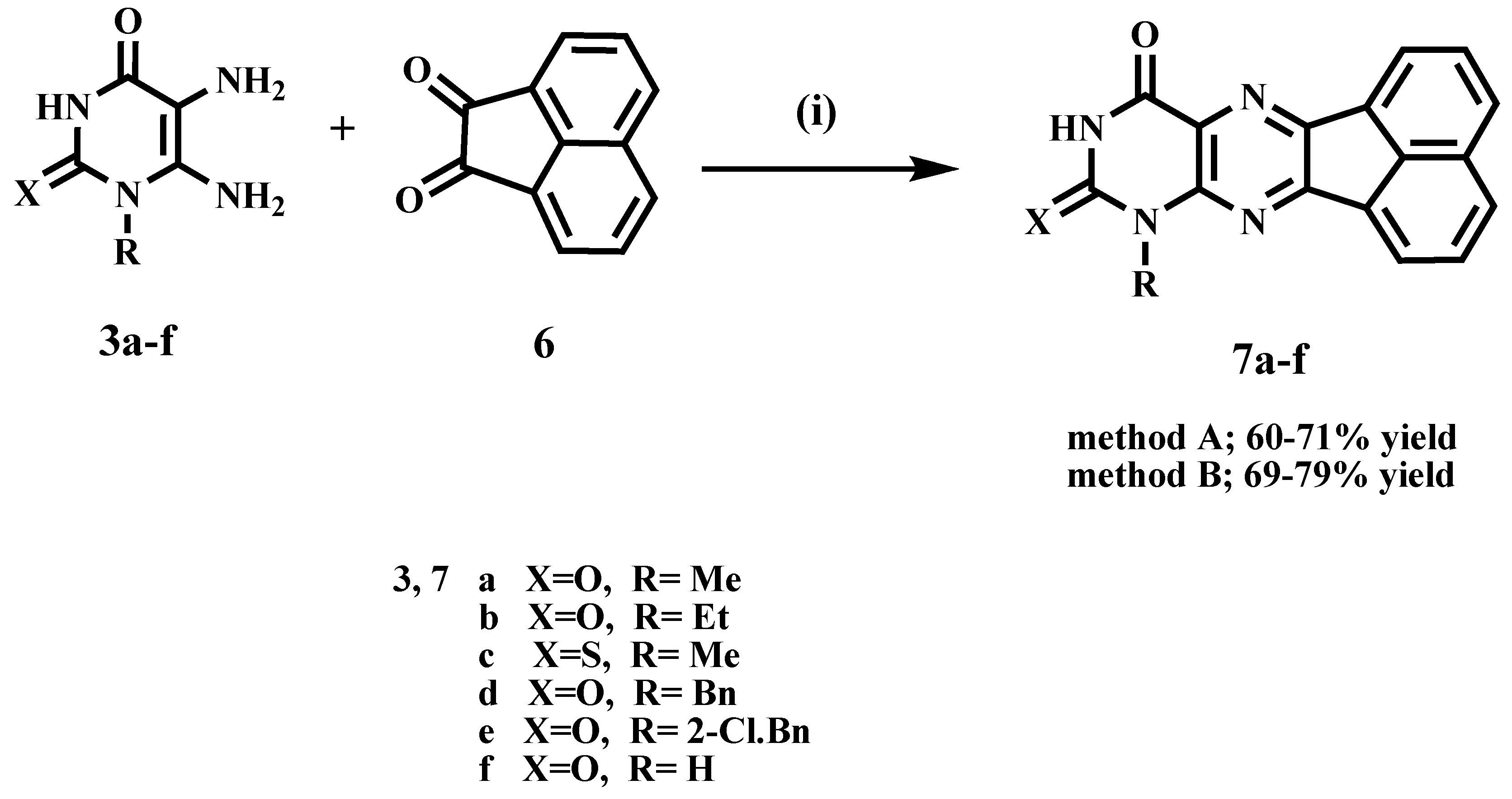
2.1. Chemistry
2.2. Biology
2.2.1. Cell Viability Assay
2.2.2. Anti-Proliferative Assay
2.2.3. EGFR Inhibitory Assay
2.2.4. BRAFV600E Inhibitory Assay
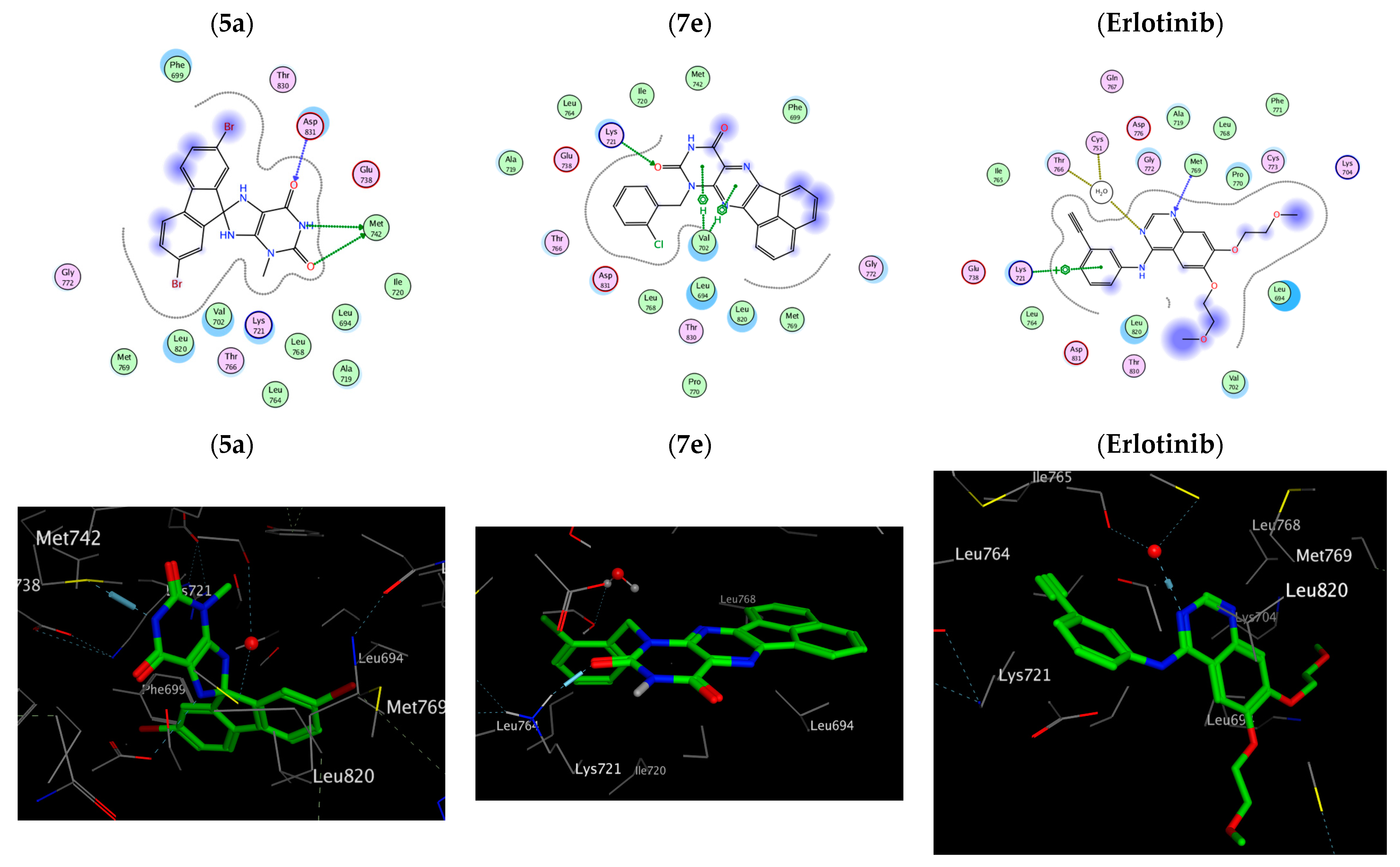
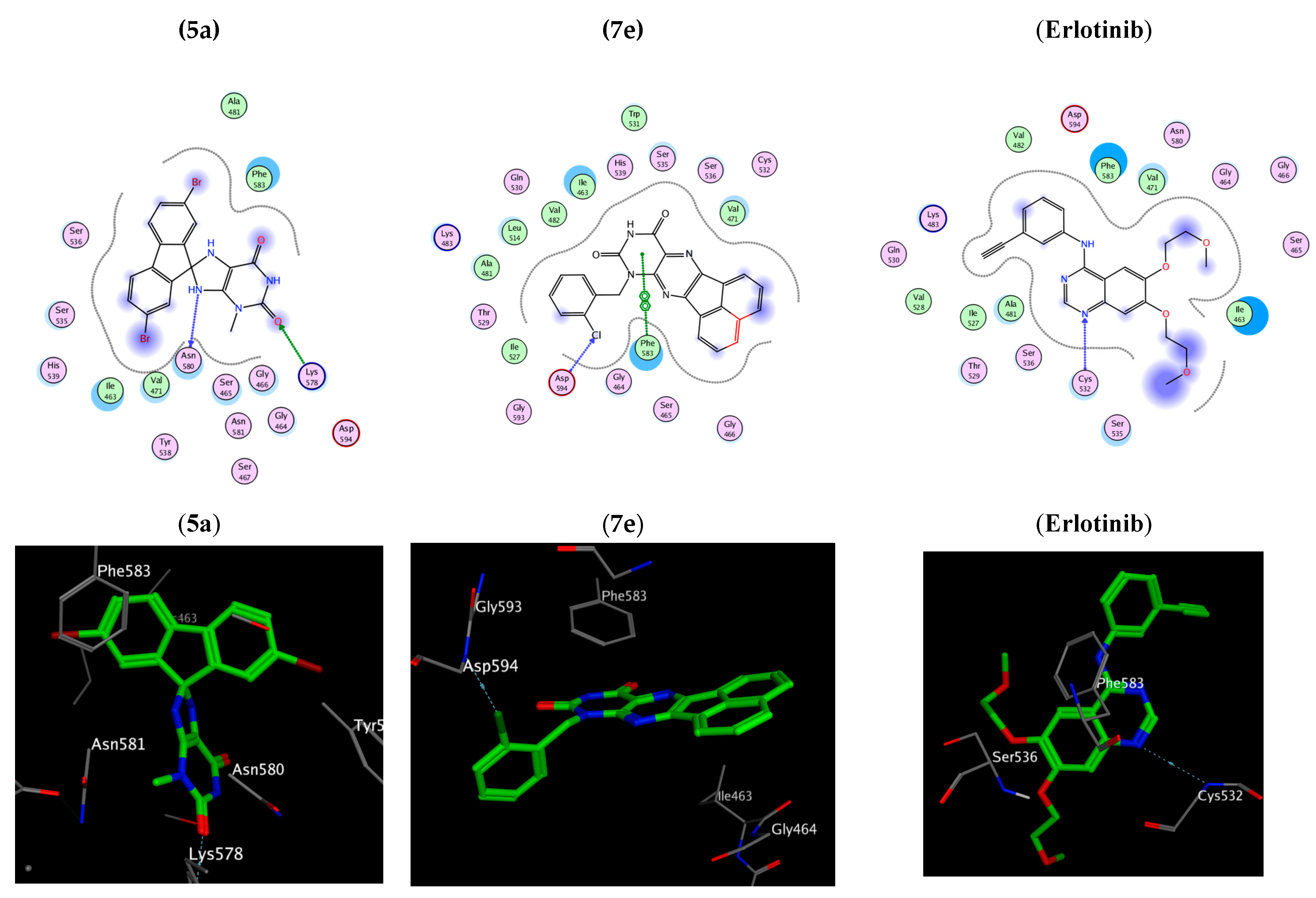
2.3. Docking Study
3. Experimental
3.1. Chemistry
3.1.1. General Procedures for the Synthesis of 2,7-Dibromo-3′-ethyl-7′,9′-dihydrospiro-[fluorene-9,8′-purines] 5a–e
- 2,7-Dibromo-3′-methyl-7′,9′-dihydrospiro[fluorene-9,8′-purine]-2′,6′(1′H,3′H)-dione (5a)
- 2,7-Dibromo-3′-ethyl-7′,9′-dihydrospiro[fluorene-9,8′-purine]-2′,6′(1′H,3′H)-dione (5b)
- 2,7-Dibromo-3′-methyl-2′-thioxo-2′,3′,7′,9′-tetrahydrospiro[fluorene-9,8′-purin]-6′(1′H)-one (5c)
- 3’-Benzyl-2,7-dibromo-7′,9′-dihydrospiro[fluorene-9,8′-purine]-2′,6′(1′H,3′H)-dione (5d)
- 2,7-Dibromo-3′-(2-chlorobenzyl)-7′,9′-dihydrospiro[fluorene-9,8′-purine]-2′,6′(1′H,3′H)-dione (5e)
3.1.2. General Procedures for the Synthesis of Acenaphtho[1,2-g]pteridines (7a–f)
- 8-Methylacenaphtho[1,2-g]pteridine-9,11(8H,10H)-dione (7a)
- 8-Ethylacenaphtho[1,2-g]pteridine-9,11(8H,10H)-dione (7b)
- 8-Methyl-9-thioxo-9,10-dihydroacenaphtho[1,2-g]pteridin-11(8H)-one (7c)
- 8-Benzylacenaphtho[1,2-g]pteridine-9,11(8H,10H)-dione (7d)
- 8-(2-Chlorobenzyl)acenaphtho[1,2-g]pteridine-9,11(8H,10H)-dione (7e)
- Acenaphtho [1,2-g]pteridine-9,11(8H,10H)-dione (7f)
3.2. Biology
3.2.1. Cell Viability Assay
3.2.2. Anti-Proliferative Assay
3.2.3. EGFR Inhibitory Assay
3.2.4. BRAFV600E Inhibitory Assay
3.3. Protocol of Docking Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Stanković, T.; Dinić, J.; Podolski-Renić, A.; Musso, L.; Burić, S.S.; Dallavalle, S.; Pešić, M. Dual Inhibitors as a New Challenge for Cancer Multidrug Resistance Treatment. Curr. Med. Chem. 2019, 26, 6074–6106. [Google Scholar] [CrossRef] [PubMed]
- Raghavendra, N.M.; Pingili, D.; Kadasi, S.; Mettu, A.; Prasad, S.V.U.M. Dual or multi-targeting inhibitors: The next generation anticancer agents. Eur. J. Med. Chem. 2018, 143, 1277–1300. [Google Scholar] [CrossRef] [PubMed]
