
Voltammetric Behaviour of Rhodamine B at a Screen-Printed Carbon Electrode and Its Trace Determination in Environmental Water Samples

Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Apparatus
2.3. Voltammetric Procedures
2.4. Sample Preparation
3. Results
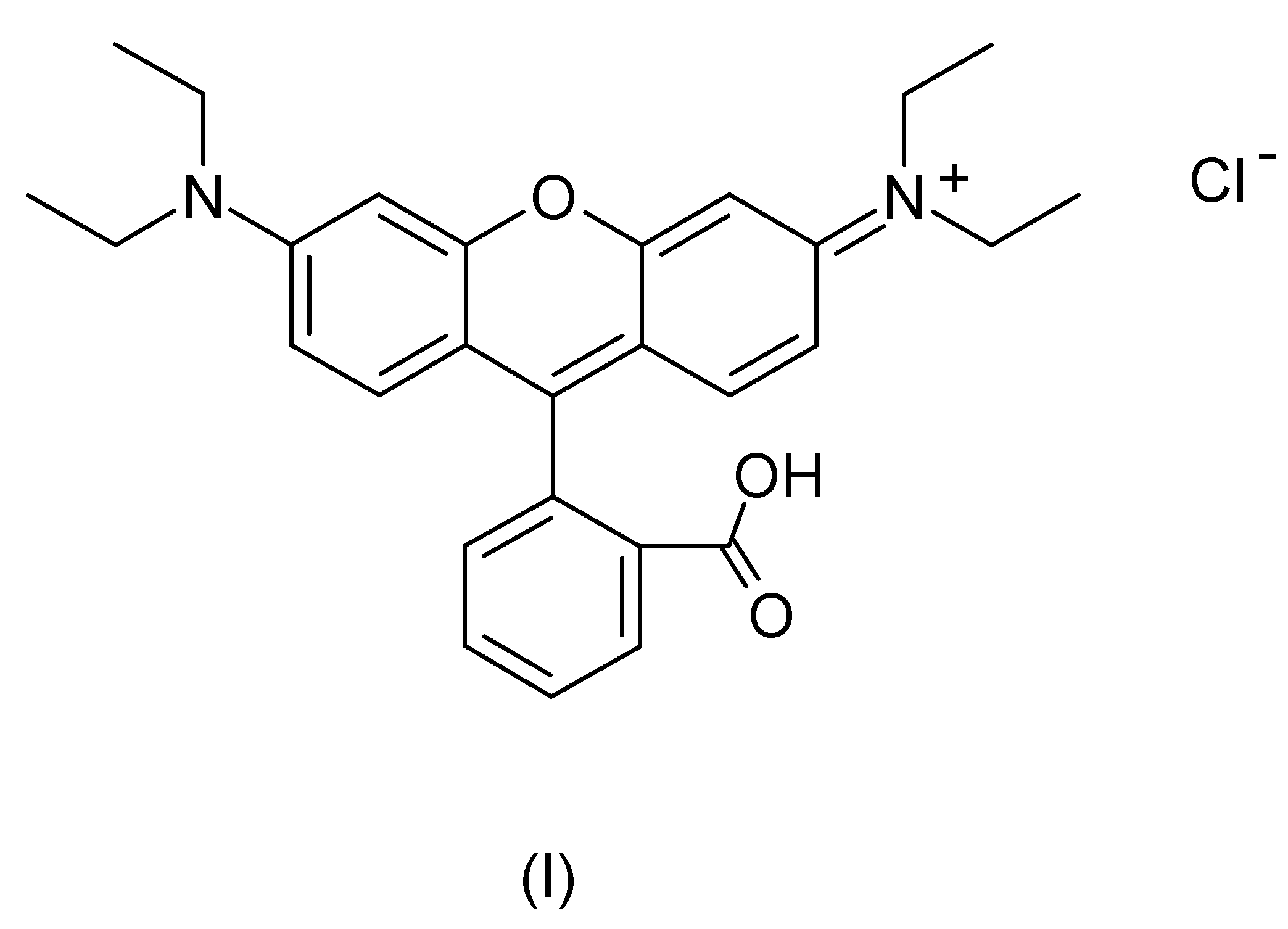
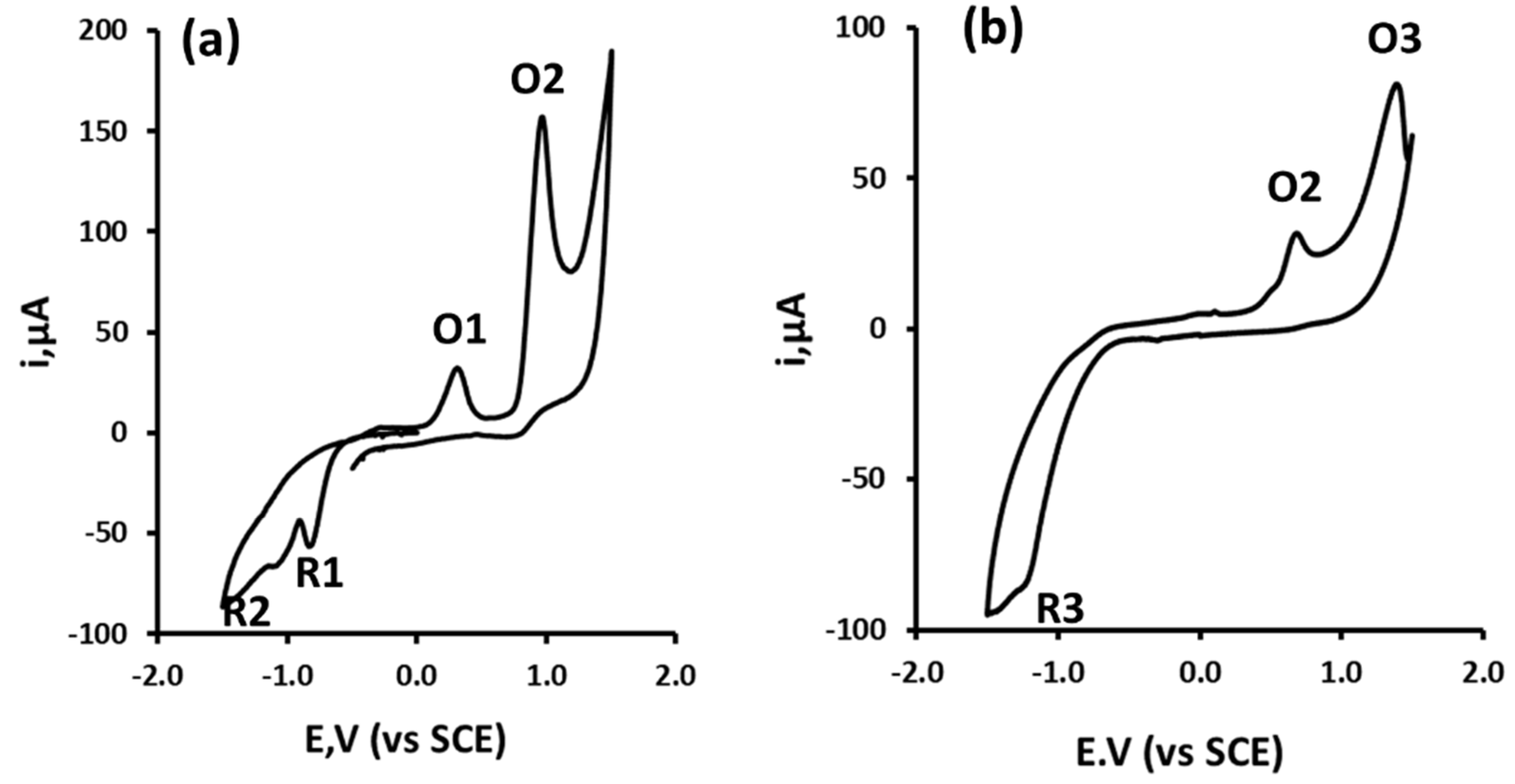
