
One-Pot Synthesis of 1-Thia-4-azaspiro[4.4/5]alkan-3-ones via Schiff Base: Design, Synthesis, and Apoptotic Antiproliferative Properties of Dual EGFR/BRAFV600E Inhibitors


Abstract
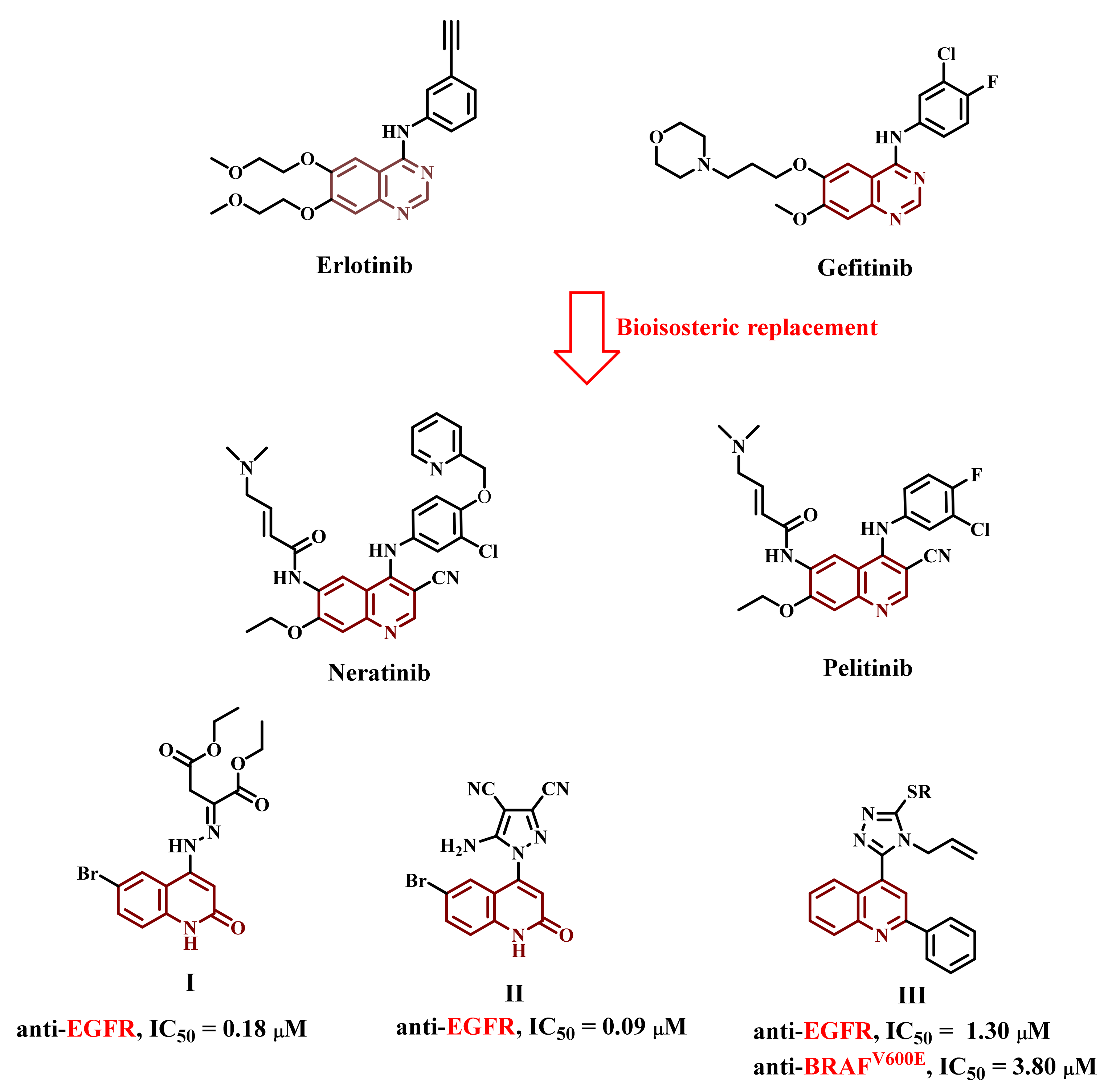
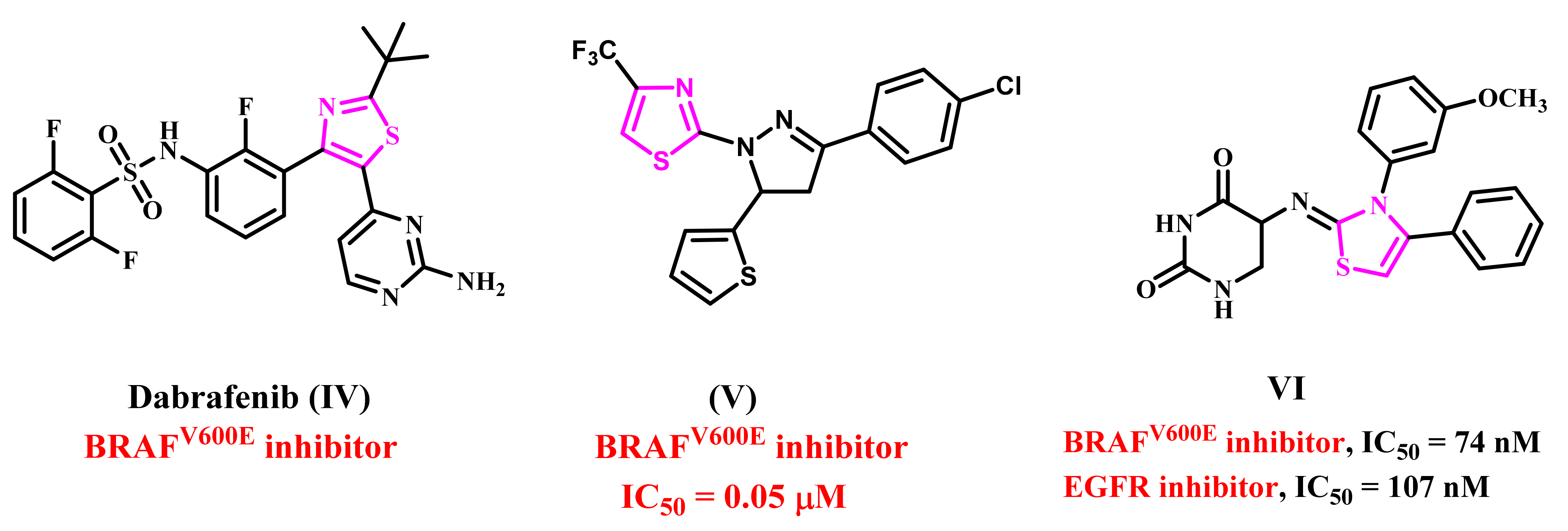
1. Introduction
2. Results and Discussion
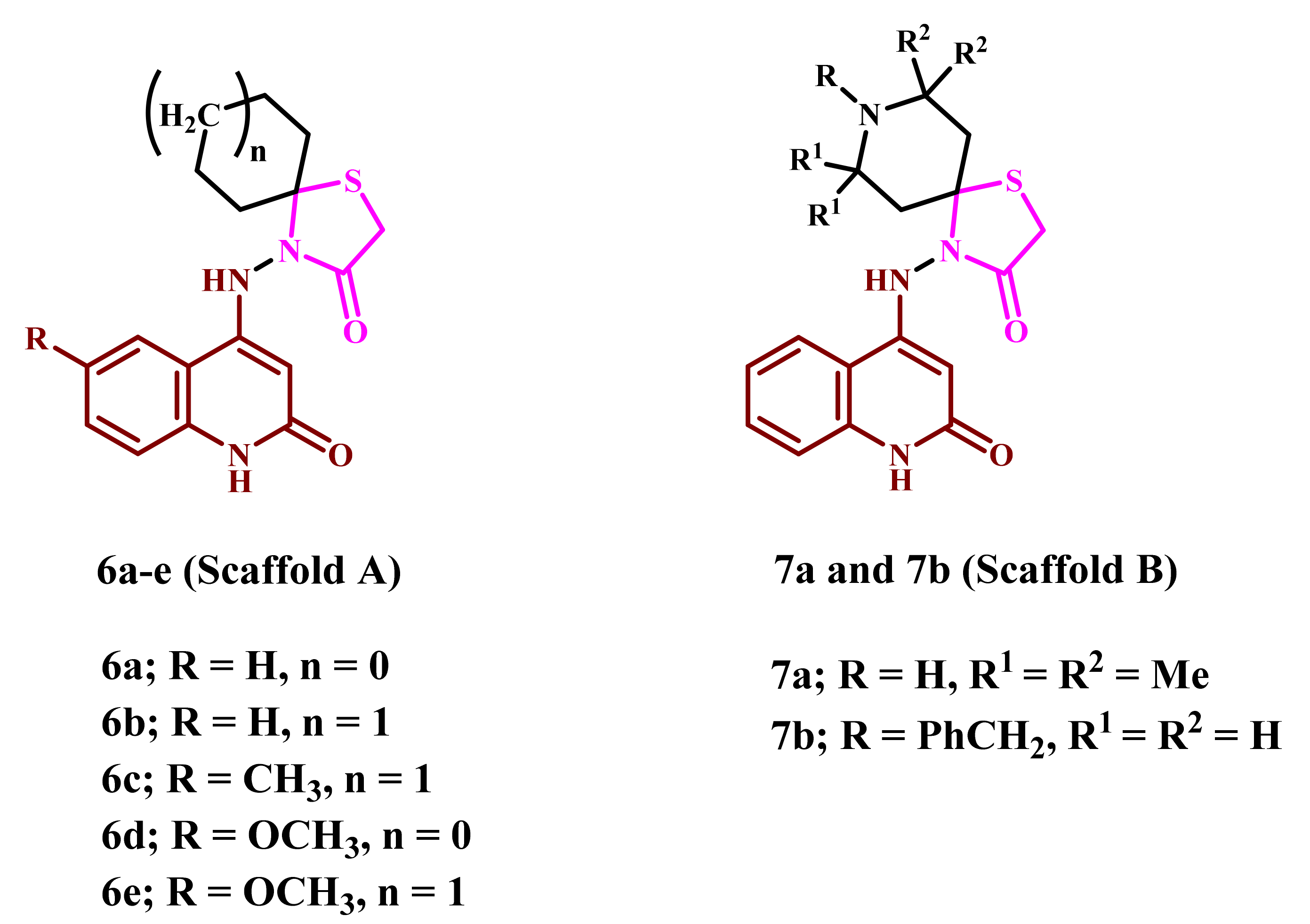
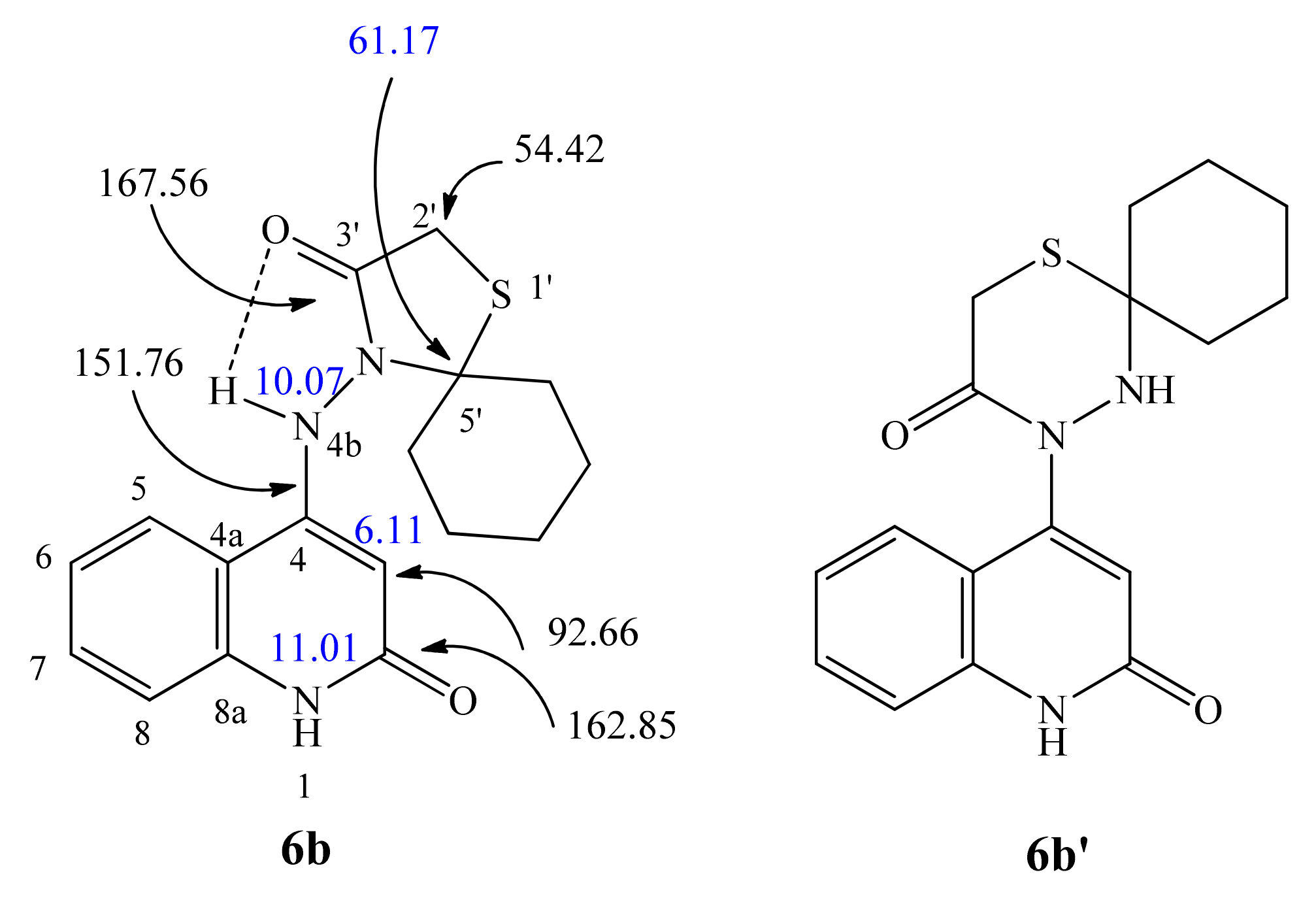
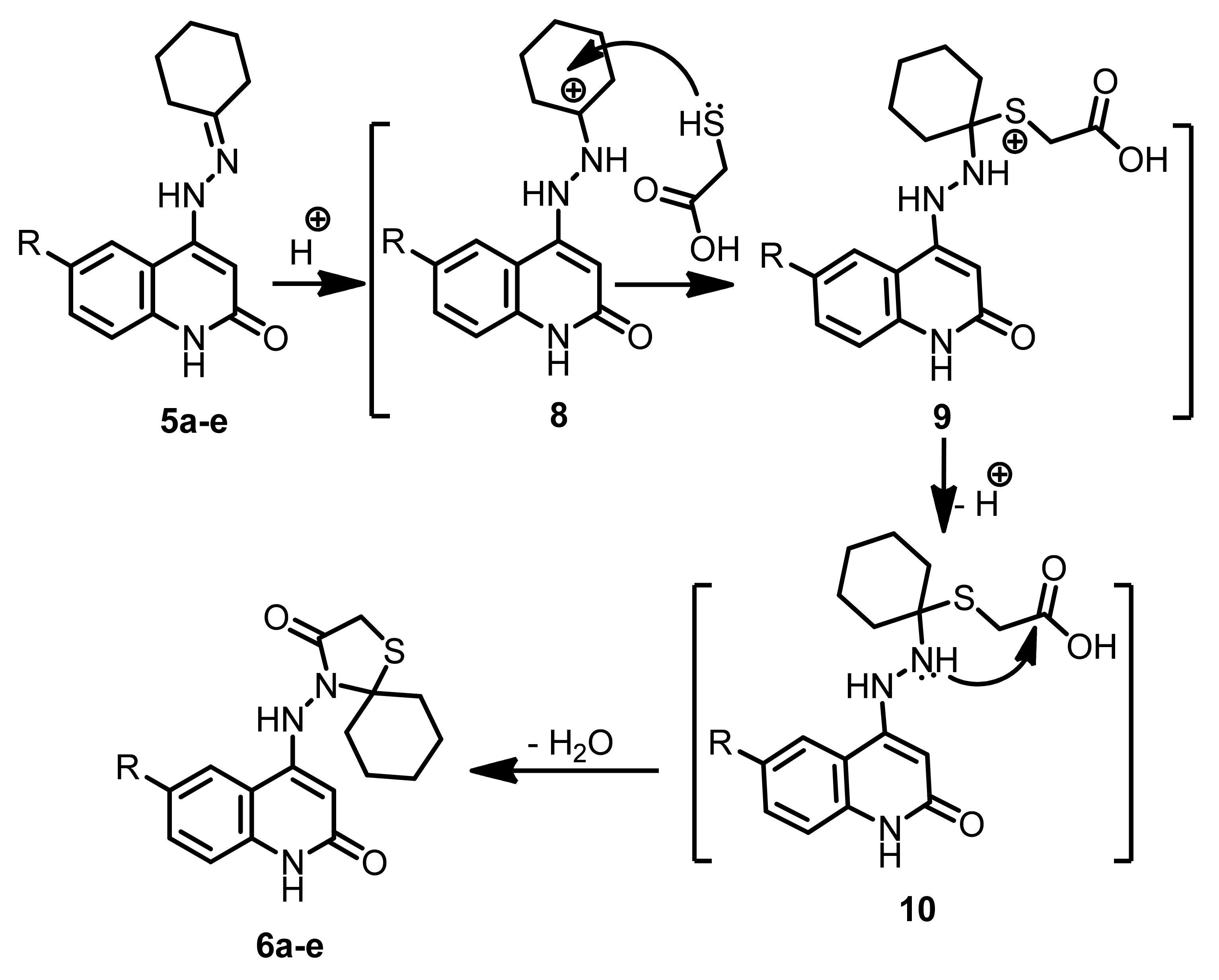
2.1. Chemistry
2.2. Biology
2.2.1. Cell Viability Assay
2.2.2. Antiproliferative Assay
2.2.3. EGFR Inhibitory Assay
2.2.4. BRAFV600E Inhibitory Assay
2.2.5. Apoptotic Markers Activation Assay
Caspase 3 Activation Assay
Caspase-8, Bax and Bcl-2 Levels Assay
3. Conclusions
4. Experimental
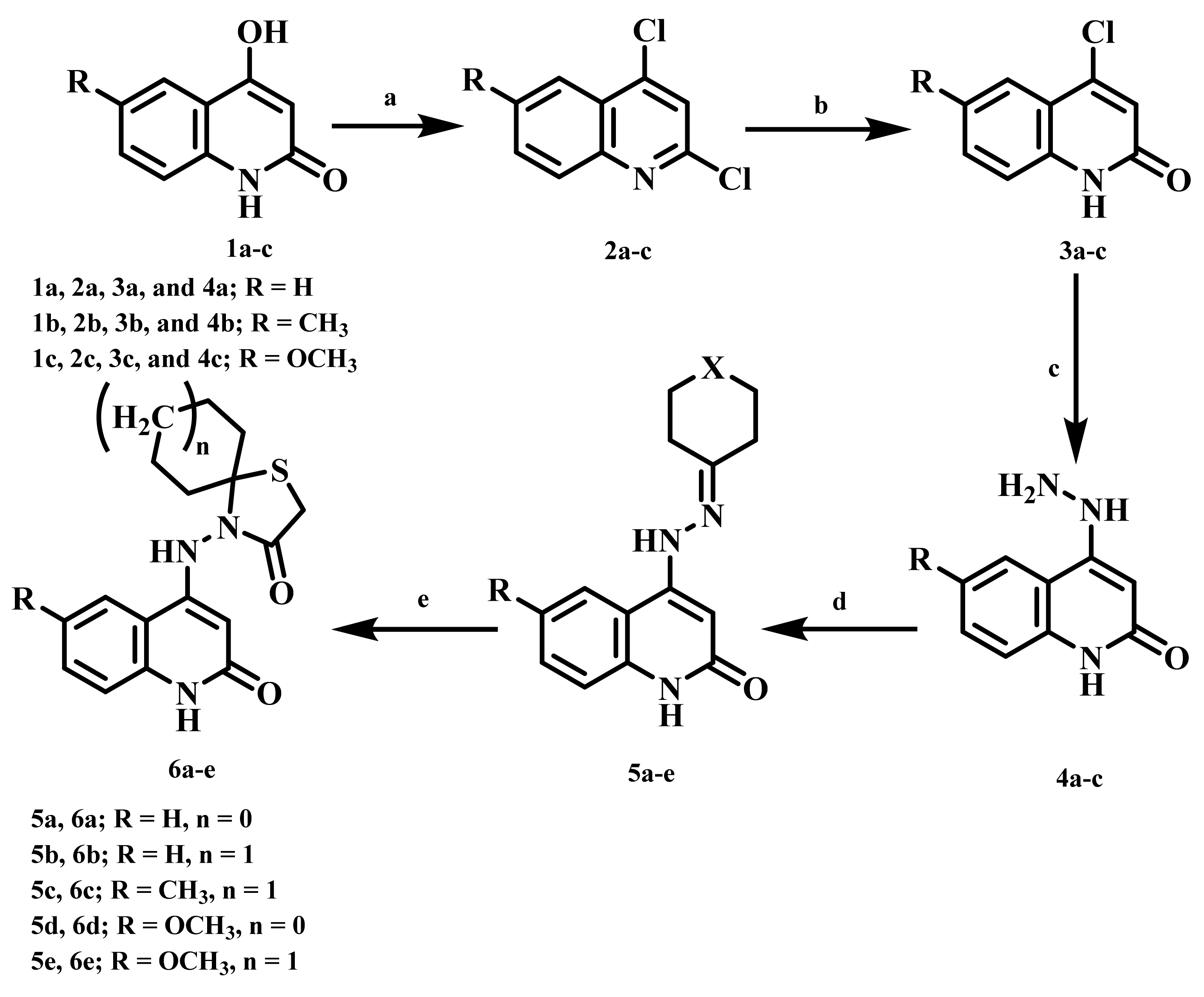
4.1. Chemistry
4.1.1. 4-((2-Oxo-1,2-dihydroquinolin-4-yl)amino)-1-thia-4-azaspiro[4.4]nonan-3-one (6a)
4.1.2. 4-((2-Oxo-1,2-dihydroquinolin-4-yl)amino)-1-thia-4-azaspiro[4.5]decan-3-one (6b)
4.1.3. 4-((6-Methyl-2-oxo-1,2-dihydroquinolin-4-yl)amino)-1-thia-4-azaspiro[4.5]decan-3-one (6c)
4.1.4. 4-((6-Methoxy-2-oxo-1,2-dihydroquinolin-4-yl)amino)-1-thia-4-azaspiro[4.4]nonan-3-one (6d)
4.1.5. 4-((6-Methoxy-2-oxo-1,2-dihydroquinolin-4-yl)amino)-1-thia-4-azaspiro[4.5]decan-3-one (6e)
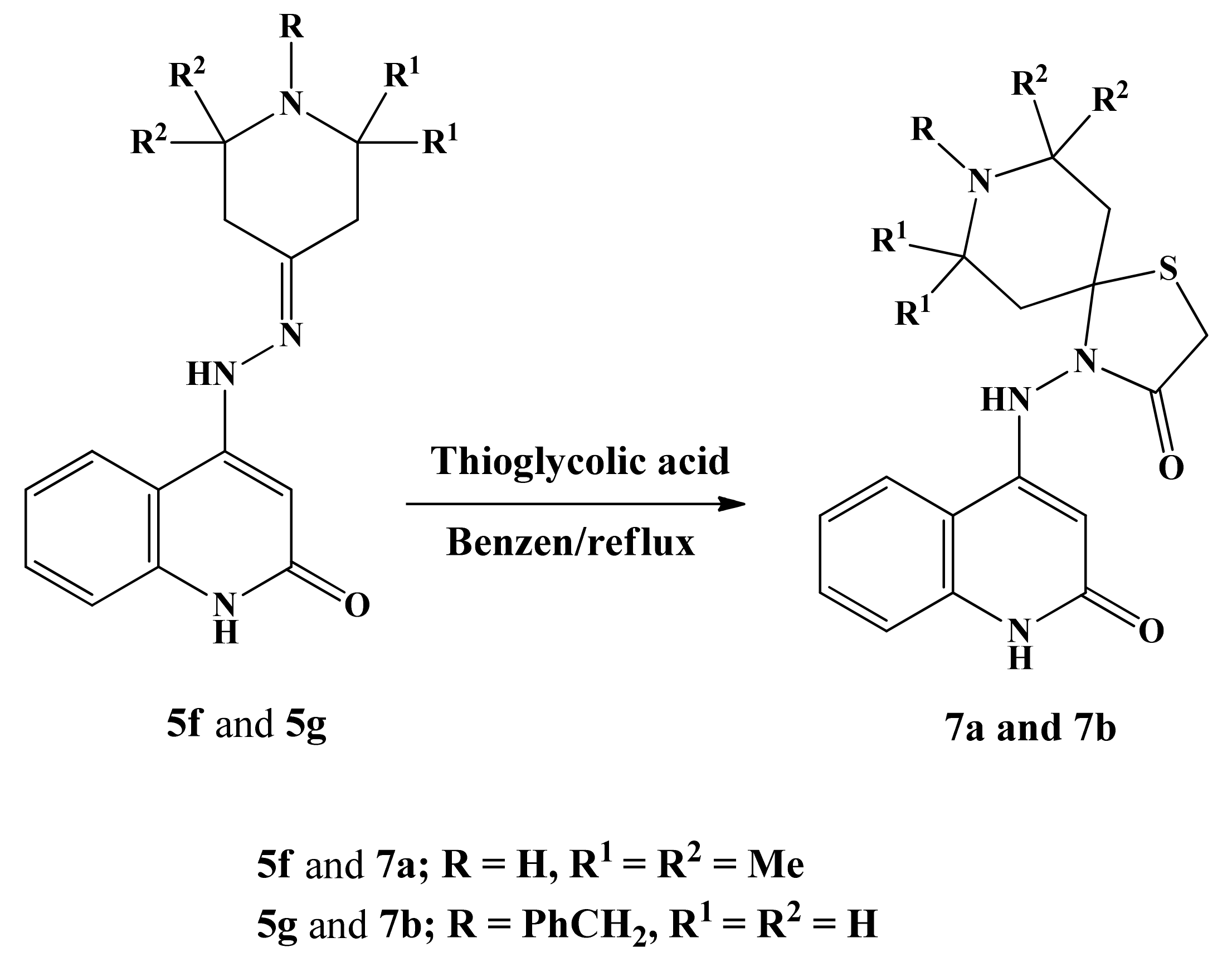
4.1.6. 7,7,9,9-Tetramethyl-4-((2-oxo-1,2-dihydroquinolin-4-yl)amino)-1-thia-4,8-diazaspiro-[4.5]decan-3-one (7a)
4.1.7. 8-Benzyl-4-((2-oxo-1,2-dihydroquinolin-4-yl)amino)-1-thia-4,8-diazaspiro[4.5]decan-3-one (7b)
4.2. Biology
4.2.1. Cell Viability Assay
4.2.2. Antiproliferative Assay
4.2.3. EGFR Inhibitory Assay
4.2.4. BRAFV600E Inhibitory Assay
4.2.5. Apoptotic Markers Assay
Caspase 3 Activation Assay
Caspase-8, Bax and Bcl-2 Levels Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stanković, T.; Dinić, J.; Podolski-Renić, A.; Musso, L.; Burić, S.S.; Dallavalle, S.; Pešić, M. Dual Inhibitors as a New Challenge for Cancer Multidrug Resistance Treatment. Curr. Med. Chem. 2019, 26, 6074–6106. [Google Scholar] [CrossRef] [PubMed]
- Raghavendra, N.M.; Pingili, D.; Kadasi, S.; Mettu, A.; Prasad, S.V.U.M. Dual or multi-targeting inhibitors: The next generation anti-cancer agents. Eur. J. Med. Chem. 2018, 143, 1277–1300. [Google Scholar] [CrossRef] [PubMed]
- Hughes, D.; Andersson, D.I. Evolutionary consequences of drug resistance: Shared principles across diverse targets and organisms. Nat. Rev. Genet. 2015, 16, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Staunton, J.E.; Jin, X.; Lee, M.S.; Zimmermann, G.R.; Borisy, A.A. Synergistic drug combinations tend to improve therapeutically relevant selectivity. Nat. Biotechnol. 2009, 27, 659–666. [Google Scholar] [CrossRef]