- Fu, R.-G.; Sun, Y.; Sheng, W.-B.; Liao, D.-F. Designing multi-targeted agents: An emerging anticancer drug discovery paradigm. Eur. J. Med. Chem. 2017, 136, 195–211. [Google Scholar] [CrossRef] [PubMed]
- Zha, G.-F.; Qin, H.-L.; Youssif, B.G.; Amjad, M.W.; Raja, M.A.G.; Abdelazeem, A.H.; Bukhari, S.N.A. Discovery of potential anticancer multi-targeted ligustrazine based cyclohexanone and oxime analogs overcoming the cancer multidrug resistance. Eur. J. Med. Chem. 2017, 135, 34–48. [Google Scholar] [CrossRef] [PubMed]
- Palmeira, A.; Sousa, E.; Vasconcelos, M.H.; Pinto, M.M. Three Decades of P-gp Inhibitors: Skimming Through Several Generations and Scaffolds. Curr. Med. Chem. 2012, 19, 1946–2025. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Zhao, Y.; Luo, Q.; Zhang, Y.; Wu, K.; Wang, F. Multi-targeted anticancer agents. Curr. Top. Med. Chem. 2017, 17, 3084–3098. [Google Scholar] [CrossRef] [PubMed]
- Mahboobi, S.; Sellmer, A.; Winkler, M.; Eichhorn, E.; Pongratz, H.; Ciossek, T.; Baer, T.; Maier, T.; Beckers, T. Novel Chimeric Histone Deacetylase Inhibitors: A Series of Lapatinib Hybrides as Potent Inhibitors of Epidermal Growth Factor Receptor (EGFR), Human Epidermal Growth Factor Receptor 2 (HER2), and Histone Deacetylase Activity. J. Med. Chem. 2010, 53, 8546–8555. [Google Scholar] [CrossRef]
- Bhullar, K.S.; Lagarón, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef]
- Ho, C.-C.; Liao, W.-Y.; Lin, C.-A.; Shih, J.-Y.; Yu, C.-J.; Yang, J.C.-H. Acquired BRAF V600E Mutation as Resistant Mechanism after Treatment with Osimertinib. J. Thorac. Oncol. 2017, 12, 567–572. [Google Scholar] [CrossRef]
- Hyman, D.M.; Puzanov, I.; Subbiah, V.; Faris, J.E.; Chau, I.; Blay, J.-Y.; Wolf, J.; Raje, N.S.; Diamond, E.L.; Hollebecque, A.; et al. Vemurafenib in Multiple Nonmelanoma Cancers with BRAF V600 Mutations. N. Engl. J. Med. 2015, 373, 726–736. [Google Scholar] [CrossRef]
