
Design of D-Amino Acids SARS-CoV-2 Main Protease Inhibitors Using the Cationic Peptide from Rattlesnake Venom as a Scaffold

 , , , , ,
, , , , ,  , and
, and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Preparation of SARS-CoV-2 3CLpro
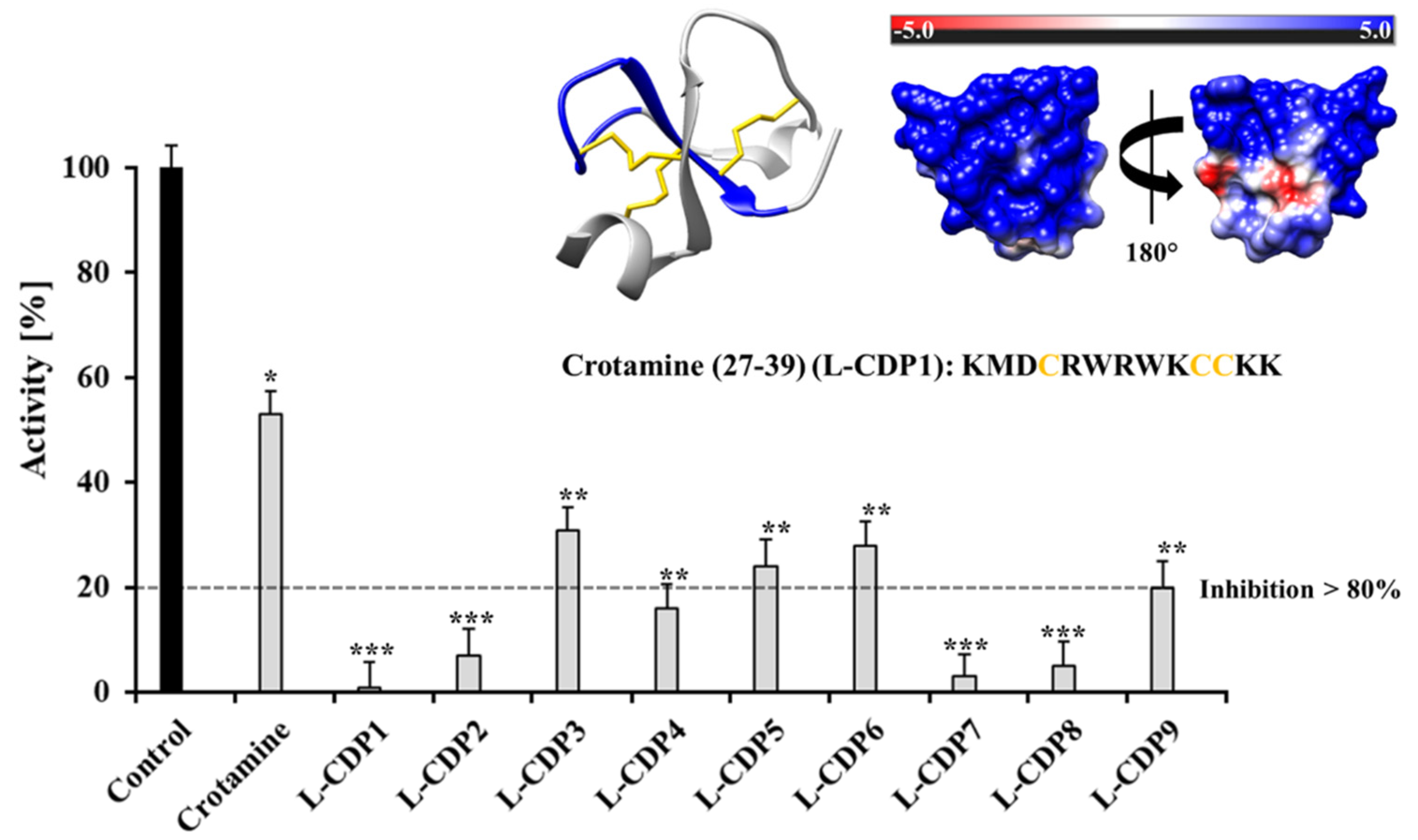
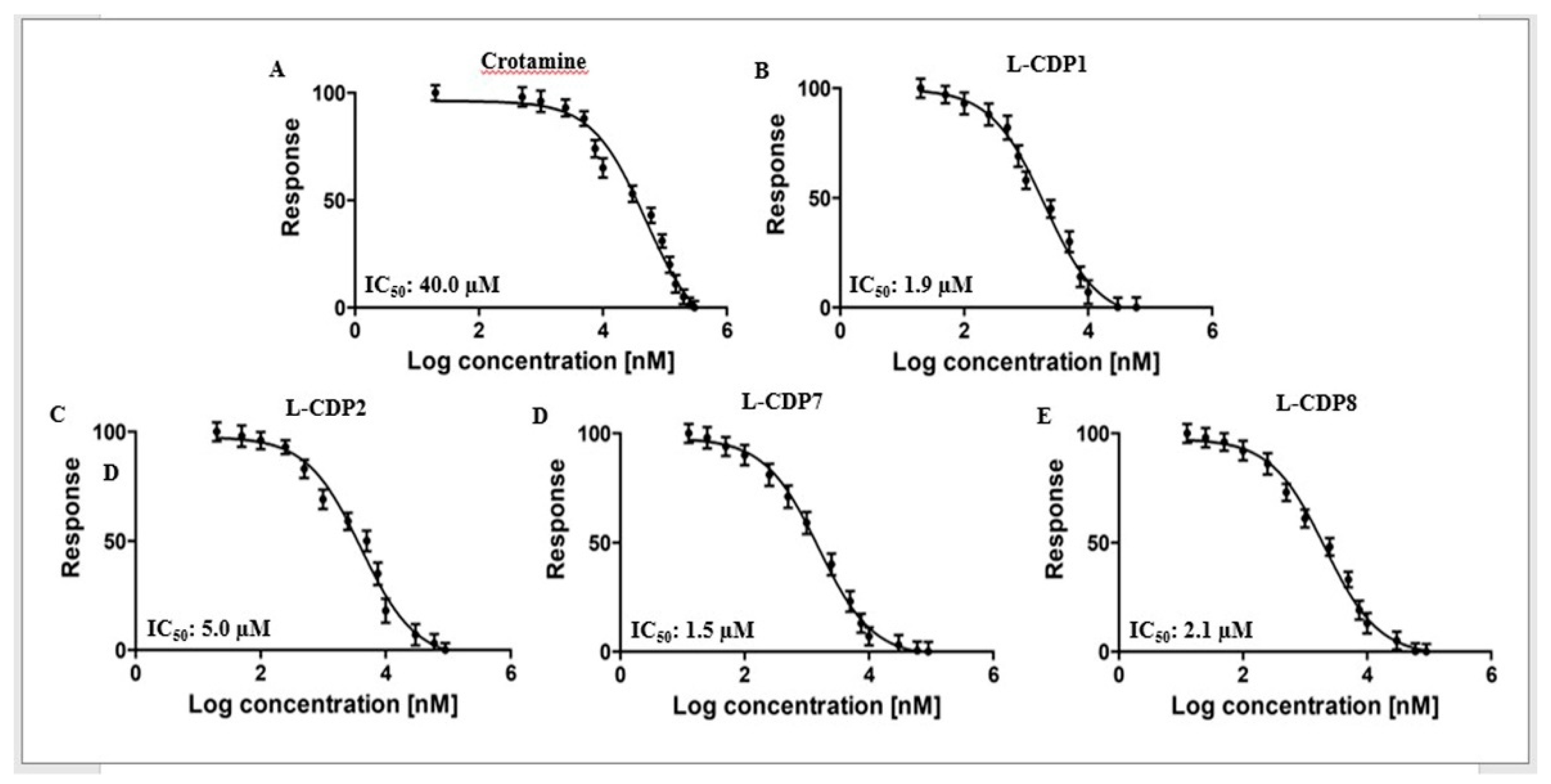
2.2. Primary Inhibition Assay of Crotamine and L-CDPs against SARS-CoV-2 3CLpro
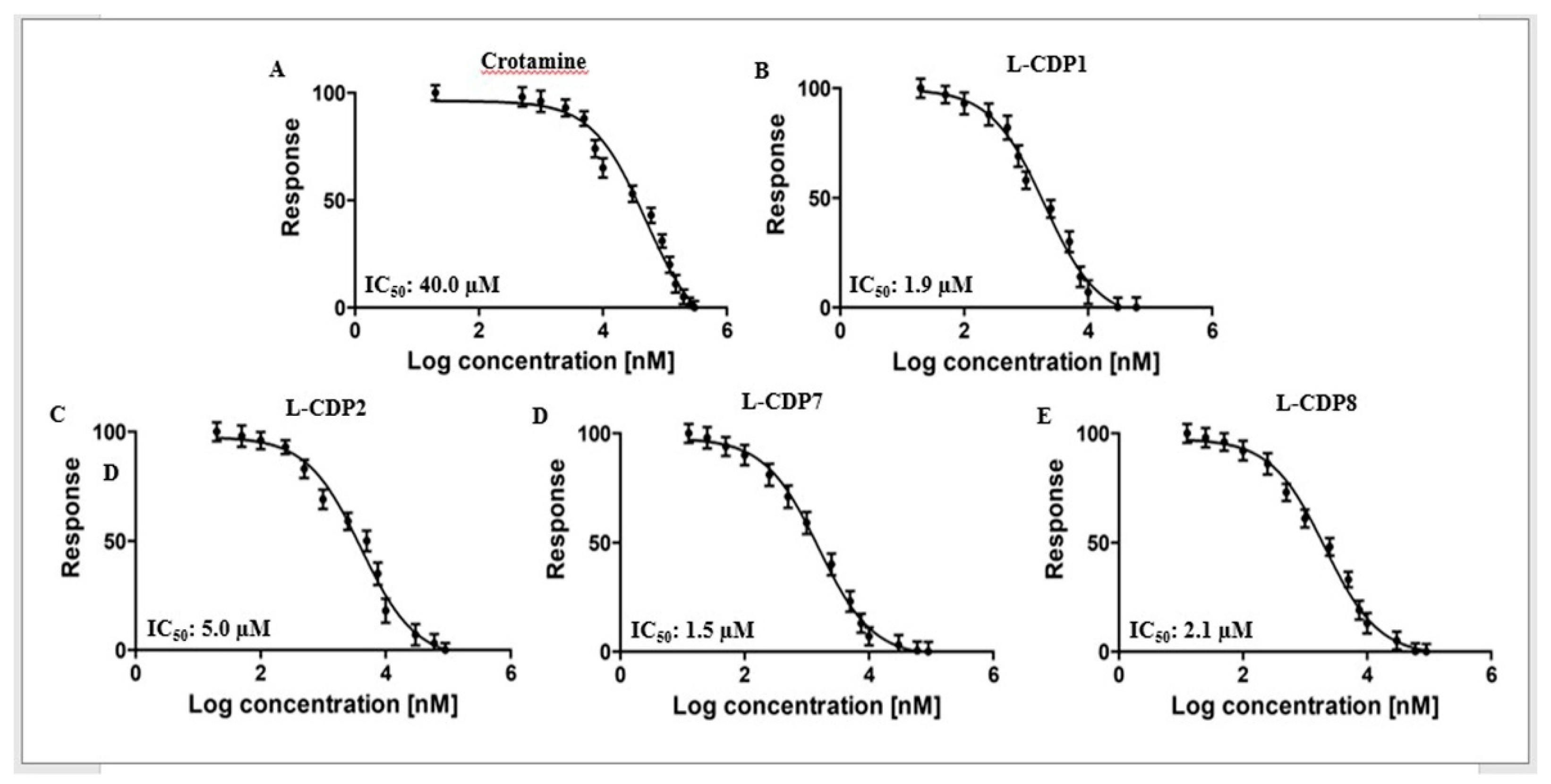
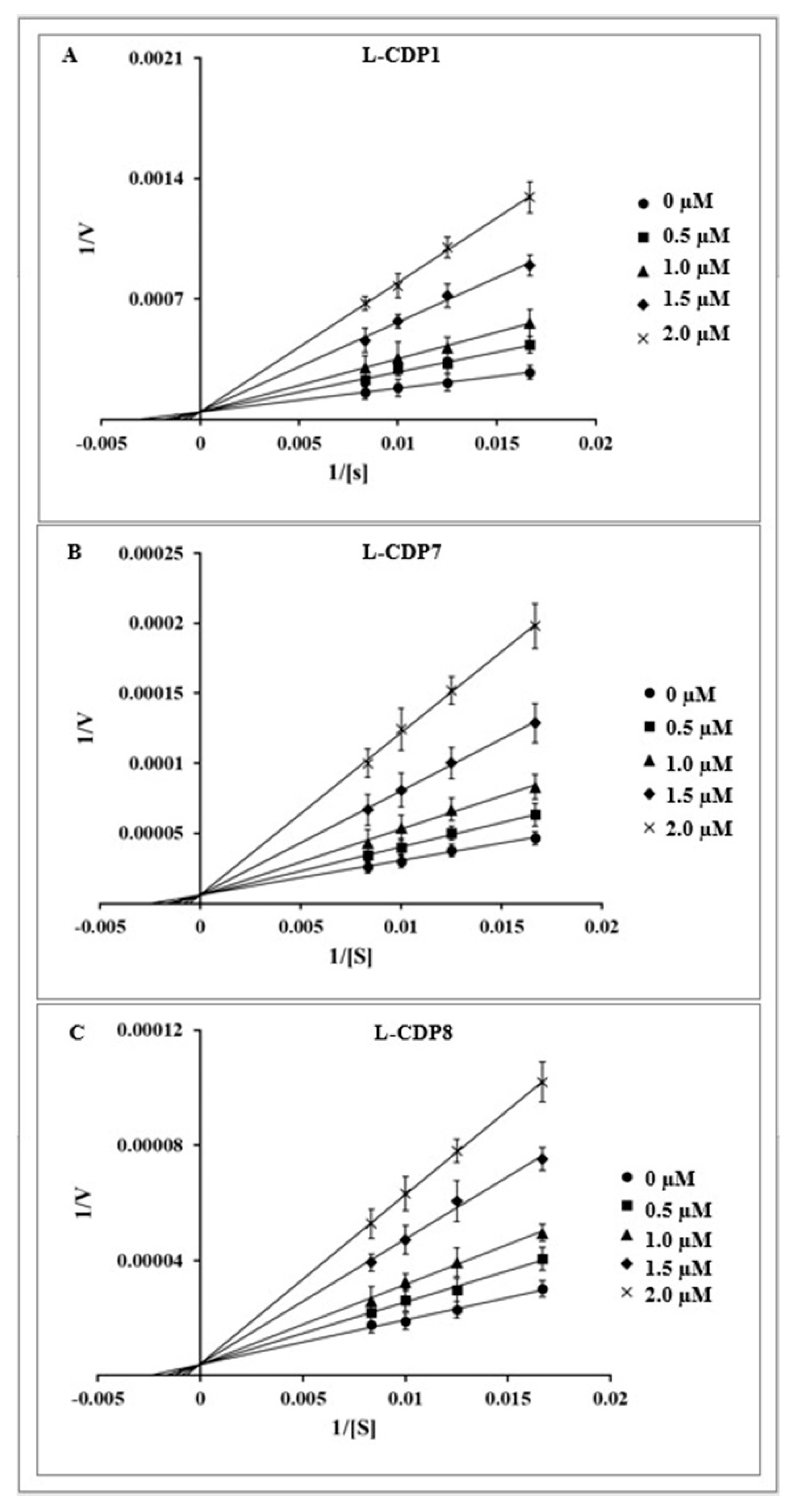
