Papain-Like Proteases as Coronaviral Drug Targets: Current Inhibitors, Opportunities, and Limitations



Abstract
1. Introduction
2. Coronaviruses
3. CoV Enzyme Inhibitors and Their Feasibility for Therapeutics
4. PLpro—A Prospective Pharmacological Target
5. A Hindsight View of Developing CoV PLpro Inhibitors
6. Current Development of Inhibitors of CoV PLpro
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Xie, M.; Chen, Q. Insight into 2019 novel coronavirus—An updated interim review and lessons from SARS-CoV and MERS-CoV. Int. J. Infect. Dis. 2020, 94, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Breban, R.; Riou, J.; Fontanet, A. Interhuman transmissibility of Middle East respiratory syndrome coronavirus: Estimation of pandemic risk. Lancet 2013, 382, 694–699. [Google Scholar] [CrossRef]
- Chan, K.H.; Poon, L.L.L.M.; Cheng, V.C.C.; Guan, Y.; Hung, I.F.N.; Kong, J.; Yam, L.Y.C.; Seto, W.H.; Yuen, K.Y.; Peiris, J.S.M. Detection of SARS Coronavirus in Patients with Suspected SARS. Emerg. Infect. Dis. 2004, 10, 294–299. [Google Scholar] [CrossRef]
- Bosch, B.J.; Smits, S.L.; Haagmans, B.L. Membrane ectopeptidases targeted by human coronaviruses. Curr. Opin. Virol. 2014, 6, 55–60. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, N.D.; Barrett, A.J.; Thomas, P.D.; Huang, X.; Bateman, A.; Finn, R.D. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 2018, 46, D624–D632. [Google Scholar] [CrossRef]
- Verma, S.; Dixit, R.; Pandey, K.C. Cysteine Proteases: Modes of Activation and Future Prospects as Pharmacological Targets. Front. Pharmacol. 2016, 7, 107. [Google Scholar] [CrossRef]
- Liu, H.; Hu, M.; Wang, Q.; Cheng, L.; Zhang, Z. Role of Papain-Like Cysteine Proteases in Plant Development. Front. Plant Sci. 2018, 9, 1717. [Google Scholar] [CrossRef]
- Zamyatnin, A.A. Plant Proteases Involved in Regulated Cell Death. Biochemistry 2015, 80. [Google Scholar] [CrossRef]
- Petushkova, A.I.; Savvateeva, L.V.; Korolev, D.O.; Zamyatnin, A.A. Cysteine Cathepsins: Potential Applications in Diagnostics and Therapy of Malignant Tumors. Biochemistry 2019, 84, 746–761. [Google Scholar] [CrossRef]
- Soond, S.M.; Kozhevnikova, M.V.; Zamyatnin, A.A.; Townsend, P.A. Cysteine cathepsin protease inhibition: An update on its diagnostic, prognostic and therapeutic potential in cancer. Pharmaceuticals 2019, 12, 87. [Google Scholar] [CrossRef]
- Rudzińska, M.; Parodi, A.; Maslova, V.D.; Efremov, Y.M.; Gorokhovets, N.V.; Makarov, V.A.; Popkov, V.A.; Golovin, A.V.; Zernii, E.Y.; Zamyatnin, A.A. Cysteine cathepsins inhibition affects their expression and human renal cancer cell phenotype. Cancers 2020, 12, 1310. [Google Scholar] [CrossRef] [PubMed]
- Branquinha, M.H.; Oliveira, S.S.C.; Sangenito, L.S.; Sodre, C.L.; Kneipp, L.F.; D’Avila-Levy, C.M.; Santos, A.L.S. Cruzipain: An Update on its Potential as Chemotherapy Target against the Human Pathogen Trypanosoma cruzi. Curr. Med. Chem. 2015, 22, 2225–2235. [Google Scholar] [CrossRef]
- Rosenthal, P.J. Falcipain cysteine proteases of malaria parasites: An update. Biochim. Biophys. Acta Proteins Proteom. 2020, 1868, 140362. [Google Scholar] [CrossRef]
- Knutson, T.P.; Velayudhan, B.T.; Marthaler, D.G. A porcine enterovirus G associated with enteric disease contains a novel papain-like cysteine protease. J. Gen. Virol. 2017, 98, 1305–1310. [Google Scholar] [CrossRef]
- Kim, E.; Myoung, J. Hepatitis E Virus Papain-Like Cysteine Protease Inhibits Type I Interferon Induction by Down-Regulating Melanoma Differentiation-Associated Gene 5. J. Microbiol. Biotechnol. 2018, 28, 1908–1915. [Google Scholar] [CrossRef] [PubMed]
- Barretto, N.; Jukneliene, D.; Ratia, K.; Chen, Z.; Mesecar, A.D.; Baker, S.C. The papain-like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. J. Virol. 2005, 79, 15189–15198. [Google Scholar] [CrossRef] [PubMed]