- Desai, J.; Markman, B.; Ananda, S.; Tebbutt, N.C.; Michael, M.; Solomon, B.J.; McArthur, G.A.; Tie, J.; Gibbs, P.; Ritchie, D.; et al. A phase I/II trial of combined BRAF and EGFR inhibition in patients (pts) with BRAF V600E mutated (BRAFm) metastatic colorectal (mCRC): The EViCT (Erlotinib and Vemurafenib in Combination Trial) study. J. Clin. Oncol. 2017, 35, 3557. [Google Scholar] [CrossRef]
- Makower, D.; Malik, U.; Novik, Y.; Wiernik, P.H. Therapeutic efficacy of theophylline in chronic lymphocytic leukemia. Med. Oncol. 1999, 16, 69–71. [Google Scholar] [CrossRef]
- Sabisz, M.; Skladanowski, A. Modulation of cellular response to anticancer treatment by caffeine: Inhibition of cell cycle checkpoints, DNA repair and more. Curr. Pharm. Biotechnol. 2008, 9, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Tenzer, A.; Pruschy, M. Potentiation of DNA-damage-induced cytotoxicity by G2checkpoint abrogators. Curr. Med. Chem. Anti-Cancer Agents 2003, 3, 35–46. [Google Scholar] [CrossRef]
- Lentini, A.; Kleinman, H.K.; Mattioli, P.; Autuori-Pezzoli, V.; Nicoli, L.; Pietrini, A.; Abbruzzese, A.; Cardinali, M.; Beninati, S. Inhibition of melanoma pulmonary metastasis by methylxanthines due to decreased invasion and proliferation. Melanoma Res. 1998, 8, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Hisham, M.; Youssif, B.G.; Osman, E.E.A.; Hayallah, A.M.; Abdel-Aziz, M. Synthesis and biological evaluation of novel xanthine derivatives as potential apoptotic antitumor agents. Eur. J. Med. Chem. 2019, 176, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Lee, H.; Jun, H.; Hong, S.-S.; Hong, S. Fluorescent phosphoinositide 3-kinase inhibitors suitable for monitoring of intracellular distribution. Bioorg. Med. Chem. 2011, 19, 2508–2516. [Google Scholar] [CrossRef]
- Lee, K.; Jeong, K.-W.; Lee, Y.; Song, J.Y.; Kim, M.S.; Lee, G.S.; Kim, Y. Pharmacophore modeling and virtual screening studies for new VEGFR-2 kinase inhibitors. Eur. J. Med. Chem. 2010, 45, 5420–5427. [Google Scholar] [CrossRef]