2.3. Characterisation of the 3CLpro Inhibition by Crotamine and L-CDPs
2.4. The Binding Affinity of the L-CDP1 and L-CDP7 Using Surface Plasmon Resonance
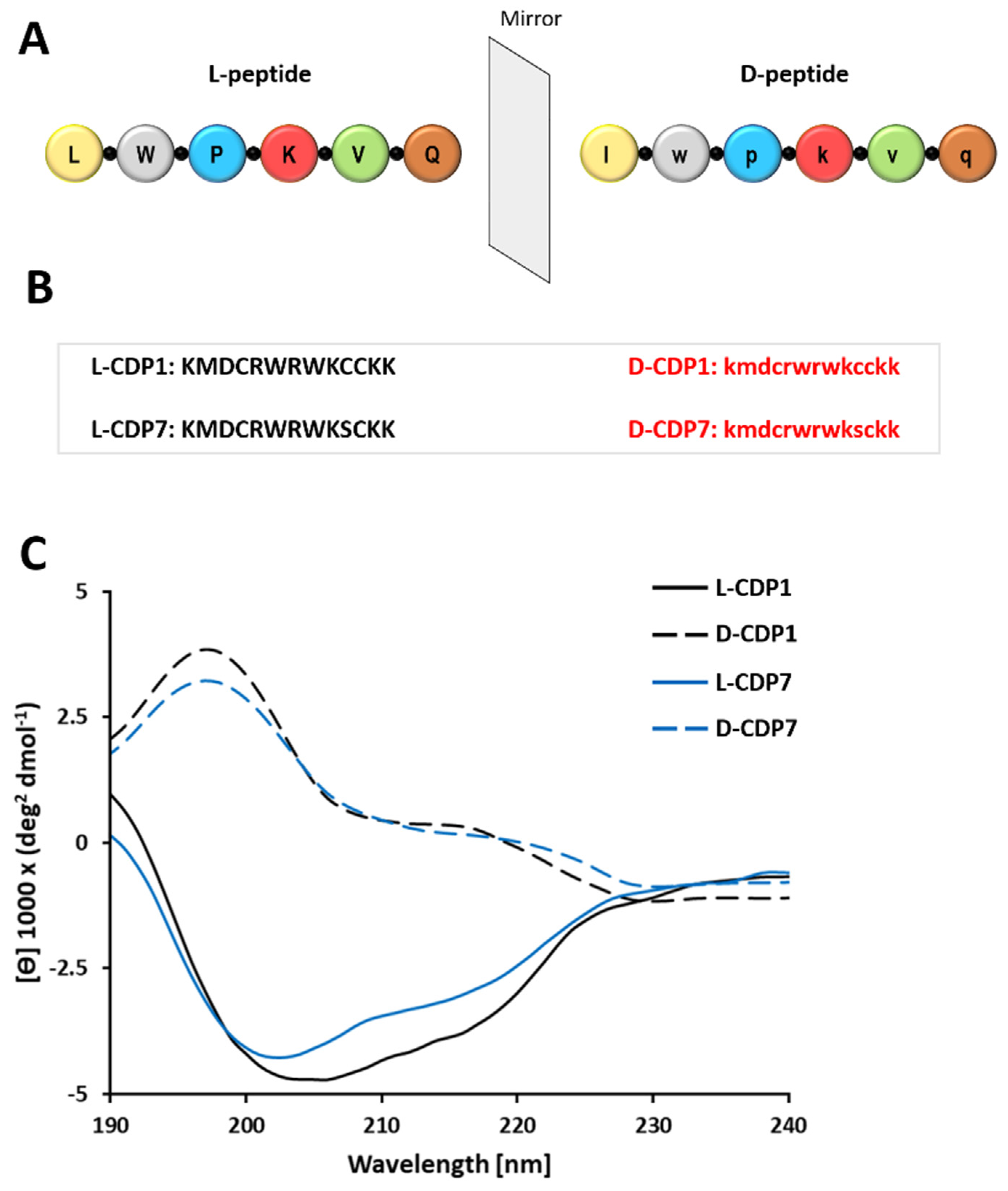
2.5. Characterization of the CDP1 and CDP7 D-Peptide Enantiomers
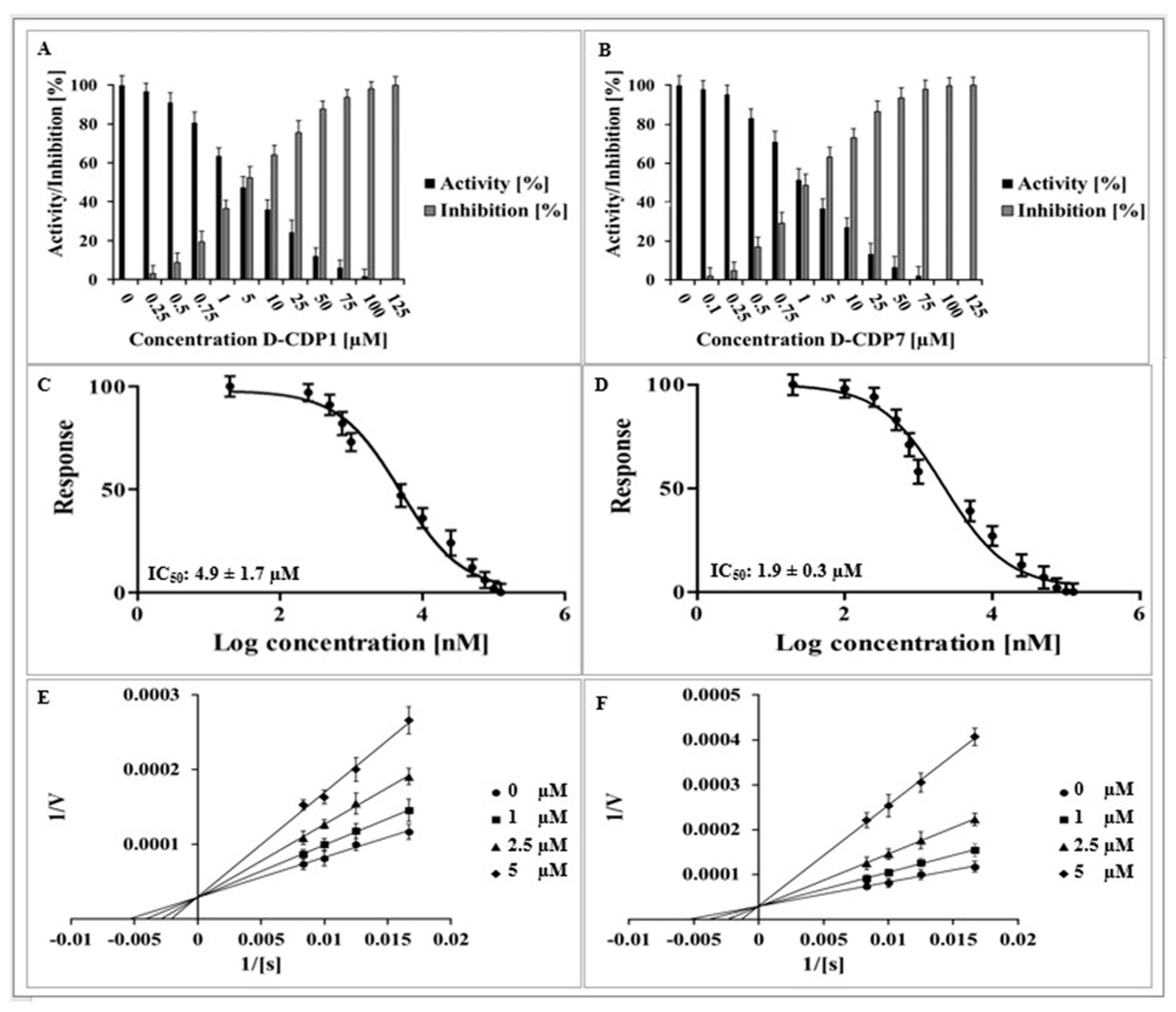
2.6. 24 h Stability and Promiscuous Assays of L/D-CDP1 and L/D-CDP7
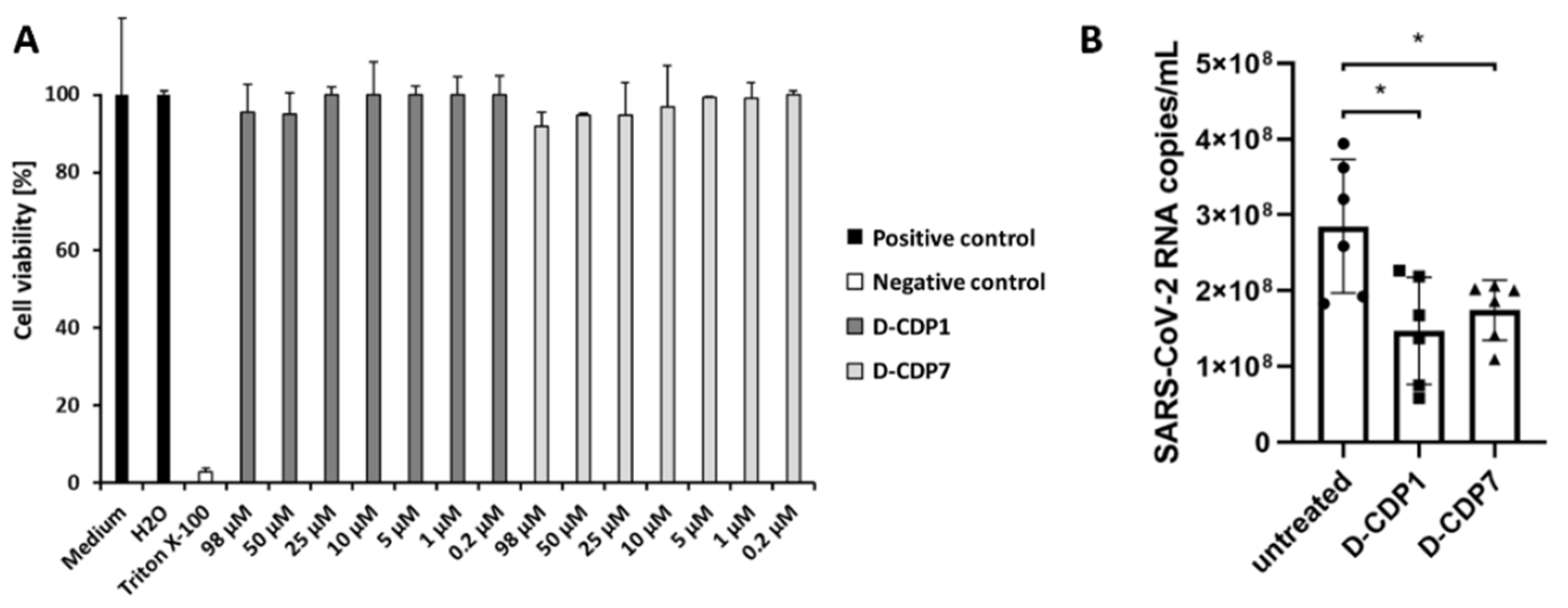
2.7. Cytotoxicity Assay of D-CDP1 and D-CDP7