- Adalja, A.; Inglesby, T. Broad-Spectrum Antiviral Agents: A Crucial Pandemic Tool. Expert Rev. Anti. Infect. Ther. 2019, 17, 467–470. [Google Scholar] [CrossRef] [PubMed]
- Siddell, S.G.; Walker, P.J.; Lefkowitz, E.J.; Mushegian, A.R.; Adams, M.J.; Dutilh, B.E.; Gorbalenya, A.E.; Harrach, B.; Harrison, R.L.; Junglen, S.; et al. Additional changes to taxonomy ratified in a special vote by the International Committee on Taxonomy of Viruses (October 2018). Arch. Virol. 2019, 164, 943–946. [Google Scholar] [CrossRef] [PubMed]
- Kahn, J.S.; McIntosh, K. History and recent advances in coronavirus discovery. Pediatr. Infect. Dis. J. 2005, 24, S223–S227. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.M.; Cavanagh, D. The molecular biology of coronaviruses. Adv. Virus Res. 1997, 48, 1–100. [Google Scholar] [PubMed]
- Gorbalenya, A.E.; Enjuanes, L.; Ziebuhr, J.; Snijder, E.J. Nidovirales: Evolving the largest RNA virus genome. Virus Res. 2006, 117, 17–37. [Google Scholar] [CrossRef] [PubMed]
- Neuman, B.W.; Adair, B.D.; Yoshioka, C.; Quispe, J.D.; Orca, G.; Kuhn, P.; Milligan, R.A.; Yeager, M.; Buchmeier, M.J. Supramolecular architecture of severe acute respiratory syndrome coronavirus revealed by electron cryomicroscopy. J. Virol. 2006, 80, 7918–7928. [Google Scholar] [CrossRef] [PubMed]
- Letko, M.; Marzi, A.; Munster, V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol. 2020, 5, 562–569. [Google Scholar] [CrossRef]
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of their replication and pathogenesis. Methods Mol. Biol. 2015, 1282, 1–23. [Google Scholar] [CrossRef]
- Zumla, A.; Chan, J.F.W.; Azhar, E.I.; Hui, D.S.C.; Yuen, K.-Y. Coronaviruses—Drug discovery and therapeutic options. Nat. Rev. Drug Discov. 2016, 15, 327–347. [Google Scholar] [CrossRef]
- Ziebuhr, J.; Snijder, E.J.; Gorbalenya, A.E. Virus-encoded proteinases and proteolytic processing in the Nidovirales. J. Gen. Virol. 2000, 81, 853–879. [Google Scholar] [CrossRef]
- Báez-Santos, Y.M.; St John, S.E.; Mesecar, A.D. The SARS-coronavirus papain-like protease: Structure, function and inhibition by designed antiviral compounds. Antivir. Res. 2015, 115, 21–38. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Q.; Guo, D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020, 92, 418–423. [Google Scholar] [CrossRef]
- Subissi, L.; Posthuma, C.C.; Collet, A.; Zevenhoven-Dobbe, J.C.; Gorbalenya, A.E.; Decroly, E.; Snijder, E.J.; Canard, B.; Imbert, I. One severe acute respiratory syndrome coronavirus protein complex integrates processive RNA polymerase and exonuclease activities. Proc. Natl. Acad. Sci. USA 2014, 111, E3900–E3909. [Google Scholar] [CrossRef]
- Domingo, E.; Escarmís, C.; Sevilla, N.; Moya, A.; Elena, S.F.; Quer, J.; Novella, I.S.; Holland, J.J. Basic concepts in RNA virus evolution. FASEB J. 1996, 10, 859–864. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, K.; Lokugamage, K.G.; Makino, S. Viral and Cellular mRNA Translation in Coronavirus-Infected Cells. Adv. Virus Res. 2016, 96, 165–192. [Google Scholar] [CrossRef] [PubMed]
- Millet, J.K.; Whittaker, G.R. Host cell proteases: Critical determinants of coronavirus tropism and pathogenesis. Virus Res. 2015, 202, 120–134. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.P.; Wang, T.; Kaletsky, R.L.; Myers, M.C.; Purvis, J.E.; Jing, H.; Huryn, D.M.; Greenbaum, D.C.; Smith, A.B.; Bates, P.; et al. A small-molecule oxocarbazate inhibitor of human cathepsin L blocks severe acute respiratory syndrome and ebola pseudotype virus infection into human embryonic kidney 293T cells. Mol. Pharmacol. 2010, 78, 319–324. [Google Scholar] [CrossRef]
- Adedeji, A.O.; Severson, W.; Jonsson, C.; Singh, K.; Weiss, S.R.; Sarafianos, S.G. Novel inhibitors of severe acute respiratory syndrome coronavirus entry that act by three distinct mechanisms. J. Virol. 2013, 87, 8017–8028. [Google Scholar] [CrossRef]