- Ruddarraju, R.R.; Murugulla, A.C.; Kotla, R.; Tirumalasetty, M.C.B.; Wudayagiri, R.; Donthabakthuni, S.; Maroju, R.; Baburao, K.; Parasa, L.S. Design, synthesis, anticancer, antimicrobial activities and molecular docking studies of theophylline containing acetylenes and theophylline containing 1,2,3-triazoles with variant nucleoside derivatives. Eur. J. Med. Chem. 2016, 123, 379–396. [Google Scholar] [CrossRef]
- Abou-Zied, H.A.; Youssif, B.G.; Mohamed, M.F.; Hayallah, A.M.; Abdel-Aziz, M. EGFR inhibitors and apoptotic inducers: Design, synthesis, anticancer activity and docking studies of novel xanthine derivatives carrying chalcone moiety as hybrid molecules. Bioorg. Chem. 2019, 89, 102997. [Google Scholar] [CrossRef]
- El-Kalyoubi, S.; Agili, F. Synthesis, In Silico Prediction and In Vitro Evaluation of Antitumor Activities of Novel Pyrido [2,3-d] pyrimidine, Xanthine and Lumazine Derivatives. Molecules 2020, 25, 5205. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Tice, C.M.; Singh, S.B. The use of spirocyclic scaffolds in drug discovery. Bioorg. Med. Chem. Lett. 2014, 24, 3673–3682. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Martínez, V.; Ruiz-Alcaraz, A.J.; Vera, M.; Guirado, A.; Martínez-Esparza, M.; García-Peñarrubia, P. Therapeutic potential of pteridine derivatives: A comprehensive review. Med. Res. Rev. 2018, 39, 461–516. [Google Scholar] [CrossRef] [PubMed]
- Kompis, I.M.; Islam, K.; Then, R.L. DNA and RNA Synthesis: Antifolates. Chem. Rev. 2005, 105, 593–620. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Liu, X.; Tu, Z.; Zhang, L.; Ku, X.; Bai, F.; Zhao, Z.; Xu, Y.; Ding, K.; Li, H. Discovery of Pteridin-7(8H)-one-Based Irreversible Inhibitors Targeting the Epidermal Growth Factor Receptor (EGFR) Kinase T790M/L858R Mutant. J. Med. Chem. 2013, 56, 7821–7837. [Google Scholar] [CrossRef]