- Shah, K.N.; Bhatt, R.; Rotow, J.; Rohrberg, J.; Olivas, V.; Wang, V.E.; Hemmati, G.; Martins, M.M.; Maynard, A.; Kuhn, J.; et al. Aurora kinase A drives the evolution of resistance to third-generation EGFR inhibitors in lung cancer. Nat. Med. 2019, 25, 111–118. [Google Scholar] [CrossRef]
- Notarangelo, T.; Sisinni, L.; Condelli, V.; Landriscina, M. Dual EGFR and BRAF blockade overcomes resistance to vemurafenib in BRAF mutated thyroid carcinoma cells. Cancer Cell Int. 2017, 17, 86–94. [Google Scholar] [CrossRef]
- Mondaca, S.; Lacouture, M.; Hersch, J.; Yaeger, R. Balancing RAF, MEK, and EGFR inhibitor doses to achieve clinical responses and modulate toxicity in BRAF V600E colorectal cancer. JCO Precis. Oncol. 2018, 10, 1–5. [Google Scholar] [CrossRef]
- Zhang, Q.; Diao, Y.; Wang, F.; Fu, Y.; Tang, F.; You, Q.; Zhou, H. Design and discovery of 4-anilinoquinazoline ureas as multikinase inhibitors targeting BRAF, VEGFR-2 and EGFR. MedChemComm 2013, 4, 979–986. [Google Scholar] [CrossRef]
- Okaniwa, M.; Hirose, M.; Imada, T.; Ohashi, T.; Hayashi, Y.; Miyazaki, T.; Arita, T.; Yabuki, M.; Kakoi, K.; Kato, J.; et al. Design and synthesis of novel DFG-out RAF/vascular endothelial growth factor receptor 2 (VEGFR2) inhibitors. 1. Exploration of [5,6]-fused bicyclic scaffolds. J. Med. Chem. 2012, 55, 3452–3478. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Properties of FDA-approved small molecule protein kinase inhibitors. Pharmacol. Res. 2019, 144, 19–50. [Google Scholar] [CrossRef]
- Prajapati, S.M.; Patel, K.D.; Vekariya, R.H.; Panchal, S.N.; Patel, H.D. Recent advances in the synthesis of quinolines: A review. RSC Adv. 2014, 4, 24463–24476. [Google Scholar] [CrossRef]
- Navneetha, O.; Deepthi, K.; Rao, A.M.; Jyostna, T.S. A review on chemotherapeutic activities of quinolone. Int. J. Pharm. Chem. Biol. Sci. 2017, 7, 364–372. [Google Scholar]
- Barlési, F.; Tchouhadjian, C.; Doddoli, C.; Villani, P.; Greillier, L.; Kleisbauer, J.P.; Thomas, P.; Astoul, P. Gefitinib (ZD1839, Iressa®) in non-small-cell lung cancer: A review of clinical trials from a daily practice perspective. Fund. Clin. Pharmacol. 2005, 19, 385–393. [Google Scholar] [CrossRef]
- Iyer, R.; Bharthuar, A. A review of erlotinib–an oral, selective epidermal growth factor receptor tyrosine kinase inhibitor. Exp. Opin. Pharmacother. 2010, 11, 311–320. [Google Scholar] [CrossRef]