- Elshabrawy, H.A.; Fan, J.; Haddad, C.S.; Ratia, K.; Broder, C.C.; Caffrey, M.; Prabhakar, B.S. Identification of a broad-spectrum antiviral small molecule against severe acute respiratory syndrome coronavirus and Ebola, Hendra, and Nipah viruses by using a novel high-throughput screening assay. J. Virol. 2014, 88, 4353–4365. [Google Scholar] [CrossRef]
- Zhou, N.; Pan, T.; Zhang, J.; Li, Q.; Zhang, X.; Bai, C.; Huang, F.; Peng, T.; Zhang, J.; Liu, C.; et al. Glycopeptide Antibiotics Potently Inhibit Cathepsin L in the Late Endosome/Lysosome and Block the Entry of Ebola Virus, Middle East Respiratory Syndrome Coronavirus (MERS-CoV), and Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV). J. Biol. Chem. 2016, 291, 9218–9232. [Google Scholar] [CrossRef]
- Kawase, M.; Shirato, K.; van der Hoek, L.; Taguchi, F.; Matsuyama, S. Simultaneous treatment of human bronchial epithelial cells with serine and cysteine protease inhibitors prevents severe acute respiratory syndrome coronavirus entry. J. Virol. 2012, 86, 6537–6545. [Google Scholar] [CrossRef]
- Zhou, Y.; Vedantham, P.; Lu, K.; Agudelo, J.; Carrion, R.; Nunneley, J.W.; Barnard, D.; Pöhlmann, S.; McKerrow, J.H.; Renslo, A.R.; et al. Protease inhibitors targeting coronavirus and filovirus entry. Antivir. Res. 2015, 116, 76–84. [Google Scholar] [CrossRef]
- Lu, C.-C.; Chen, M.-Y.; Chang, Y.-L. Potential therapeutic agents against COVID-19: What we know so far. J. Chin. Med. Assoc. 2020, 83, 534–536. [Google Scholar] [CrossRef]
- Jean, S.-S.; Lee, P.-I.; Hsueh, P.-R. Treatment options for COVID-19: The reality and challenges. J. Microbiol. Immunol. Infect. 2020, 53, 436–443. [Google Scholar] [CrossRef] [PubMed]
- Jácome, R.; Campillo-Balderas, J.A.; Ponce de León, S.; Becerra, A.; Lazcano, A. Sofosbuvir as a potential alternative to treat the SARS-CoV-2 epidemic. Sci. Rep. 2020, 10, 9294. [Google Scholar] [CrossRef] [PubMed]
- Ko, W.; Rolain, J.; Lee, N.; Chen, P.; Huang, C. Arguments in favour of remdesivir for treating SARS-CoV-2 infections. Int. J. Antimicrob Agents 2020, 55, 105933. [Google Scholar] [CrossRef] [PubMed]
- Briguglio, I.; Piras, S.; Corona, P.; Carta, A. Inhibition of RNA Helicases of ssRNA(+) Virus Belonging to Flaviviridae, Coronaviridae and Picornaviridae Families. Int. J. Med. Chem. 2011, 2011, 213135. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.-S.; Lee, J.; Lee, J.M.; Kim, Y.; Chin, Y.-W.; Jee, J.-G.; Keum, Y.-S.; Jeong, Y.-J. Identification of myricetin and scutellarein as novel chemical inhibitors of the SARS coronavirus helicase, nsP13. Bioorg. Med. Chem. Lett. 2012, 22, 4049–4054. [Google Scholar] [CrossRef]
- Adedeji, A.O.; Singh, K.; Calcaterra, N.E.; DeDiego, M.L.; Enjuanes, L.; Weiss, S.; Sarafianos, S.G. Severe acute respiratory syndrome coronavirus replication inhibitor that interferes with the nucleic acid unwinding of the viral helicase. Antimicrob. Agents Chemother. 2012, 56, 4718–4728. [Google Scholar] [CrossRef]
- Yu, X.; Chen, S.; Hou, P.; Wang, M.; Chen, Y.; Guo, D. VHL negatively regulates SARS coronavirus replication by modulating nsp16 ubiquitination and stability. Biochem. Biophys. Res. Commun. 2015, 459, 270–276. [Google Scholar] [CrossRef]
- Lee, J.-M.; Cho, J.-B.; Ahn, H.-C.; Jung, W.; Jeong, Y.-J. A Novel Chemical Compound for Inhibition of SARS Coronavirus Helicase. J. Microbiol. Biotechnol. 2017, 27, 2070–2073. [Google Scholar] [CrossRef]
- Zaher, N.H.; Mostafa, M.I.; Altaher, A.Y. Design, synthesis and molecular docking of novel triazole derivatives as potential CoV helicase inhibitors. Acta Pharm. 2020, 70, 145–159. [Google Scholar] [CrossRef]
- Snijder, E.J.; Decroly, E.; Ziebuhr, J. The Nonstructural Proteins Directing Coronavirus RNA Synthesis and Processing. Adv. Virus Res. 2016, 96, 59–126. [Google Scholar] [CrossRef]