- El-Kalyoubi, S.; Agili, F.; Zordok, W.A.; El-Sayed, A.S.A. Synthesis, In Silico Prediction and In Vitro Evaluation of Antimicrobial Activity, DFT Calculation and Theoretical Investigation of Novel Xanthines and Uracil Containing Imidazolone Derivatives. Int. J. Mol. Sci. 2021, 22, 10979. [Google Scholar] [CrossRef]
- El-Kalyoubi, S.; Agili, F.; Adel, I.; Tantawy, M.A. Novel uracil derivatives depicted potential anticancer agents: In Vitro, molecular docking, and ADME study. Arab. J. Chem. 2022, 15, 103669. [Google Scholar] [CrossRef]
- Ram, V.J.; Pandey, H.K.; Vlietinck, A.J. Synthesis of 2,4-substituted 6,7-phenanthreno-and 6,7-acenaphthenopteridines. J. Heterocycl. Chem. 1981, 18, 55–57. [Google Scholar] [CrossRef]
- Gomaa, H.A.; Shaker, M.E.; Alzarea, S.I.; Hendawy, O.; Mohamed, F.A.; Gouda, A.M.; Ali, A.T.; Morcoss, M.M.; Abdelrahman, M.H.; Trembleau, L.; et al. Optimization and SAR investigation of novel 2,3-dihydropyrazino[1,2-a]indole-1,4-dione derivatives as EGFR and BRAFV600E dual inhibitors with potent antiproliferative and antioxidant activities. Bioorg. Chem. 2022, 120, 105616. [Google Scholar] [CrossRef]
- Youssif, B.G.; Gouda, A.M.; Moustafa, A.H.; Abdelhamid, A.A.; Gomaa, H.A.; Kamal, I.; Marzouk, A.A. Design and synthesis of new triarylimidazole derivatives as dual inhibitors of BRAFV600E/p38α with potential antiproliferative activity. J. Mol. Struct. 2021, 1253, 132218. [Google Scholar] [CrossRef]
- Mahmoud, M.A.; Mohammed, A.F.; Salem, O.I.; Gomaa, H.A.; Youssif, B.G. New 1,3,4-oxadiazoles linked with the 1,2,3-triazole moiety as antiproliferative agents targeting the EGFR tyrosine kinase. Arch. Pharm. 2022, 355, 2200009. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, M.A.; Mohammed, A.F.; Salem, O.I.; Rabea, S.M.; Youssif, B.G. Design, synthesis, and antiproliferative properties of new 1,2,3-triazole-carboximidamide derivatives as dual EGFR/VEGFR-2 inhibitors. J. Mol. Struct. 2023, 1282, 135165. [Google Scholar] [CrossRef]