- Bao, B.; Mitrea, C.; Wijesinghe, P.; Marchetti, L.; Girsch, E.; Farr, R.L.; Boerner, J.L.; Mohammad, R.; Dyson, G.; Terlecky, S.R.; et al. Treating triple negative breast cancer cells with erlotinib plus a select antioxidant overcomes drug resistance by targeting cancer cell heterogeneity. Sci. Rep. 2017, 7, 44125. [Google Scholar] [CrossRef]
- Stamos, J.; Sliwkowski, M.X.; Eigenbrot, C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J. Biol. Chem. 2002, 277, 46265–46272. [Google Scholar] [CrossRef]
- Wissner, A.; Berger, D.M.; Boschelli, D.H.; Floyd, M.B.; Greenberger, L.M.; Gruber, B.C.; Johnson, B.D.; Mamuya, N.; Nilakantan, R.; Reich, M.F.; et al. 4-Anilino-6,7-dialkoxyquinoline-3-carbonitrile inhibitors of epidermal growth factor receptor kinase and their bioisosteric relationship to the 4-anilino-6,7-dialkoxyquinazoline inhibitors. J. Med. Chem. 2000, 43, 3244–3256. [Google Scholar] [CrossRef]
- Wissner, A.; Mansour, T.S. The development of HKI-272 and related compounds for the treatment of cancer. Arch. Pharm. 2008, 341, 465–477. [Google Scholar] [CrossRef]
- Kiesel, B.F.; Parise, R.A.; Wong, A.; Keyvanjah, K.; Jacobs, S.; Beumer, J.H. LC–MS/MS assay for the quantitation of the tyrosine kinase inhibitor neratinib in human plasma. J. Pharm. Biomed. Anal. 2017, 134, 130–136. [Google Scholar] [CrossRef]
- Pisaneschi, F.; Nguyen, Q.-D.; Shamsaei, E.; Glaser, M.; Robins, E.; Kaliszczak, M.; Smith, G.; Spivey, A.C.; Aboagye, E.O. Development of a new epidermal growth factor receptor positron emission tomography imaging agent based on the 3-cyanoquinoline core: Synthesis and biological evaluation. Bioorg. Med. Chem. 2010, 18, 6634–6645. [Google Scholar] [CrossRef]
- Lü, S.; Zheng, W.; Ji, L.; Luo, Q.; Hao, X.; Li, X.; Wang, F. Synthesis, characterization, screening and docking analysis of 4-anilinoquinazoline derivatives as tyrosine kinase inhibitors. Eur. J. Med. Chem. 2013, 61, 84–94. [Google Scholar] [CrossRef]
- Luethi, D.; Durmus, S.; Schinkel, A.H.; Schellens, J.H.M.; Beijnen, J.H.; Sparidans, R.W. Liquid chromatography–tandem mass spectrometry assay for the EGFR inhibitor pelitinib in plasma. J. Chromatogr. B 2013, 934, 22–25. [Google Scholar] [CrossRef] [PubMed]
- Pawar, V.G.; Sos, M.L.; Rode, H.B.; Rabiller, M.; Heynck, S.; van Otterlo, W.A.L.; Thomas, R.K.; Rauh, D. Synthesis and biological evaluation of 4-anilinoquinolines as potent inhibitors of epidermal growth factor receptor. J. Med. Chem. 2010, 53, 2892–2901. [Google Scholar] [CrossRef] [PubMed]