- Chi, X.; Wang, M.; Pan, Y.; Jiang, J.; Jiang, T.; Yan, H.; Wu, R.; Wang, X.; Gao, X.; Niu, J. Inosine triphosphate pyrophosphatase polymorphisms are predictors of anemia in Chinese patients with chronic hepatitis C during therapy with ribavirin and interferon. J. Gastroenterol. Hepatol. 2020, 35, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Naqvi, A.A.T.; Fatima, K.; Mohammad, T.; Fatima, U.; Singh, I.K.; Singh, A.; Atif, S.M.; Hariprasad, G.; Hasan, G.M.; Hassan, M.I. Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: Structural genomics approach. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165878. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Kusov, Y.; Hilgenfeld, R. Nsp3 of coronaviruses: Structures and functions of a large multi-domain protein. Antivir. Res. 2018, 149, 58–74. [Google Scholar] [CrossRef] [PubMed]
- Wojdyla, J.A.; Manolaridis, I.; van Kasteren, P.B.; Kikkert, M.; Snijder, E.J.; Gorbalenya, A.E.; Tucker, P.A. Papain-like protease 1 from transmissible gastroenteritis virus: Crystal structure and enzymatic activity toward viral and cellular substrates. J. Virol. 2010, 84, 10063–10073. [Google Scholar] [CrossRef]
- Neuman, B.W. Bioinformatics and functional analyses of coronavirus nonstructural proteins involved in the formation of replicative organelles. Antivir. Res. 2016, 135, 97–107. [Google Scholar] [CrossRef]
- Lei, J.; Hilgenfeld, R. RNA-virus proteases counteracting host innate immunity. FEBS Lett. 2017, 591, 3190–3210. [Google Scholar] [CrossRef]
- Shokri, S.; Mahmoudvand, S.; Taherkhani, R.; Farshadpour, F. Modulation of the immune response by Middle East respiratory syndrome coronavirus. J. Cell. Physiol. 2019, 234, 2143–2151. [Google Scholar] [CrossRef]
- Matthews, K.; Schäfer, A.; Pham, A.; Frieman, M. The SARS coronavirus papain like protease can inhibit IRF3 at a post activation step that requires deubiquitination activity. Virol. J. 2014, 11, 209. [Google Scholar] [CrossRef]
- Yuen, C.-K.; Lam, J.-Y.; Wong, W.-M.; Mak, L.-F.; Wang, X.; Chu, H.; Cai, J.-P.; Jin, D.-Y.; To, K.K.-W.; Chan, J.F.-W.; et al. SARS-CoV-2 nsp13, nsp14, nsp15 and orf6 function as potent interferon antagonists. Emerg. Microbes Infect. 2020, 9, 1418–1428. [Google Scholar] [CrossRef]
- Békés, M.; van der Heden van Noort, G.J.; Ekkebus, R.; Ovaa, H.; Huang, T.T.; Lima, C.D. Recognition of Lys48-Linked Di-ubiquitin and Deubiquitinating Activities of the SARS Coronavirus Papain-like Protease. Mol. Cell 2016, 62, 572–585. [Google Scholar] [CrossRef]
- Lei, J.; Hilgenfeld, R. Structural and mutational analysis of the interaction between the Middle-East respiratory syndrome coronavirus (MERS-CoV) papain-like protease and human ubiquitin. Virol. Sin. 2016, 31, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.-S.; Chang, G.-G.; Juo, C.-G.; Lee, H.-J.; Yeh, S.-H.; Hsu, J.T.-A.; Chen, X. Papain-like protease 2 (PLP2) from severe acute respiratory syndrome coronavirus (SARS-CoV): Expression, purification, characterization, and inhibition. Biochemistry 2005, 44, 10349–10359. [Google Scholar] [CrossRef] [PubMed]
- Skalny, A.V.; Rink, L.; Ajsuvakova, O.P.; Aschner, M.; Gritsenko, V.A.; Alekseenko, S.I.; Svistunov, A.A.; Petrakis, D.; Spandidos, D.A.; Aaseth, J.; et al. Zinc and respiratory tract infections: Perspectives for COVID-19 (Review). Int. J. Mol. Med. 2020, 46, 17–26. [Google Scholar] [CrossRef]
- Park, J.-Y.; Kim, J.H.; Kim, Y.M.; Jeong, H.J.; Kim, D.W.; Park, K.H.; Kwon, H.-J.; Park, S.-J.; Lee, W.S.; Ryu, Y.B. Tanshinones as selective and slow-binding inhibitors for SARS-CoV cysteine proteases. Bioorg. Med. Chem. 2012, 20, 5928–5935. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-Y.; Jeong, H.J.; Kim, J.H.; Kim, Y.M.; Park, S.-J.; Kim, D.; Park, K.H.; Lee, W.S.; Ryu, Y.B. Diarylheptanoids from Alnus japonica Inhibit Papain-Like Protease of Severe Acute Respiratory Syndrome Coronavirus. Biol. Pharm. Bull. 2012, 35, 2036–2042. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.K.; Curtis-Long, M.J.; Lee, K.H.; Kim, D.W.; Ryu, H.W.; Yuk, H.J.; Park, K.H. Geranylated flavonoids displaying SARS-CoV papain-like protease inhibition from the fruits of Paulownia tomentosa. Bioorg. Med. Chem. 2013, 21, 3051–3057. [Google Scholar] [CrossRef]