2.8. Antiviral Activity of D-CDP1 and D-CDP7
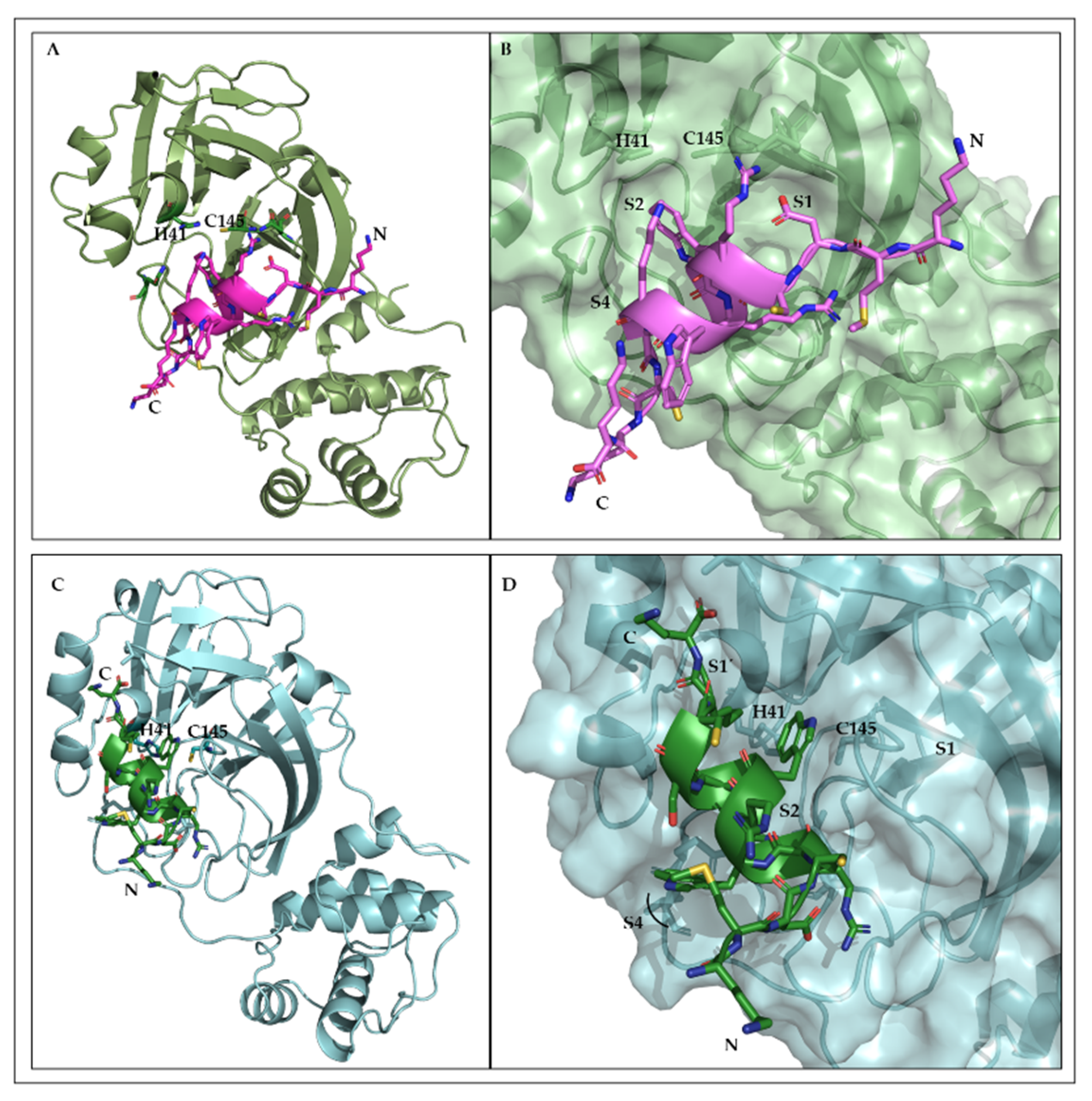
2.9. Structural Interfaces of the Predicted 3CLpro/L-CDP1 Complex
3. Materials and Methods
3.1. Crotamine Purification
3.2. Crotamine Derivative Peptides (CDP) Synthesis
3.3. Cloning, Expression and Purification of SARS-CoV-2 3CLpro
3.4. Activity Assay of SARS-CoV-2 3CLpro
3.5. Inhibition Assay of SARS-CoV-2 3CLpro
3.6. Determination of Inhibition Mode
3.7. Inhibitor Stability over 24 h
3.8. Assays to Exclude L-CDPs and D-CDPs as Promiscuous Inhibitors
3.9. L- and D-Peptide Stability against Pepsin Hydrolysis
3.10. Determination of Dissociation Constant Using Surface Plasmon Resonance
3.11. Determination of Dissociation Constant Using Microscale Thermophoresis (MST)
3.12. Circular Dichroism (CD) Spectroscopy
3.13. Cell Viability Assay
3.14. SARS-CoV-2 Infection Assay
3.15. Statistical Analysis
3.16. System Preparation
3.17. Simulation Setup
3.18. Molecular Dynamics Analysis and Interaction Energy Calculation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lu, H.; Stratton, C.W.; Tang, Y.W. Outbreak of pneumonia of unknown etiology in Wuhan, China: The mystery and the miracle. J. Med. Virol. 2020, 92, 401–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO. World Health Organization, Coronavirus Disease 2019 (COVID-19) Dashboard. 2022. Available online: https://covid19.who.int/ (accessed on 25 February 2022).