- Elbastawesy, M.A.I.; Aly, A.A.; Ramadan, M.; Elshaier, Y.A.M.M.; Youssif, B.G.M.; Brown, A.B.; Abuo-Rahma, G.E.A. Novel Pyrazoloquinolin-2-ones: Design, Synthesis, Docking Studies, and Biological Evaluation as Antiproliferative EGFR- TK Inhibitors. Bioorg. Chem. 2019, 90, 103045–103060. [Google Scholar] [CrossRef] [PubMed]
- Mohassab, A.M.; Hassan, H.A.; Abdelhamid, D.; Gouda, A.M.; Youssif, B.G.M.; Tateishi, H.; Fujita, M.; Otsuka, M.; Abdel-Aziz, M. Design and Synthesis of Novel quinoline/chalcone/1,2,4-triazole hybrids as potent antiproliferative agent targeting EGFR and BRAFV600E kinases. Bioorg. Chem. 2021, 106, 104510. [Google Scholar] [CrossRef]
- Zheng, Y.; Tice, C.M.; Singh, S.B. The use of spirocyclic scaffolds in drug discovery. Bioorg. Med. Chem. Lett. 2014, 24, 3673–3682. [Google Scholar] [CrossRef]
- Batista, V.F.; Pinto, D.C.G.A.; Silva, A.M.S. Recent in vivo advances of spirocyclic scaffolds for drug discovery. Expert Opin. Drug Discov. 2022, 17, 603–618. [Google Scholar] [CrossRef]
- Hiesinger, K.; Dar’in, D.; Proschak, E.; Krasavin, M. Spirocyclic Scaffolds in Medicinal Chemistry. J. Med. Chem. 2021, 64, 150–183. [Google Scholar] [CrossRef]
- Ayati, A.; Emami, S.; Asadipour, A.; Shafiee, A.; Foroumadi, A. Recent applications of 1,3-thiazole core structure in the identification of new lead compounds and drug discovery. Eur. J. Med. Chem. 2015, 97, 699–718. [Google Scholar] [CrossRef]
- Sharma, P.C.; Jain, A.; Yar, M.S.; Pahwa, R.; Singh, J.; Chanalia, P. Novel fluoroquinolone derivatives bearing N-thiomide linkage with 6-substituted-2-aminobenzothiazoles: Synthesis and antibacterial evaluation. Arab. J. Chem. 2017, 10, S568–S575. [Google Scholar] [CrossRef]
- Petrou, A.; Fesatidou, M.; Geronikaki, A. Thiazole Ring—A Biologically Active Scaffold. Molecules 2021, 26, 3166. [Google Scholar] [CrossRef]
- Othman, I.M.M.; Alamshany, Z.M.; Tashkandi, N.Y.; Gad-Elkareem, M.A.M.; Abd El-Karim, S.S.; Nossier, E.S. Synthesis and biological evaluation of new derivatives of thieno-thiazole and dihydrothiazolo-thiazole scaffolds integrated with a pyrazoline nucleus as anticancer and multi-targeting kinase inhibitor. RSC Adv. 2022, 12, 561–577. [Google Scholar] [CrossRef]
- Nafie, M.S.; Kishk, S.M.; Mahgoub, S.; Amer, A.M. Quinoline-based thiazolidinone derivatives as potent cytotoxic and apoptosis-inducing agents through EGFR inhibition. Chem. Biol. Drug Des. 2022, 99, 547–560. [Google Scholar] [CrossRef]
- Xiong, L.; He, H.; Fan, M.; Hu, L.; Wang, F.; Song, X.; Shi, S.; Qi, B. Discovery of novel conjugates of quinoline and thiazolidinone urea as potential anti-colorectal cancer agent. J. Enzym. Inhib. Med. Chem. 2022, 37, 2334–2347. [Google Scholar] [CrossRef]