- Park, J.-Y.; Ko, J.-A.; Kim, D.W.; Kim, Y.M.; Kwon, H.-J.; Jeong, H.J.; Kim, C.Y.; Park, K.H.; Lee, W.S.; Ryu, Y.B. Chalcones isolated from Angelica keiskei inhibit cysteine proteases of SARS-CoV. J. Enzym. Inhib. Med. Chem. 2016, 31, 23–30. [Google Scholar] [CrossRef]
- Park, J.-Y.; Yuk, H.J.; Ryu, H.W.; Lim, S.H.; Kim, K.S.; Park, K.H.; Ryu, Y.B.; Lee, W.S. Evaluation of polyphenols from Broussonetia papyrifera as coronavirus protease inhibitors. J. Enzym. Inhib. Med. Chem. 2017, 32, 504–515. [Google Scholar] [CrossRef]
- Lee, H.; Lei, H.; Santarsiero, B.D.; Gatuz, J.L.; Cao, S.; Rice, A.J.; Patel, K.; Szypulinski, M.Z.; Ojeda, I.; Ghosh, A.K.; et al. Inhibitor recognition specificity of MERS-CoV papain-like protease may differ from that of SARS-CoV. ACS Chem. Biol. 2015, 10, 1456–1465. [Google Scholar] [CrossRef]
- Cheng, K.-W.; Cheng, S.-C.; Chen, W.-Y.; Lin, M.-H.; Chuang, S.-J.; Cheng, I.-H.; Sun, C.-Y.; Chou, C.-Y. Thiopurine analogs and mycophenolic acid synergistically inhibit the papain-like protease of Middle East respiratory syndrome coronavirus. Antivir. Res. 2015, 115, 9–16. [Google Scholar] [CrossRef]
- Chu, H.-F.; Chen, C.-C.; Moses, D.C.; Chen, Y.-H.; Lin, C.-H.; Tsai, Y.-C.; Chou, C.-Y. Porcine epidemic diarrhea virus papain-like protease 2 can be noncompetitively inhibited by 6-thioguanine. Antivir. Res. 2018, 158, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.-H.; Moses, D.C.; Hsieh, C.-H.; Cheng, S.-C.; Chen, Y.-H.; Sun, C.-Y.; Chou, C.-Y. Disulfiram can inhibit MERS and SARS coronavirus papain-like proteases via different modes. Antivir. Res. 2018, 150, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Zaza, G.; Cheok, M.; Krynetskaia, N.; Thorn, C.; Stocco, G.; Hebert, J.M.; McLeod, H.; Weinshilboum, R.M.; Relling, M.V.; Evans, W.E.; et al. Thiopurine pathway. Pharmacogenet. Genom. 2010, 20, 573–574. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Zhao, H.; Yu, Y.; Su, H. Formation of Guanine-6-sulfonate from 6-Thioguanine and Singlet Oxygen: A Combined Theoretical and Experimental Study. J. Am. Chem. Soc. 2013, 135, 4509–4515. [Google Scholar] [CrossRef]
- Johansson, B. A review of the pharmacokinetics and pharmacodynamics of disulfiram and its metabolites. Acta Psychiatr. Scand. 1992, 86, 15–26. [Google Scholar] [CrossRef]
- Fischer, A.; Sellner, M.; Neranjan, S.; Smieško, M.; Lill, M.A. Potential Inhibitors for Novel Coronavirus Protease Identified by Virtual Screening of 606 Million Compounds. Int. J. Mol. Sci. 2020, 21, 3626. [Google Scholar] [CrossRef]
- Dong, S.; Sun, J.; Mao, Z.; Wang, L.; Lu, Y.; Li, J. A guideline for homology modeling of the proteins from newly discovered betacoronavirus, 2019 novel coronavirus (2019-nCoV). J. Med. Virol. 2020. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef]
- Kouznetsova, V.; Huang, D.; Tsigelny, I.F. Potential COVID-19 Protease Inhibitors: Repurposing FDAapproved Drugs. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Henderson, J.A.; Verma, N.; Shen, J. Assessment of Proton-Coupled Conformational Dynamics of SARS and MERS Coronavirus Papain-like Proteases: Implication for Designing Broad-Spectrum Antiviral Inhibitors. J. Chem. Phys. 2020, 153, 115101. [Google Scholar] [CrossRef]
- Amin, S.A.; Ghosh, K.; Gayen, S.; Jha, T. Chemical-informatics approach to COVID-19 drug discovery: Monte Carlo based QSAR, virtual screening and molecular docking study of some in-house molecules as papain-like protease (PLpro) inhibitors. J. Biomol. Struct. Dyn. 2020, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 2020. [Google Scholar] [CrossRef] [PubMed]