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Marc, G.P.; Moreira, E.D.; Zerbini, C.; et al. Safety and efficacy of the BNT162b2 mRNA COVID-19 vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef] [PubMed]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Voysey, M.; Clemens, S.A.C.; Madhi, S.A.; Weckx, L.Y.; Folegatti, P.M.; Aley, P.K.; Bijker, E. Safety and efficacy of the ChAdOx1 nCoV-19 vaccine (AZD1222) against SARS-CoV-2: An interim analysis of four randomised controlled trials in Brazil, South Africa, and the UK. Lancet 2021, 397, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Livingston, E.H.; Malani, P.N.; Creech, C.B. The Johnson & Johnson Vaccine for COVID-19. JAMA 2021, 325, 1575. [Google Scholar] [CrossRef]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the Treatment of COVID-19-Final Report. N. Engl. J. Med. 2020, 338, 1813–1826. [Google Scholar] [CrossRef]
- Hosseinzadeh, M.H.; Shamshirian, A.; Ebrahimzadeh, M.A. Dexamethasone vs. COVID-19: An Experimental Study in Line with the Preliminary Findings of a Large Trial. Int. J. Clin. Pract. 2020, 75, e13943. [Google Scholar] [CrossRef]
- The Japanese Association for Infectious Diseases. Treatment of Novel Coronavirus Disease in Japan. Available online: https://www.kansensho.or.jp (accessed on 12 July 2021).
- French Resuscitation Society. Expert Recommendations on Treating Patients during SARS-CoV-2 Epidemic. Available online: https://www.eahp.eu/ (accessed on 11 July 2021).
- Italian Society of Infectious and Tropical Diseases (SIMIT). Guidelines for the Treatment and Support Management of Patients with COVID-19 Coronavirus Infection. Available online: https://covid-19.ebscomedical.com/ (accessed on 13 July 2021).
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef]
- Ramajayam, R.; Tan, K.P.; Liang, P.H. Recent development of 3C and 3CL protease inhibitors for anti-coronavirus and anti-picornavirus drug discovery. Biochem. Soc. Trans. 2011, 39, 1371–1375. [Google Scholar] [CrossRef]
- Ren, Z.; Yan, L.; Zhang, N.; Guo, Y.; Yang, C.; Lou, Z.; Rao, Z. The newly emerged SARS-like coronavirus HCoV-EMC also has an “Achilles’ heel”: Current effective inhibitor targeting a 3C-like protease. Protein Cell. 2013, 4, 248–250. [Google Scholar] [CrossRef]
- Anand, K.; Palm, G.J.; Mesters, J.R.; Siddell, S.G.; Ziebuhr, J.; Hilgenfeld, R. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra α-helical domain. EMBO J. 2002, 21, 3213–3224. [Google Scholar] [CrossRef] [PubMed]
- Anand, K.; Ziebuhr, J.; Wadhwani, P.; Mesters, J.R.; Hilgenfeld, R. Coronavirus main proteinase (3CLpro) structure: Basis for design of anti-SARS drugs. Science 2003, 300, 1763–1767. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Yang, M.; Ding, Y.; Liu, Y.; Lou, Z.; Zhou, Z.; Sun, L.; Mo, L.; Ye, S.; Pang, H.; et al. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. USA 2003, 100, 13190–13195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pillaiyar, T.; Manickam, M.; Namasivayam, V.; Hayashi, Y.; Jung, S.H. An overview of severe acute respiratory syndrome–coronavirus (SARS-CoV) 3CL protease inhibitors: Peptidomimetics and small molecule chemotherapy. J. Med. Chem. 2016, 59, 6595–6628. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.; Siman-Tov, G.; Hall, G.; Bhalla, N.; Narayanan, A. Human antimicrobial peptides as therapeutics for viral infections. Viruses 2019, 11, 704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boas, L.C.P.V.; Campos, M.L.; Berlanda, R.L.A.; de Carvalho Neves, N.; Franco, O.L. Antiviral peptides as promising therapeutic drugs. Cell Mol. Life Sci. 2019, 76, 3525–3542. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, I.N.; Hartshorn, K.L. The role of antimicrobial peptides in influenza virus infection and their potential as antiviral and immunomodulatory therapy. Pharmaceuticals 2016, 9, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonçalves, J.M.; Polson, A. The electrophoretic analysis of snake venom. Arch. Biochem. 1947, 13, 253–259. [Google Scholar]
- Gonçalves, J.M.; Vieira, L.G. Estudos sobre venenos de serpentes brasileiras I. Análise eletroforética. An. Acad. Bras. Cienc. 1950, 22, 141–150. [Google Scholar]
- Hayashi, M.A.; Oliveira, E.B.; Kerkis, I.; Karpel, R.L. Crotamine: A novel cell-penetrating polypeptide nanocarrier with potential anti-cancer and biotechnological applications. Methods Mol. Biol. 2012, 906, 337–352. [Google Scholar] [CrossRef]
- Yamane, E.S.; Bizerra, F.C.; Oliveira, E.B.; Moreira, J.T.; Rajabi, M.; Nunes, G.L.; de Souza, A.O.; da Silva, I.D.; Yamane, T.; Karpel, R.L.; et al. Unraveling the antifungal activity of a South American rattlesnake toxin crotamine. Biochimie 2013, 95, 231–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nascimento, F.D.; Hayashi, M.A.; Kerkis, A.; Oliveira, V.; Oliveira, E.B.; Rádis-Baptista, G.; Nader, H.B.; Yamane, T.; Tersariol, I.L.S.; Kerkis, I. Crotamine mediates gene delivery into cells through the binding to heparan sulfate proteoglycans. J. Biol. Chem. 2007, 282, 21349–21360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, M.A.; Nascimento, F.D.; Kerkis, A.; Oliveira, V.; Oliveira, E.B.; Pereira, A.; Rádis-Baptista, G.; Nader, H.B.; Yamane, T.; Kerkis, I.; et al. Cytotoxic effects of crotamine are mediated through lysosomal membrane permeabilisation. Toxicon 2008, 52, 508–517. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.C.; Hayashi, M.A.; Oliveira, E.B.; Karpel, R.L. DNA-interactive properties of crotamine, a cell-penetrating polypeptide and a potential drug carrier. PLoS ONE 2012, 7, 48913. [Google Scholar] [CrossRef] [Green Version]