- Yadav, J.; Chaudhary, R.P. A review on advances in synthetic methodology and biological profile of spirothiazolidin-4-ones. J. Heterocycl. Chem. 2022, 59, 1839–1878. [Google Scholar] [CrossRef]
- Lozynskyi, A.; Zimenkovsky, B.; Lesyk, R. Synthesis and Anticancer Activity of New Thiopyrano[2,3-d]thiazoles Based on Cinnamic Acid Amides. Sci. Pharm. 2014, 82, 723–733. [Google Scholar] [CrossRef]
- Hu-Lieskovan, S.; Mok, S.; Homet Moreno, B.; Tsoi, J.; Robert, L.; Goedert, L.; Pinheiro, E.; Koya, R.; Graeber, T.; Comin-Anduix, B.; et al. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAFV600E melanoma. Sci. Transl. Med. 2015, 7, 279ra41. [Google Scholar] [CrossRef]
- Abdel-Maksoud, M.S.; Kim, M.-R.; El-Gamal, M.I.; Gamal El-Din, M.M.; Tae, J.; Choi, H.S.; Lee, K.-T.; Yoo, K.H.; Oh, C.-H. Design, synthesis, in vitro antiproliferative evaluation, and kinase inhibitory effects of a new series of imidazo[2,1-b]thiazole derivatives. Eur. J. Med. Chem. 2015, 95, 453–463. [Google Scholar] [CrossRef]
- Aly, A.A.; Alshammari, M.B.; Ahmad, A.; Gomaa, H.A.M.; Youssif, B.G.M.; Bräse, S.; Ibrahim, M.A.A.; Mohamed, A.H. Design, synthesis, docking, and mechanistic studies of new thiazolyl/thiazolidinylpyrimidine-2,4-dione antiproliferative agents. Arab. J. Chem. 2023, 16, 104612. [Google Scholar] [CrossRef]
- Al-Wahaibi, L.H.; Gouda, A.M.; Abou-Ghadir, O.F.; Salem, O.I.A.; Ali, A.T.; Farghaly, H.S.; Abdelrahman, M.H.; Trembleau, L.; Abdu-Allah, H.H.M.; Youssif, B.G.M. Design and synthesis of novel 2,3-dihydropyrazino[1,2-a]indole-1,4-dione derivatives as antiproliferative EGFR and BRAFV600E dual inhibitors. Bioorg. Chem. 2020, 104, 104260. [Google Scholar] [CrossRef]
- Gomaa, H.A.M.; Shaker, M.E.; Alzarea, S.I.; Hendawy, O.M.; Mohamed, F.A.M.; Gouda, A.M.; Ali, A.T.; Morcoss, M.M.; Abdelrahman, M.H.; Trembleau, L.; et al. Optimization and SAR investigation of novel 2,3-dihydropyrazino[1,2-a]indole-1,4-dione derivatives as EGFR and BRAFV600E dual inhibitors with potent antiproliferative and antioxidant activities. Bioorg. Chem. 2022, 120, 105616. [Google Scholar] [CrossRef] [PubMed]
- Alshammari, M.B.; Aly, A.A.; Youssif, B.G.M.; Bräse, S.; Ahmad, A.; Brown, A.B.; Ibrahim, M.A.A.; Mohamed, A.H. Design and synthesis of new thiazolidinone/uracil derivatives as antiproliferative agents targeting EGFR and/or BRAFV600E. Front. Chem. 2022, 10, 1076383. [Google Scholar] [CrossRef] [PubMed]
- Buckle, D.R.; Cantello, B.C.C.; Smith, H.; Spicer, B.A. 4-Hydroxy-3-nitro-2-quinolones and related compounds as inhibitors of allergic reactions. J. Med. Chem. 1975, 18, 726–732. [Google Scholar] [CrossRef] [PubMed]