- Arya, R.; Das, A.; Prashar, V.; Kumar, M. Potential inhibitors against papain-like protease of novel coronavirus (SARS-CoV-2) from FDA approved drugs. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Colson, P.; Rolain, J.-M.; Raoult, D. Chloroquine for the 2019 novel coronavirus SARS-CoV-2. Int. J. Antimicrob. Agents 2020, 55, 105923. [Google Scholar] [CrossRef]
- Rolain, J.-M.; Colson, P.; Raoult, D. Recycling of chloroquine and its hydroxyl analogue to face bacterial, fungal and viral infections in the 21st century. Int. J. Antimicrob. Agents 2007, 30, 297–308. [Google Scholar] [CrossRef]
- Edelstein, C.L.; Venkatachalam, M.A.; Dong, Z. Autophagy inhibition by chloroquine and hydroxychloroquine could adversely affect acute kidney injury and other organ injury in critically ill patients with COVID-19. Kidney Int. 2020, 98, 234–235. [Google Scholar] [CrossRef]
- Elfiky, A.; Ibrahim, N.S. Anti-SARS and Anti-HCV Drugs Repurposing Against the Papain-like Protease of the Newly Emerged Coronavirus (2019-nCoV). Res. Sq. 2020. [Google Scholar] [CrossRef]
- Elfiky, A.; Ibrahim, N.; Elshemey, W. Drug repurposing against MERS CoV and SARS-COV-2 PLpro in silico. Res. Sq. 2020. [Google Scholar] [CrossRef]
- Zhang, D.; Wu, K.; Zhang, X.; Deng, S.; Peng, B. In silico screening of Chinese herbal medicines with the potential to directly inhibit 2019 novel coronavirus. J. Integr. Med. 2020, 18, 152–158. [Google Scholar] [CrossRef]
- Naidoo, D.; Roy, A.; Kar, P.; Mutanda, T.; Anandraj, A. Cyanobacterial metabolites as promising drug leads against the M pro and PL pro of SARS-CoV-2: An in silico analysis. J. Biomol. Struct. Dyn. 2020, 1–13. [Google Scholar] [CrossRef]
- Sasidharan, S.; Selvaraj, C.; Singh, S.K.; Dubey, V.K.; Kumar, S.; Fialho, A.M.; Saudagar, P. Bacterial protein azurin and derived peptides as potential anti-SARS-CoV-2 agents: Insights from molecular docking and molecular dynamics simulations. J. Biomol. Struct. Dyn. 2020, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Kandeel, M.; Abdelrahman, A.H.M.; Oh-Hashi, K.; Ibrahim, A.; Venugopala, K.N.; Morsy, M.A.; Ibrahim, M.A.A. Repurposing of FDA-approved antivirals, antibiotics, anthelmintics, antioxidants, and cell protectives against SARS-CoV-2 papain-like protease. J. Biomol. Struct. Dyn. 2020, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Quimque, M.T.J.; Notarte, K.I.R.; Fernandez, R.A.T.; Mendoza, M.A.O.; Liman, R.A.D.; Lim, J.A.K.; Pilapil, L.A.E.; Ong, J.K.H.; Pastrana, A.M.; Khan, A.; et al. Virtual screening-driven drug discovery of SARS-CoV2 enzyme inhibitors targeting viral attachment, replication, post-translational modification and host immunity evasion infection mechanisms. J. Biomol. Struct. Dyn. 2020, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Höltje, H.-D.; Folkers, G. Molecular Modeling: Basic Principles and Applications; Wiley: Hoboken, NJ, USA, 2008. [Google Scholar]
- Chen, Y.-C. Beware of docking! Trends Pharmacol. Sci. 2015, 36, 78–95. [Google Scholar] [CrossRef]
- Freitas, B.T.; Durie, I.A.; Murray, J.; Longo, J.E.; Miller, H.C.; Crich, D.; Hogan, R.J.; Tripp, R.A.; Pegan, S.D. Characterization and noncovalent inhibition of the deubiquitinase and deISGylase activity of SARS-CoV-2 papain-like protease. ACS Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Shin, D.; Mukherjee, R.; Grewe, D.; Bojkova, D.; Baek, K.; Bhattacharya, A.; Schulz, L.; Widera, M.; Mehdipour, A.R.; Tascher, G.; et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 2020. [Google Scholar] [CrossRef]
- Rut, W.; Lv, Z.; Zmudzinski, M.; Patchett, S.; Nayak, D.; Snipas, S.J.; El Oualid, F.; Huang, T.T.; Bekes, M.; Drag, M.; et al. Activity profiling and structures of inhibitor-bound SARS-CoV-2-PLpro protease provides a framework for anti-COVID-19 drug design. bioRxiv 2020. [Google Scholar] [CrossRef]
- Węglarz-Tomczak, E.; Tomczak, J.M.; Talma, M.; Brul, S. Ebselen as a highly active inhibitor of PLProCoV2. BioRxiv 2020. [Google Scholar] [CrossRef]