- Kerkis, A.; Kerkis, I.; Radis-Baptista, G.; Oliveira, E.B.; Vianna-Morgante, A.M.; Pereira, L.V.; Yamane, T. Crotamine is a novel cell-penetrating protein from the venom of rattlesnake Crotalus durissus terrificus. FASEB J. 2004, 18, 1407–1409. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.G.; Sayers, E.J.; He, L.; Narayan, R.; Williams, T.L.; Mills, E.M.; Allemann, R.K.; Luk, L.Y.P.; Jones, A.T.; Tsai, Y.H. Cell-penetrating peptide sequence and modification dependent uptake and subcellular distribution of green fluorescent protein in different cell lines. Sci. Rep. 2019, 9, 6298. [Google Scholar] [CrossRef]
- Eberle, R.J.; Olivier, D.S.; Amaral, M.S.; Gering, I.; Willbold, D.; Arni, R.K.; Coronado, M.A. The Repurposed Drugs Suramin and Quinacrine Cooperatively Inhibit SARS-CoV-2 3CLpro In Vitro. Viruses 2021, 13, 873. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Lin, D.; Kusov, Y.; Nian, Y.; Ma, Q.; Wang, J.; De Wilde, A. α-Ketoamides as broad-spectrum inhibitors of coronavirus and enterovirus replication: Structure-based design, synthesis, and activity assessment. J. Med. Chem. 2020, 63, 4562–4578. [Google Scholar] [CrossRef]
- Ma, C.; Sacco, M.D.; Hurst, B.; Townsend, J.A.; Hu, Y.; Szeto, T.; Zhang, X.; Tarbet, B.; Marty, M.T.; Chen, Y.; et al. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020, 30, 678–692. [Google Scholar] [CrossRef]
- Jha, D.; Mishra, R.; Gottschalk, S.; Wiesmüller, K.H.; Ugurbil, K.; Maier, M.E.; Engelmann, J. CyLoP-1: A novel cysteine-rich cell-penetrating peptide for cytosolic delivery of cargoes. Bioconjug. Chem. 2011, 22, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Van Regenmortel, M.H.; Muller, S. D-peptides as immunogens and diagnostic reagents. Curr. Opin. Biotechnol. 1998, 9, 377–382. [Google Scholar] [CrossRef]
- Sadowski, M.; Pankiewicz, J.; Scholtzova, H.; Ripellino, J.A.; Li, Y.; Schmidt, S.D.; Mathews, P.M.; Fryer, J.D.; Holtzman, D.M.; Sigurdsson, E.M.; et al. A synthetic peptide blocking the apolipoprotein E/beta-amyloid binding mitigates beta amyloidtoxicity and fibril formation in vitro and reduces beta-amyloid plaques in transgenic mice. Am. J. Pathol. 2004, 165, 937–948. [Google Scholar] [CrossRef]
- Dintzis, H.M.; Symer, D.E.; Dintzis, R.Z.; Zawadzke, L.E.; Berg, J.M. A comparison of the immunogenicity of a pair of enantiomeric proteins. Proteins Struct. Funct. Bioinform. 1993, 16, 306–308. [Google Scholar] [CrossRef] [PubMed]
- Welch, B.D.; VanDemark, A.P.; Heroux, A.; Hill, C.P.; Kay, M.S. Potent D-peptide inhibitors of HIV-1 entry. Proc. Natl. Acad. Sci. USA 2007, 104, 16828–16833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, G.; de Leeuw, E.; Pazgier, M.; Yuan, W.; Zou, G.; Wang, J.; Ericksen, B.; Lu, W.Y.; Lehrer, R.I.; Lu, W. Through the looking glass, mechanistic insights from enantiomeric human defensins. J. Biol. Chem. 2009, 284, 29180–29192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, L.; Sheeley, S.; Sweedler, J.V. Analysis of endogenous D-amino acid-containing peptides in metazoa. Bioanal. Rev. 2009, 1, 7–24. [Google Scholar] [CrossRef]
- Johnston, P.A. Redox cycling compounds generate H2O2 in HTS buffers containing strong reducing reagents real hits or promiscuous artifacts? Curr. Opin. Chem. Biol. 2011, 15, 174–182. [Google Scholar] [CrossRef] [Green Version]
- Feng, B.Y.; Shoichet, B.K. A detergent-based assay for the detection of promiscuous inhibitors. Nat. Protoc. 2006, 1, 550–553. [Google Scholar] [CrossRef]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [Green Version]
- González, J.E.H.; Eberle, R.J.; Willbold, D.; Coronado, M.A. A Computer-Aided Approach for the Discovery of D-Peptides as Inhibitors of SARS-CoV-2 Main Protease. Front. Mol. Biosci. 2021, 8, 816166. [Google Scholar] [CrossRef] [PubMed]
- Coronado, M.A.; Georgieva, D.; Buck, F.; Gabdoulkhakov, A.H.; Ullah, A.; Spencer, P.J.; Arni, R.K.; Betzel, C. Purification, crystallisation and preliminary X-ray diffraction analysis of crotamine, a myotoxic polypeptide from the Brazilian snake Crotalus durissus terrificus. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2012, 68, 1052–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, A.; Lim, L.; Srivastava, S.; Lu, Y.; Song, J. Solution conformations of Zika NS2B-NS3pro and its inhibition by natural products from edible plants. PLoS ONE 2017, 12, e0180632. [Google Scholar] [CrossRef] [PubMed]
- Motulsky, H.; Christopoulos, A. Fitting Models to Biological Data Using Linear and Nonlinear Regression: A Practical Guide to Curve Fitting; Oxford University Press: Oxford, UK, 2004. [Google Scholar]