- Bhudevi, B.; Ramana, P.V.; Mudiraj, A.; Reddy, A.R. Synthesis of 4-hydroxy-3-formylideneamino-lH/methyl/phenylquinolin2-ones. Indian J. Chem. B 2009, 48, 255–260. [Google Scholar]
- Yang, Z.; Sun, P. Compare of three ways of synthesis of simple Schiff bas. Molbank 2006, 2006, M514. [Google Scholar] [CrossRef]
- Mahmoud, M.A.; Mohammed, A.F.; Salem, O.I.A.; Gomaa, H.A.M.; Youssif, B.G.M. New 1,3,4-oxadiazoles linked 1,2,3-triazole moiety as antiproliferative agents targeting EGFR-TK. Arch. Der Pharm. 2022, 355, e2200009. [Google Scholar] [CrossRef]
- Ramadan, M.; Abd El-Aziz, M.; Elshaier, Y.A.M.M.; Youssif, B.G.M.; Brown, A.B.; Fathy, H.M.; Aly, A.A. Design and synthesis of new pyranoquinolinone heteroannulated to triazolopyrimidine of potential apoptotic antiproliferative activity. Bioorg. Chem. 2020, 105, 104392. [Google Scholar] [CrossRef]
- Al-Sanea, M.M.; Gotina, L.; Mohamed, M.F.A.; Parambi, D.G.T.; Gomaa, H.A.M.; Mathew, B.; Youssif, B.G.M.; Alharbi, K.S.; Elsayed, Z.M.; Abdelgawad, M.A.; et al. Design, Synthesis and Biological Evaluation of New HDAC1 and HDAC2 Inhibitors Endowed with Ligustrazine as a Novel Cap Moiety. Drug Des. Dev. Ther. 2020, 14, 497–508. [Google Scholar] [CrossRef]
- Hisham, M.; Hassan, H.A.; Gomaa, H.A.M.; Youssif, B.G.M.; Hayallah, A.M.; Abdel-Aziz, M. Structure-based design, synthesis and antiproliferative action of new quinazoline-4-one/chalcone hybrids as EGFR inhibitors. J. Mol. Struct. 2022, 1254, 132422. [Google Scholar] [CrossRef]
- Mohamed, F.A.M.; Gomaa, H.A.M.; Hendawy, O.M.; Ali, A.T.; Farghaly, H.S.; Gouda, A.M.; Abdelazeem, A.H.; Abdelrahman, M.H.; Trembleau, L.; Youssif, B.G.M. Design, synthesis, and biological evaluation of novel EGFR inhibitors containing 5-chloro-3-hydroxymethyl-indole-2-carboxamide scaffold with apoptotic antiproliferative activity. Bioorg. Chem. 2021, 112, 104960. [Google Scholar] [CrossRef]
- Abou-Zied, H.A.; Beshr, E.A.M.; Gomaa, H.A.M.; Mostafa, Y.A.; Youssif, B.G.M.; Hayallah, A.M.; Abdel-Aziz, M. Discovery of new cyanopyridine/chalcone hybrids as dual inhibitors of EGFR/BRAFV600E with promising antiproliferative properties. Arch. Der Pharm. 2022, 355, e2200464. [Google Scholar] [CrossRef]
- Hisham, M.; Youssif, B.G.M.; Osman, E.E.A.; Hayallah, A.M.; Abdel-Aziz, M. Synthesis and biological evaluation of novel xanthine derivatives as potential apoptotic antitumor agents. Eur. J. Med. Chem. 2019, 176, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.J. Caspases: Executioners of apoptosis. Pathobiol. Hum. Dis. 2014, 145, 145–152. [Google Scholar]