- Mekheimer, R.A.; Allam, S.M.; Al-Sheikh, M.A.; Moustafa, M.S.; Al-Mousawi, S.M.; Mostafa, Y.A.; Youssif, B.G.; Gomaa, H.A.; Hayallah, A.M.; Abdelaziz, M.; et al. Discovery of new pyrimido [5,4-c] quinolines as potential antiproliferative agents with multitarget actions: Rapid synthesis, docking, and ADME studies. Bioorg. Chem. 2022, 121, 105693. [Google Scholar] [CrossRef]
- Abdel-Aziz, S.A.; Taher, E.S.; Lan, P.; Asaad, G.F.; Gomaa, H.A.; El-Koussi, N.A.; Youssif, B.G. Design, synthesis, and biological evaluation of new pyrimidine-5-carbonitrile derivatives bearing 1,3-thiazole moiety as novel anti-inflammatory EGFR inhibitors with cardiac safety profile. Bioorg. Chem. 2021, 111, 104890. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, F.A.; Gomaa, H.A.; Hendawy, O.; Ali, A.T.; Farghaly, H.S.; Gouda, A.M.; Abdelazeem, A.H.; Abdelrahman, M.H.; Trembleau, L.; Youssif, B.G. Design, synthesis, and biological evaluation of novel EGFR inhibitors containing 5-chloro-3-hydroxymethyl-indole-2-carboxamide scaffold with apoptotic antiproliferative activity. Bioorg. Chem. 2021, 112, 104960. [Google Scholar] [CrossRef] [PubMed]
- El-Sherief, H.A.; Youssif, B.G.; Abdelazeem, A.H.; Abdel-Aziz, M.; Abdel-Rahman, H.M. Design, synthesis and antiproliferative evaluation of novel 1, 2, 4-triazole/schiff base hybrids with EGFR and B-RAF inhibitory activities. Anti-Cancer Agents Med. Chem. 2019, 19, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, T.S.; Bokhtia, R.; Al-Mahmoudy, A.M.; Taher, E.S.; AlAwadh, M.; Elagawany, M.; Abdel-Aal, E.H.; Panda, S.; Gouda, A.M.; Asfour, H.Z.; et al. Design, synthesis and biological evaluation of novel 5-((substituted quinolin-3-yl/1-naphthyl) methylene)-3-substituted imidazolidin-2,4-dione as HIV-1 fusion inhibitors. Bioorg. Chem. 2020, 99, 103782. [Google Scholar] [CrossRef]