- Klemm, T.; Ebert, G.; Calleja, D.J.; Allison, C.C.; Richardson, L.W.; Bernardini, J.P.; Lu, B.G.; Kuchel, N.W.; Grohmann, C.; Shibata, Y.; et al. Mechanism and inhibition of the papain-like protease, PLpro, of SARS-CoV-2. EMBO J. 2020, e106275. [Google Scholar] [CrossRef]
- Tsai, Y.-C.; Lee, C.-L.; Yen, H.-R.; Chang, Y.-S.; Lin, Y.-P.; Huang, S.-H.; Lin, C.-W. Antiviral Action of Tryptanthrin Isolated from Strobilanthes cusia Leaf against Human Coronavirus NL63. Biomolecules 2020, 10, 366. [Google Scholar] [CrossRef]
- Swaim, C.D.; Perng, Y.-C.; Zhao, X.; Canadeo, L.A.; Harastani, H.H.; Darling, T.L.; Boon, A.C.M.; Lenschow, D.J.; Huibregtse, J.M. 6-Thioguanine blocks SARS-CoV-2 replication by inhibition of PLpro protease activities. BioRxiv 2020. [Google Scholar] [CrossRef]
- Zou, X.; Chen, K.; Zou, J.; Han, P.; Hao, J.; Han, Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front. Med. 2020, 14, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Jiang, Y.; Qiu, H.; Gao, T.; Zeng, Y.; Guo, Y.; Yu, H.; Li, J.; Kou, Z.; Du, L.; et al. Multi-Organ Damage in Human Dipeptidyl Peptidase 4 Transgenic Mice Infected with Middle East Respiratory Syndrome-Coronavirus. PLoS ONE 2015, 10, e0145561. [Google Scholar] [CrossRef]
- Peterfreund, R.A.; Philip, J.H. Critical parameters in drug delivery by intravenous infusion. Expert Opin. Drug Deliv. 2013, 10, 1095–1108. [Google Scholar] [CrossRef] [PubMed]
- Patra, J.K.; Das, G.; Fraceto, L.F.; Campos, E.V.R.; Rodriguez-Torres, M.D.P.; Acosta-Torres, L.S.; Diaz-Torres, L.A.; Grillo, R.; Swamy, M.K.; Sharma, S.; et al. Nano based drug delivery systems: Recent developments and future prospects. J. Nanobiotechnol. 2018, 16, 71. [Google Scholar] [CrossRef]
- Sohrab, S.S.; El-Kafrawy, S.A.; Mirza, Z.; Kamal, M.A.; Azhar, E.I. Design and Delivery of Therapeutic siRNAs: Application to MERS-Coronavirus. Curr. Pharm. Des. 2018, 24, 62–77. [Google Scholar] [CrossRef]
- Silva, S.; Almeida, A.J.; Vale, N. Combination of Cell-Penetrating Peptides with Nanoparticles for Therapeutic Application: A Review. Biomolecules 2019, 9, 22. [Google Scholar] [CrossRef]
- Zeitlinger, M.; Koch, B.C.P.; Bruggemann, R.; De Cock, P.; Felton, T.; Hites, M.; Le, J.; Luque, S.; MacGowan, A.P.; Marriott, D.J.E.; et al. Pharmacokinetics/Pharmacodynamics of Antiviral Agents Used to Treat SARS-CoV-2 and Their Potential Interaction with Drugs and Other Supportive Measures: A Comprehensive Review by the PK/PD of Anti-Infectives Study Group of the European Society of Antimicr. Clin. Pharmacokinet. 2020, 1–22. [Google Scholar] [CrossRef]
- Klemm, T.; Ebert, G.; Calleja, D.J.; Allison, C.C.; Richardson, L.W.; Bernardini, J.P.; Lu, B.G.C.; Kuchel, N.W.; Grohmann, C.; Shibata, Y.; et al. Mechanism and inhibition of SARS-CoV-2 PLpro. BioRxiv 2020. [Google Scholar] [CrossRef]
- Chou, C.-Y.; Chien, C.-H.; Han, Y.-S.; Prebanda, M.T.; Hsieh, H.-P.; Turk, B.; Chang, G.-G.; Chen, X. Thiopurine analogues inhibit papain-like protease of severe acute respiratory syndrome coronavirus. Biochem. Pharmacol. 2008, 75, 1601–1609. [Google Scholar] [CrossRef]



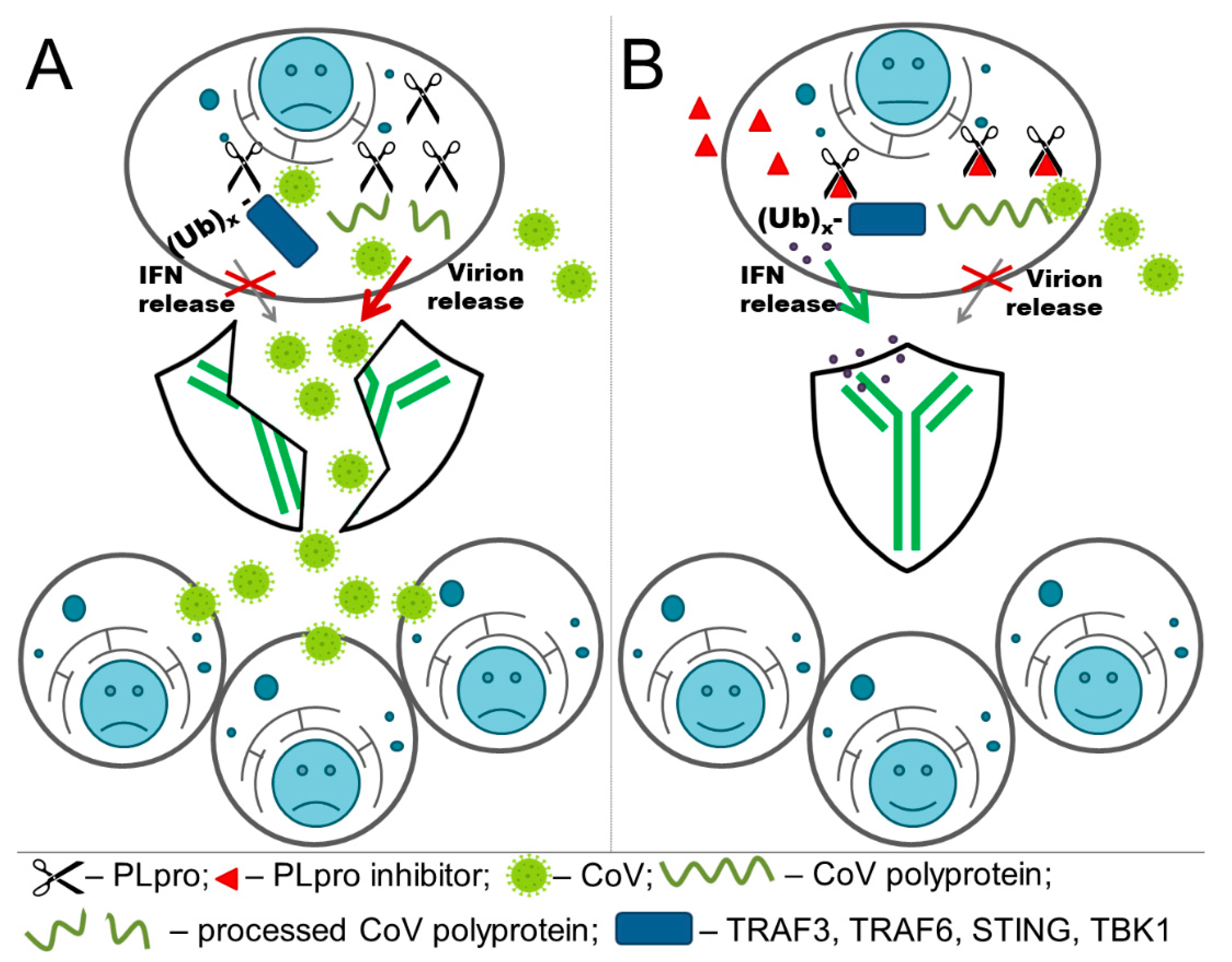{kind=link}
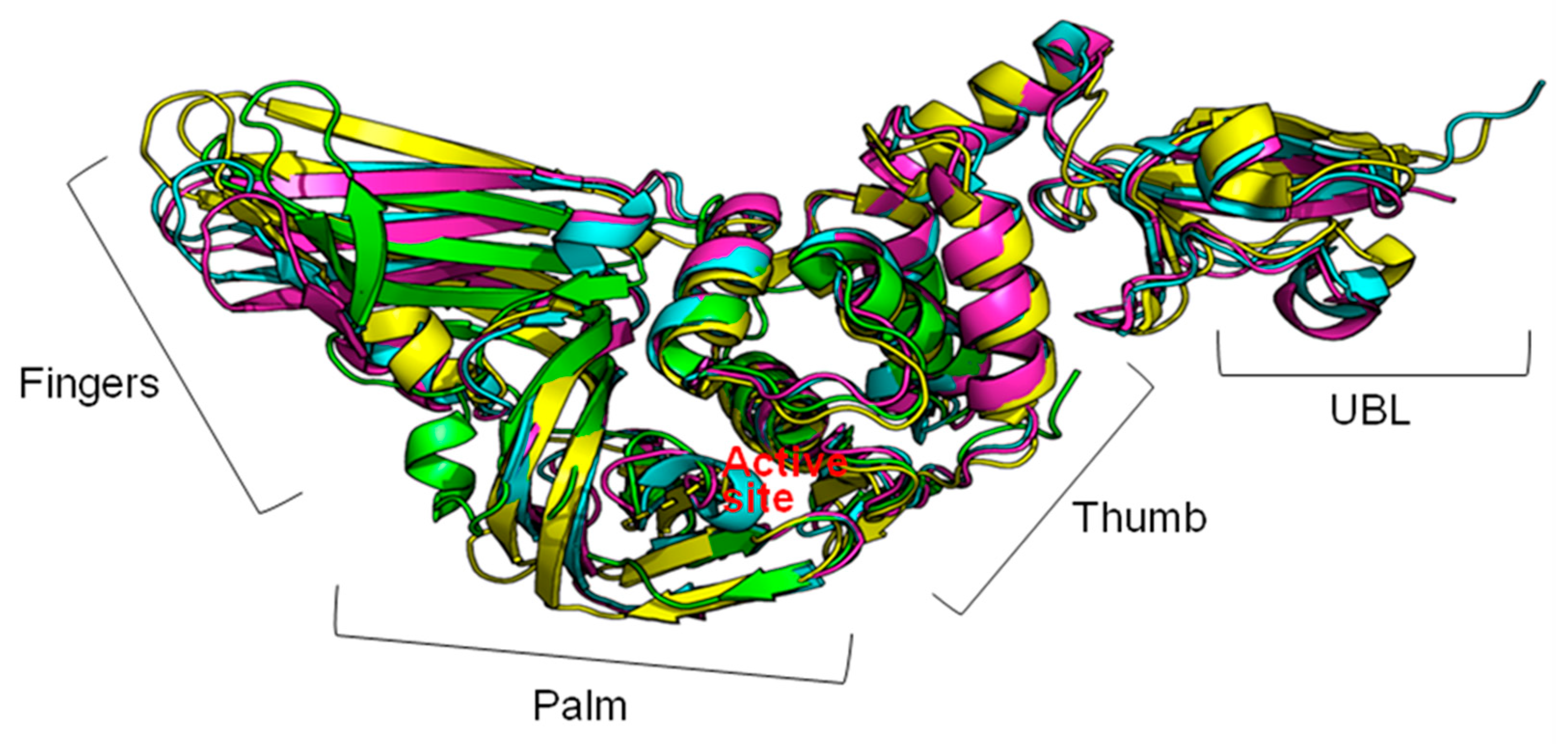{kind=link}
| Enzyme | Function | |
|---|---|---|
| Viral enzymes | ||
| Endoribonucleases 1 | Nsp1 | In complex with a 40S ribosome induces endonucleolytic cleavage near the 5′ untranslated region (UTR) of the host mRNA |
| Nsp15 | Possesses endoribonuclease (RNase) activity and enables the evasion of viral dsRNA from the host dsRNA sensors | |
| Proteases | Nsp3 (up to two PLpro) | Process the viral polyproteins, deubiquitinate IRF3 and NF-κB, deISGylate IFN-stimulated gene 15 product (ISG15) |
| Nsp5 (3CLpro) | Processes the viral polyproteins, signal transducer and activator of transcription 2 (STAT2), and NF-κB essential modulator (NEMO) | |
| Primase | Nsp8 | In complex with nsp7 synthesizes short RNA primers de novo |
| RNA-dependent RNA polymerase | Nsp12 | In complex with nsp7 and nsp8 synthesizes RNA and enables replication and transcription of the viral genome |