- Abdelbaset, M.S.; Abdel-Aziz, M.; Abuo-Rahma, G.E.A.; Abdelrahman, M.H.; Ramadan, M.; Youssif, B.G.M. Novel quinoline derivatives carrying nitrones/oximes nitric oxide donors: Design, synthesis, antiproliferative and caspase-3 activation activities. Arch. Der Pharm. 2018, 352, 1800270. [Google Scholar] [CrossRef] [PubMed]
- Abou-Zied, H.A.; Youssif, B.G.M.; Mohamed, M.F.A.; Hayallah, A.M.; Abdel-Aziz, M. EGFR inhibitors and apoptotic inducers: Design, synthesis, anticancer activity and docking studies of novel xanthine derivatives carrying chalcone moiety as hybrid molecules. Bioorg. Chem. 2019, 89, 102997. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
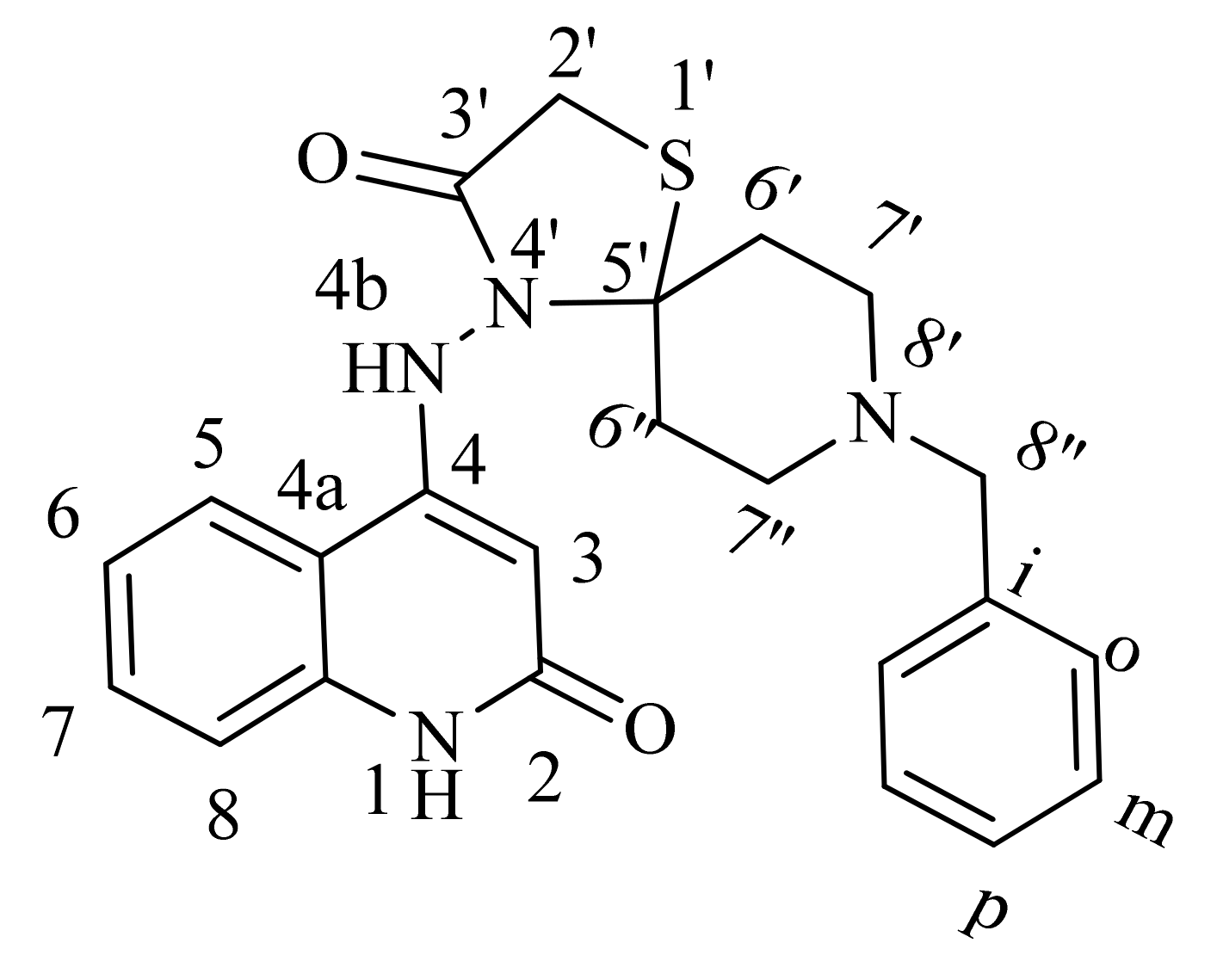
| 1H NMR | 1H-1H COSY | Assignment | |
|---|---|---|---|
| 11.01 (bs; 1H) | 6.08 | NH-1 | |
| 10.09 (s; 1H) | NH-4b | ||
| 7.98 (d, J = 8.1; 1H) | 7.46, 7.12 | H-5 | |
| 7.46 (dd, J = 7.7, 7.5; 1H) | 7.98, 7.28, 7.12 | H-7 | |
| 7.35 (m; 4H) | 7.28 | H-o, m | |
| 7.28 (m; 3H) | 7.46, 7.35, 7.12 | H-8, p | |
| 7.12 (dd, J = 7.7, 7.4; 1H) | 7.98, 7.46, 7.28 | H-6 | |
| 6.08 (s; 1H) | 11.01 | H-3 | |
| 3.71 (s; 2H) | H-2′ | ||
| 3.56 (s; 2H) | H-8″ | ||
| 2.67 (t, J = 5.6; 2H) | 2.55, 2.44 | H-6′/6″ | |
| 2.55 (m; 4H) | 2.67, 2.44 | H-7′,7″ | |
| 2.44 (t, J = 5.4; 2H) | 2.67, 2.55 | H-6″/6′ | |
| 13C NMR | HSQC | HMBC | Assignment |
| 167.33 | 10.09, 3.71 | C-3′ | |
| 162.90 | 10.09, 6.08 | C-2 | |
| 149.51 | 10.09, 7.98, 7.28, 6.08 | C-4 | |
| 139.23 | 7.98, 7.46 | C-8a | |
| 138.31 | 7.35, 7.28, 3.56 | C-i | |
| 130.13 | 7.46 | 7.98, 7.46, 7.28 | C-7 |
| 128.73 | 7.35 | 7.35, 7.35, 3.56 | C-o |
| 128.16 | 7.35 | 7.35, 7.35 | C-m |
| 126.93 | 7.28 | 7.28 | C-p |
| 122.44 | 7.98 | 7.98, 7.46, 6.08 | C-5 |
| 120.31 | 7.12 | 7.28, 7.12 | C-6 |
| 115.45 | 7.28 | 7.46, 7.12 | C-8 |
| 112.26 | 10.09, 7.98, 7.28, 7.12, 6.08 | C-4a | |
| 92.33 | 6.08 | 10.09, 6.08 | C-3 |
| 61.36 | 3.56, 2.55 | C-8″ | |
| 54.11 | 3.71 | 2.67, 244 | C-2′ |
| 53.23, 52.07 | 2.55 | 3.56, 2.67, 2.55, 2.55, 2.44 | C-7′,7″ |
| 47.33 | 3.56 | 2.67, 2.44 | C-5′ |
| 34.46 | 2.44 | 2.67, 2.55 | C-6′/6″ |
| 27.26 | 2.67 | 2.67, 2.55, 2.55 | C-6″/6′ |
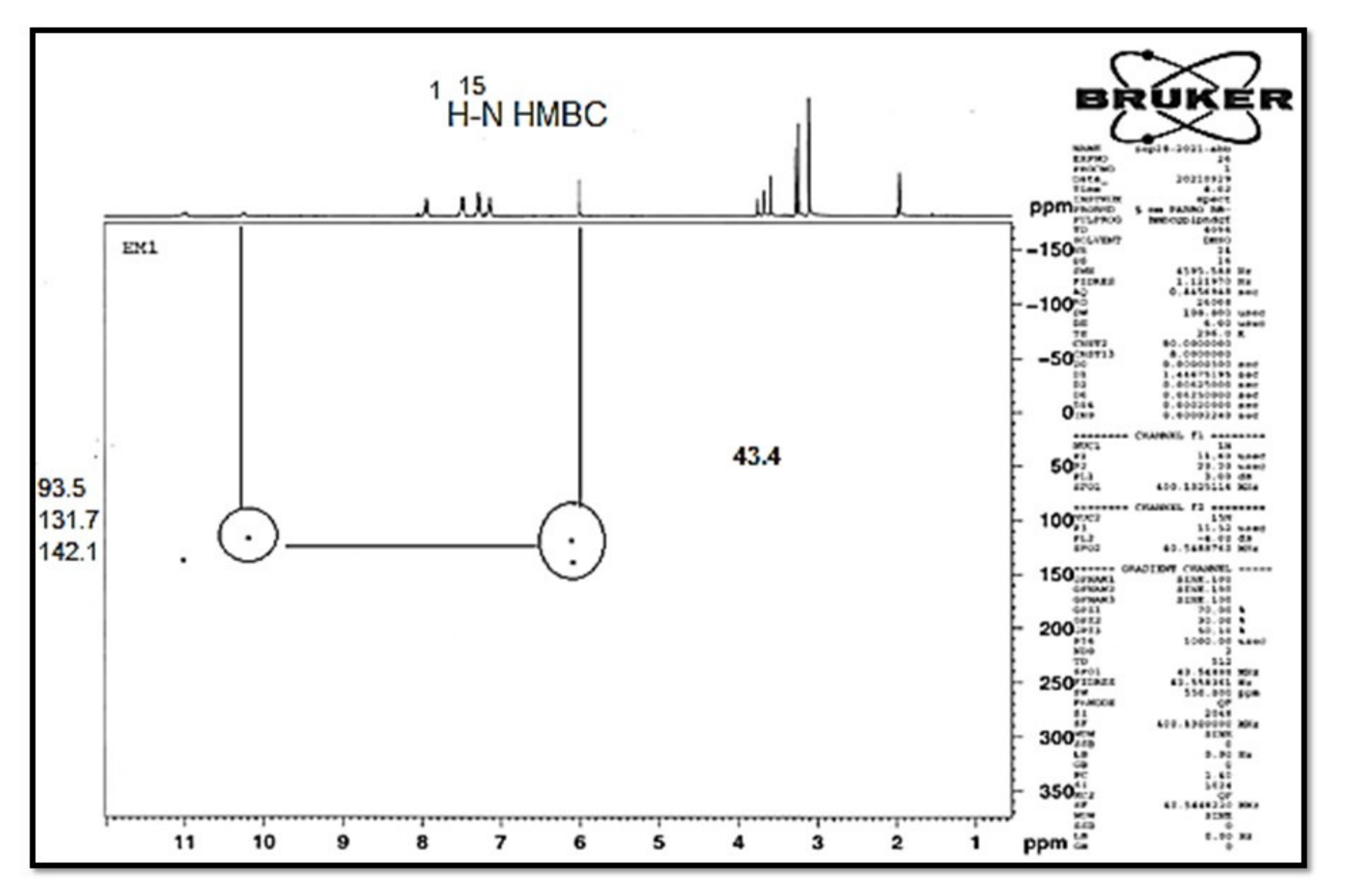
| 15N NMR: | HSQC | HMBC | Assignment |
| 141.9 | 11.00 | 7.28, 6.08 | N-1 |
| 132.0 | 10.09 | 10.09, 6.08 | N-4b |
| 49.7 | 2.67, 2.44 | N-8′ | |
| Compd. | Cell Viability % | Antiproliferative Activity IC50 ± SEM (µM) | ||||
|---|---|---|---|---|---|---|
| A-549 | MCF-7 | Panc-1 | HT-29 | Average (GI50) | ||
| 6a | 90 | 40 ± 4 | 44 ± 4 | 46 ± 4 | 48 ± 4 | 45 |