- Shaykoon, M.S.; Marzouk, A.A.; Soltan, O.M.; Wanas, A.S.; Radwan, M.M.; Gouda, A.M.; Youssif, B.G.; Abdel-Aziz, M. Design, synthesis and antitrypanosomal activity of heteroaryl-based 1,2,4-triazole and 1,3,4-oxadiazole derivatives. Bioorg. Chem. 2020, 100, 103933. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Comp. | R | X | Cell Viability % | Anti-Proliferative Activity IC50 ± SEM (nM) | ||||
|---|---|---|---|---|---|---|---|---|
| A-549 | MCF-7 | Panc-1 | HT-29 | Average (GI50) | ||||
| 5a | Me | O | 89 | 36 ± 3 | 40 ± 3 | 38 ± 3 | 38 ± 3 | 38 |
| 5b | Et | O | 91 | 85 ± 8 | 88 ± 8 | 86 ± 8 | 86 ± 8 | 86 |
| 5c | Me | S | 90 | 98 ± 9 | 103 ± 10 | 100 ± 9 | 102 ± 10 | 101 |
| 5d | Bn | O | 91 | 52 ± 5 | 55 ± 5 | 54 ± 5 | 52 ± 5 | 53 |
| 5e | 2-Cl-Bn | O | 89 | 44 ± 4 | 48 ± 4 | 46 ± 4 | 46 ± 4 | 46 |
| 7a | Me | O | 92 | 56 ± 5 | 60 ± 6 | 58± 5 | 58 ± 5 | 58 |
| 7b | Et | O | 90 | 64 ± 6 | 69 ± 6 | 66 ± 6 | 68 ± 6 | 67 |
| 7c | Me | S | 89 | 80 ± 8 | 83 ± 8 | 80 ± 8 | 80 ± 8 | 81 |
| 7d | Bn | O | 91 | 90 ± 9 | 96 ± 9 | 90 ± 9 | 92 ± 9 | 92 |
| 7e | 2-Cl-Bn | O | 88 | 41 ± 4 | 46 ± 4 | 44 ± 4 | 44 ± 4 | 44 |
| 7f | H | O | 90 | 76 ± 7 | 79 ± 7 | 75 ± 7 | 75 ± 7 | 76 |
| Erlotinib | - | - | ND | 30 ± 3 | 40 ± 3 | 30 ± 3 | 30 ± 3 | 33 |
| Compd. | EGFR Inhibition IC50 ± SEM (nM) | BRAFV600E Inhibition IC50 ± SEM (nM) |
|---|---|---|
| 5a | 87 ± 07 | 92 ± 07 |
| 5d | 105 ± 09 | 164 ± 15 |
| 5e | 98 ± 08 | 137 ± 12 |
| 7a | 112 ± 10 | 183 ± 17 |
| 7e | 92 ± 07 | 109 ± 09 |
| Erlotinib | 80 ± 05 | 60 ± 05 |
| 5a | 5c | 5d | 5e | 7a | 7c | 7e | Erlotinib | |
|---|---|---|---|---|---|---|---|---|
| EGFR (PDB ID): 1M17 | ||||||||
| S (kcal/mol) | −7.05 | −5.53 | −6.34 | −4.75 | −6.28 | −4.9 | −6.70 | −7.06 |
| RMSD (Å) | 1.44 | 1.53 | 1.39 | 1.9 | 1.43 | 2. | 1.84 | 0.75 |
| Amino acids residues binding interactions and their bond length (Å) | 2Met 742 (3.61) c Asp 831 (3.0) a | Asp 776 (3.53) c Gly 695 (3.37) a | Asp 776 (3.57) c Gly 695 (3.41) b | Leu 694 (4.13) b | Glu 738 (3.43) c Met 742 (4.11) c | Lys 721 (2.97) a, 2 Val 702 (4.26) b | Lys 721 (2.80) a Val 702 (3.98) b | Met 769 (2.70) a H2O 10 (2.78) a Lys 721 (4.62) b |