| Helicase | Nsp13 | Unwinds both double-stranded deoxyribonucleic acid (dsDNA) and dsRNA in a 5′-to-3′ direction and hydrolyzes deoxyribonucleotide triphosphates (dNTPs) and NTPs |
| Exoribonuclease, guanine-N7-methyltransferase | Nsp14 | In complex with nsp10 possesses exoribonuclease and (guanine-N7)-methyltransferase (MTase) activities and methylates the RNA cap |
| 2′-O-methyltransferase | Nsp16 | In complex with nsp10 possesses 2′-O-MTase activity and methylates the RNA cap |
| Host enzymes | ||
| Ribosomes | Together with factors involved in translation are recruited to translate viral genomic and subgenomic RNAs | |
| Proteases | Serine (TMPRSS2, TMPRSS4, TMPRSS11a, TMPRSS13, HAT, trypsin, DESC1, elastase, factor Xa, plasmin, furin) | Cleave the S protein to promote the invasion of CoVs |
| Cysteine (cathepsins B, L, and S) | ||
| Inhibitor | CoV | Inhibition of (IC50, µM) | Ref. | ||
|---|---|---|---|---|---|
| Pro 1 | Ub 2 | ISG15 3 | |||
Naphthalene inhibitors | SARS-CoV-2 | 2.4 ± 0.02 | 0.74 ± 0.07 | 1.50 ± 0.08 | [28,96,97,110] |
| SARS-CoV | 0.15 ± 0.01 | 0.66 ± 0.08 | 0.66 ± 0.09 | ||
| MERS-CoV | N.d. 4 | N.d. | N.d. | ||
Ebselen | SARS-CoV-2 | N.d. | 2.02 ± 1.02 | N.d. | [99] |
| SARS-CoV | N.d. | 8.45 ± 0.96 | N.d. | ||
| MERS-CoV | N.d. | N.d. | N.d.. | ||
Tanshinones from S. miltiorrhiza | SARS-CoV-2 | N.d. | N.d. | N.d. | [64] |
| SARS-CoV | 0.8 ± 0.2 | 0.7 ± 0.2 | N.d. | ||
| MERS-CoV | N.d. | N.d. | N.d. | ||
Chalcones and coumarins from A. keiskei | SARS-CoV-2 | N.d. | N.d. | N.d. | [67] |
| SARS-CoV | 1.2 ± 0.4 | 2.6 ± 0.7 | 1.1 ± 0.6 | ||
| MERS-CoV | N.d. | N.d. | N.d. | ||
Zn2+-ion and conjugates | SARS-CoV-2 | N.d. | N.d. | N.d. | [62] |
| SARS-CoV | 1.3 ± 0.2 | N.d. | N.d. | ||
| MERS-CoV | N.d. | N.d. | N.d. | ||
Diarylheptanoids from A. japonica | SARS-CoV-2 | N.d. | N.d. | N.d. | [65] |
| SARS-CoV | 4.1 ± 0.3 | 3.0 ± 1.1 | N.d. | ||
| MERS-CoV | N.d. | N.d. | N.d. | ||
Polyphenols from B. papyrifera | SARS-CoV-2 | N.d. | N.d. | N.d. | [68] |
| SARS-CoV | 3.7 ± 1.6 | 7.6 ± 0.4 | 8.5 ± 1.2 | ||
| MERS-CoV | 39.5 ± 5.1 | N.d. | N.d. | ||
N-Ethylmaleimide | SARS-CoV-2 | N.d. | N.d. | N.d. | [70,111] |
| SARS-CoV | N.d. | 4.4 ± 1.0 | N.d. | ||
| MERS-CoV | 45.0 ± 10.1 | N.d. | N.d. | ||
Geranylated flavonoids from P. tomentosa | SARS-CoV-2 | N.d. | N.d. | N.d. | [66] |
| SARS-CoV | 5.0 ± 0.06 | N.d. | N.d. | ||
| MERS-CoV | N.d. | N.d. | N.d. | ||
Thiopurine compounds 5 | SARS-CoV-2 | N.d. | N.d. | N.d. | [70,111] |
| SARS-CoV | N.d. | 5.0 ± 1.7 | N.d. | ||
| MERS-CoV | 24.4 ± 4.3 | 12.4 ± 1.9 | N.d. | ||
8-(Trifluoromethyl)-9H-purin-6-amine | SARS-CoV-2 | N.d. | N.d. | N.d. | [69] |
| SARS-CoV | 10.9 ± 0.9 | N.d. | N.d. | ||
| MERS-CoV | 6.2 ± 0.9 | N.d. | N.d. | ||
Disulfiram | SARS-CoV-2 | N.d. | N.d. | N.d. | [72] |
| SARS-CoV | 14.2 ± 0.5 | 24.1 ± 1.8 | N.d. | ||
| MERS-CoV | 22.7 ± 0.5 | 14.6 ± 0.3 | N.d. | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petushkova, A.I.; Zamyatnin, A.A., Jr. Papain-Like Proteases as Coronaviral Drug Targets: Current Inhibitors, Opportunities, and Limitations. Pharmaceuticals 2020, 13, 277. https://doi.org/10.3390/ph13100277
Petushkova AI, Zamyatnin AA Jr. Papain-Like Proteases as Coronaviral Drug Targets: Current Inhibitors, Opportunities, and Limitations. Pharmaceuticals. 2020; 13(10):277. https://doi.org/10.3390/ph13100277
Chicago/Turabian StylePetushkova, Anastasiia I., and Andrey A. Zamyatnin, Jr. 2020. "Papain-Like Proteases as Coronaviral Drug Targets: Current Inhibitors, Opportunities, and Limitations" Pharmaceuticals 13, no. 10: 277. https://doi.org/10.3390/ph13100277
APA StylePetushkova, A. I., & Zamyatnin, A. A., Jr. (2020). Papain-Like Proteases as Coronaviral Drug Targets: Current Inhibitors, Opportunities, and Limitations. Pharmaceuticals, 13(10), 277. https://doi.org/10.3390/ph13100277