| 6b | 89 | 33 ± 3 | 35 ± 3 | 36 ± 3 | 36 ± 3 | 35 |
| 6c | 91 | 72 ± 7 | 74 ± 7 | 73 ± 7 | 72 ± 7 | 73 |
| 6d | 91 | 48 ± 4 | 52 ± 5 | 54 ± 5 | 54 ± 5 | 52 |
| 6e | 92 | 36 ± 3 | 40 ± 3 | 42 ± 4 | 42 ± 4 | 40 |
| 7a | 89 | 80 ± 8 | 82 ± 8 | 81 ± 8 | 81 ± 8 | 81 |
| 7b | 91 | 30 ± 3 | 34 ± 3 | 32 ± 3 | 32 ± 3 | 32 |
| Erlotinib | ND | 30 ± 3 | 40 ± 3 | 30 ± 3 | 30 ± 3 | 33 |
| Compd. | EGFR Inhibition IC50 ± SEM (nM) | BRAFV600E Inhibition IC50 ± SEM (nM) |
|---|---|---|
| 6a | 97 ± 07 | 137 ± 12 |
| 6b | 84 ± 06 | 108 ± 09 |
| 6c | 135 ± 11 | 164 ± 15 |
| 6d | 104 ± 08 | 159 ± 14 |
| 6e | 92 ± 07 | 129 ± 10 |
| 7a | 149 ± 12 | 187 ± 16 |
| 7b | 78 ± 05 | 96 ± 8 |
| Erlotinib | 80 ± 05 | 60 ± 05 |
| Compound Number | Caspase-3 | |
|---|---|---|
| Conc (Pg/mL) | Fold Change | |
| 6b | 487.50 ± 4 | 7.50 |
| 7b | 544.50 ± 5 | 8.5 |
| Staurosporine | 503.00 ± 4 | 8.0 |
| Control | 65.50 | 1 |
| Compound Number | Caspase-8 | Bax | Bcl-2 | |||
|---|---|---|---|---|---|---|
| Conc (ng/mL) | Fold Change | Conc (Pg/mL) | Fold Change | Conc (ng/mL) | Fold Reduction | |
| 6b | 1.50 | 17 | 220 | 28 | 1.30 | 4 |
| 7b | 1.90 | 21 | 295 | 37 | 1.00 | 5 |
| Staurosporine | 1.80 | 20 | 280 | 35 | 1.10 | 5 |
| Control | 0.09 | 1 | 8 | 1 | 5 | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Wahaibi, L.H.; El-Sheref, E.M.; Hammouda, M.M.; Youssif, B.G.M. One-Pot Synthesis of 1-Thia-4-azaspiro[4.4/5]alkan-3-ones via Schiff Base: Design, Synthesis, and Apoptotic Antiproliferative Properties of Dual EGFR/BRAFV600E Inhibitors. Pharmaceuticals 2023, 16, 467. https://doi.org/10.3390/ph16030467
Al-Wahaibi LH, El-Sheref EM, Hammouda MM, Youssif BGM. One-Pot Synthesis of 1-Thia-4-azaspiro[4.4/5]alkan-3-ones via Schiff Base: Design, Synthesis, and Apoptotic Antiproliferative Properties of Dual EGFR/BRAFV600E Inhibitors. Pharmaceuticals. 2023; 16(3):467. https://doi.org/10.3390/ph16030467
Chicago/Turabian StyleAl-Wahaibi, Lamya H., Essmat M. El-Sheref, Mohamed M. Hammouda, and Bahaa G. M. Youssif. 2023. "One-Pot Synthesis of 1-Thia-4-azaspiro[4.4/5]alkan-3-ones via Schiff Base: Design, Synthesis, and Apoptotic Antiproliferative Properties of Dual EGFR/BRAFV600E Inhibitors" Pharmaceuticals 16, no. 3: 467. https://doi.org/10.3390/ph16030467
APA StyleAl-Wahaibi, L. H., El-Sheref, E. M., Hammouda, M. M., & Youssif, B. G. M. (2023). One-Pot Synthesis of 1-Thia-4-azaspiro[4.4/5]alkan-3-ones via Schiff Base: Design, Synthesis, and Apoptotic Antiproliferative Properties of Dual EGFR/BRAFV600E Inhibitors. Pharmaceuticals, 16(3), 467. https://doi.org/10.3390/ph16030467