| 5a | 5c | 5d | 5e | 7a | 7c | 7e | Erlotinib | |
|---|---|---|---|---|---|---|---|---|
| BRAFV600E (PDB ID: 5JRQ) | ||||||||
| S (kcal/mol) | −6.69 | −3.88 | −5.03 | −5.34 | −5.82 | −4.92 | −6.30 | −8.02 |
| RMSD (Å) | 0.89 | 1.95 | 2.02 | 1.87 | 1.26 | 1.79 | 1.55 | 1.27 |
| Amino acid residues’ binding interactions and their bond length (Å) | Asn 580 (2.94) c Lys 578 (3.36) a | Ile463 (4.52) b Gln 461 (4.38) b | Ser 536 (2.74) a Phe 583 (3.72) b | Ile 463 (3.36) c | Thr 529 (3.31) c Cys 532 (2.80) a Val 471 (4.62) b | Ser 535 (4.62) b Phe 583 (3.88) b | Asp 594 (3.1) a Phe 583 (3.85) b | Cys 532 (2.95) a |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El-Kalyoubi, S.A.; Gomaa, H.A.M.; Abdelhafez, E.M.N.; Ramadan, M.; Agili, F.; Youssif, B.G.M. Design, Synthesis, and Anti-Proliferative Action of Purine/Pteridine-Based Derivatives as Dual Inhibitors of EGFR and BRAFV600E. Pharmaceuticals 2023, 16, 716. https://doi.org/10.3390/ph16050716
El-Kalyoubi SA, Gomaa HAM, Abdelhafez EMN, Ramadan M, Agili F, Youssif BGM. Design, Synthesis, and Anti-Proliferative Action of Purine/Pteridine-Based Derivatives as Dual Inhibitors of EGFR and BRAFV600E. Pharmaceuticals. 2023; 16(5):716. https://doi.org/10.3390/ph16050716
Chicago/Turabian StyleEl-Kalyoubi, Samar A., Hesham A. M. Gomaa, Elshimaa M. N. Abdelhafez, Mohamed Ramadan, Fatimah Agili, and Bahaa G. M. Youssif. 2023. "Design, Synthesis, and Anti-Proliferative Action of Purine/Pteridine-Based Derivatives as Dual Inhibitors of EGFR and BRAFV600E" Pharmaceuticals 16, no. 5: 716. https://doi.org/10.3390/ph16050716
APA StyleEl-Kalyoubi, S. A., Gomaa, H. A. M., Abdelhafez, E. M. N., Ramadan, M., Agili, F., & Youssif, B. G. M. (2023). Design, Synthesis, and Anti-Proliferative Action of Purine/Pteridine-Based Derivatives as Dual Inhibitors of EGFR and BRAFV600E. Pharmaceuticals, 16(5), 716. https://doi.org/10.3390/ph16050716