- Elfgen, A.; Santiago-Schubel, B.; Gremer, L.; Kutzsche, J.; Willbold, D. Surprisingly high stability of the Abeta oligomer eliminating all-d-enantiomeric peptide D3 in media simulating the route of orally administered drugs. Off. J. Eur. Fed. Pharm. Sci. 2017, 107, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Elfgen, A.; Hupert, M.; Bochinsky, K.; Tusche, M.; González de San Román Martin, E.; Gering, I.; Sacchi, S.; Pollegioni, L.; Huesgen, P.F.; Hartmann, R.; et al. Metabolic resistance of the D-peptide RD2 developed for direct elimination of amyloid-β oligomers. Sci. Rep. 2019, 9, 5715. [Google Scholar] [CrossRef] [Green Version]
- Micsonai, A.; Wien, F.; Bulyáki, E.; Kun, J.; Moussong, E.; Lee, Y.H.; Goto, Y.; Réfrégiers, M.; Kardos, J. BeStSel: A web server for accurate protein secondary structure prediction and fold recognition from the circular dichroism spectra. Nucl. Acids Res. 2018, 46, W315–W322. [Google Scholar] [CrossRef]
- Ramani, A.; Müller, L.; Ostermann, P.N.; Gabriel, E.; Abida-Islam, P.; Müller-Schiffmann, A.; Mariappan, A.; Goureau, O.; Gruell, H.; Walker, A.; et al. SARS-CoV-2 targets neurons of 3D human brain organoids. EMBO J. 2020, 39, e106230. [Google Scholar] [CrossRef]
- Dominguez, C.; Boelens, R.; Bonvin, A.M.J.J. HADDOCK: A Protein-Protein Docking Approach Based on Biochemical or Biophysical Information. J. Am. Chem. Soc. 2003, 125, 1731–1737. [Google Scholar] [CrossRef] [Green Version]
- Gordon, J.C.; Myers, J.B.; Folta, T.; Shoja, V.; Heath, L.S.; Onufriev, A. H++: A server for estimating pKas and adding missing hydrogens to macromolecules. Nucl. Acids Res. 2005, 33, W368–W371. [Google Scholar] [CrossRef]
- Case, D.A.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Greene, D.; Homeyer, N.; et al. AMBER 2018; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Tian, C.; Kasavajhala, K.; Belfon, K.A.A.; Raguette, L.; Huang, H.; Migues, A.N.; Bickel, J.; Wang, Y.; Pincay, J.; Wu, Q.; et al. ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J. Chem. Theory Comput. 2020, 16, 528–552. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089. [Google Scholar] [CrossRef] [Green Version]
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, H.; Roe, D.R.; Simmerling, C. Improved Generalized Born Solvent Model Parameters for Protein Simulations. J. Chem. Theory Comput. 2013, 9, 2020–2034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Case, D.A.; Belfon, K.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E.; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Giambasu, G.; et al. AMBER 2020; University of California: San Francisco, CA, USA, 2020. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | Sequence | IC50 [µM] ± STD | Inhibition Type 1 |
|---|---|---|---|
| Crotamine | 40 ± 3.1 | − | |
| L-CDP1 | KMDCRWRWKCCKK | 1.9 ± 0.3 | Competitive |
| L-CDP2 | KMDSRWRWKSSKK | 5.0 ± 0.8 | − |
| L-CDP3 | KMDCRWRWKSSKK | 8.5 ± 1.0 | − |
| L-CDP4 | KMDSRWRWKCCKK | 5.1 ± 1.1 | − |
| L-CDP5 | KMDSRWRWKSCKK | 6.2 ± 0.5 | − |
| L-CDP6 | KMDSRWRWKCSKK | 7.5 ± 0.7 | − |
| L-CDP7 | KMDCRWRWKSCKK | 1.5 ± 0.4 | Competitive |
| L-CDP8 | KMDCRWRWKCSKK | 2.1 ± 0.4 | Competitive |
| L-CDP9 | RWRWKCCKK | 4.9 ± 0.6 | − |
| Molecule | Sequence | KD [nM] ± STD |
|---|---|---|
| L-CDP1 | KMDCRWRWKCCKK | 65 ± 20.1 |
| L-CDP7 | KMDCRWRWKSCKK | 304 ± 70.3 |
| Molecule | IC50 [µM] ± STD | Inhibition Type | KD [nM] ± STD |
|---|---|---|---|
| D-CDP1 | 4.9 ± 1.7 | Competitive | 185.7 ± 17.8 |
| D-CDP7 | 1.9 ± 0.3 | Competitive | 1951.3 ± 87.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eberle, R.J.; Gering, I.; Tusche, M.; Ostermann, P.N.; Müller, L.; Adams, O.; Schaal, H.; Olivier, D.S.; Amaral, M.S.; Arni, R.K.; et al. Design of D-Amino Acids SARS-CoV-2 Main Protease Inhibitors Using the Cationic Peptide from Rattlesnake Venom as a Scaffold. Pharmaceuticals 2022, 15, 540. https://doi.org/10.3390/ph15050540
Eberle RJ, Gering I, Tusche M, Ostermann PN, Müller L, Adams O, Schaal H, Olivier DS, Amaral MS, Arni RK, et al. Design of D-Amino Acids SARS-CoV-2 Main Protease Inhibitors Using the Cationic Peptide from Rattlesnake Venom as a Scaffold. Pharmaceuticals. 2022; 15(5):540. https://doi.org/10.3390/ph15050540
Chicago/Turabian StyleEberle, Raphael J., Ian Gering, Markus Tusche, Philipp N. Ostermann, Lisa Müller, Ortwin Adams, Heiner Schaal, Danilo S. Olivier, Marcos S. Amaral, Raghuvir K. Arni, and et al. 2022. "Design of D-Amino Acids SARS-CoV-2 Main Protease Inhibitors Using the Cationic Peptide from Rattlesnake Venom as a Scaffold" Pharmaceuticals 15, no. 5: 540. https://doi.org/10.3390/ph15050540
APA StyleEberle, R. J., Gering, I., Tusche, M., Ostermann, P. N., Müller, L., Adams, O., Schaal, H., Olivier, D. S., Amaral, M. S., Arni, R. K., Willbold, D., & Coronado, M. A. (2022). Design of D-Amino Acids SARS-CoV-2 Main Protease Inhibitors Using the Cationic Peptide from Rattlesnake Venom as a Scaffold. Pharmaceuticals, 15(5), 540. https://doi.org/10.3390/ph15050540