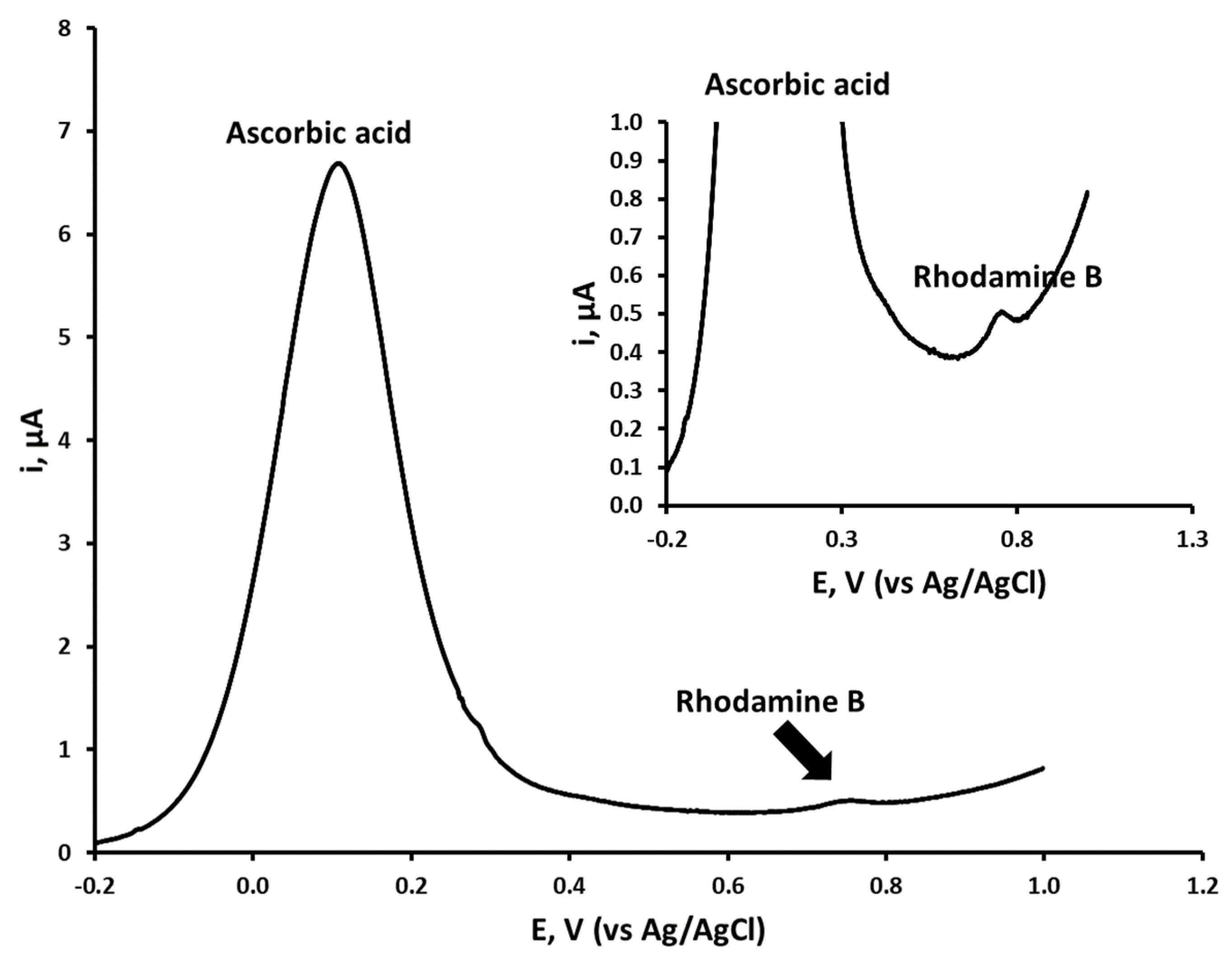
3.1. Cyclic Voltammetric Investigations
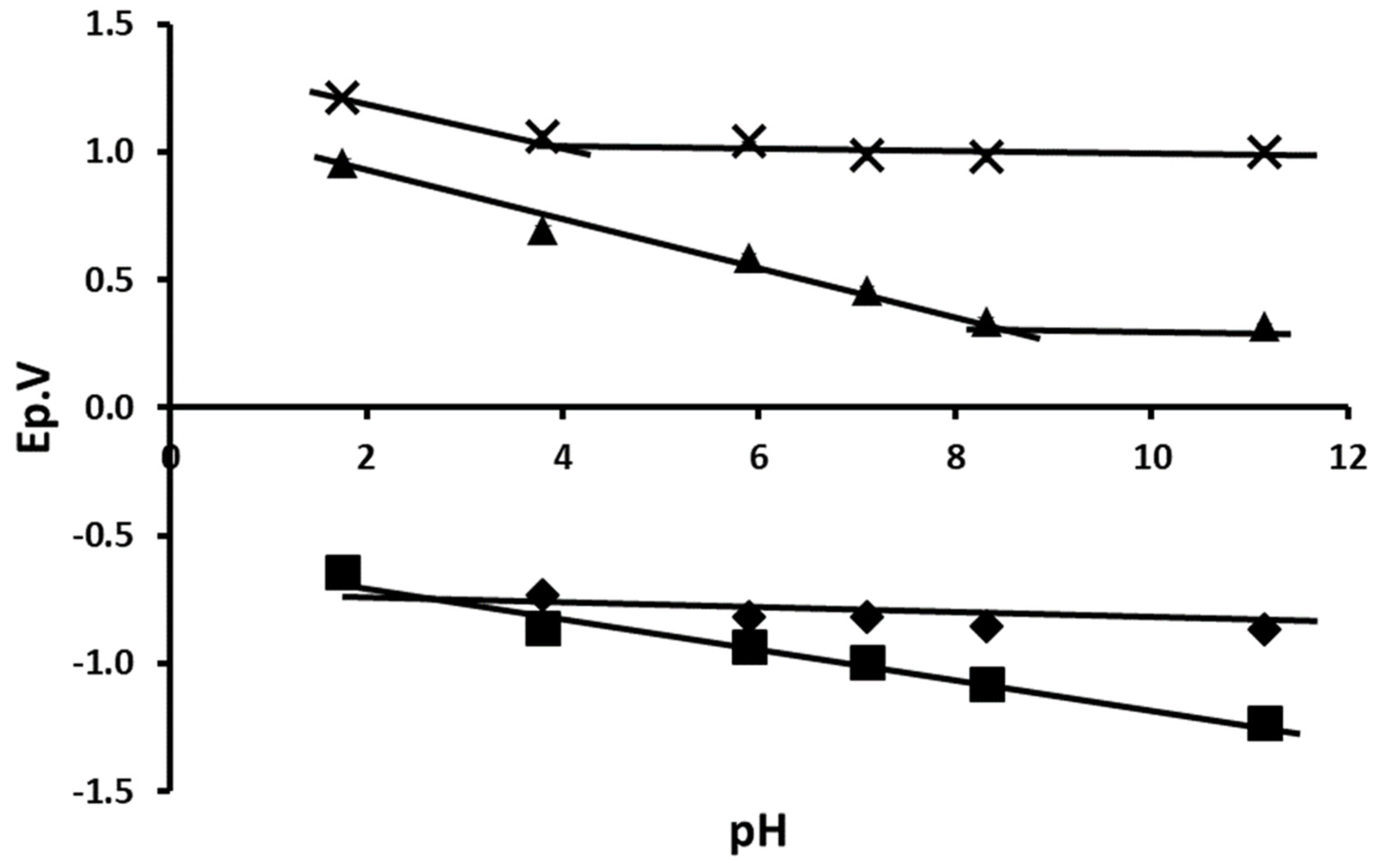
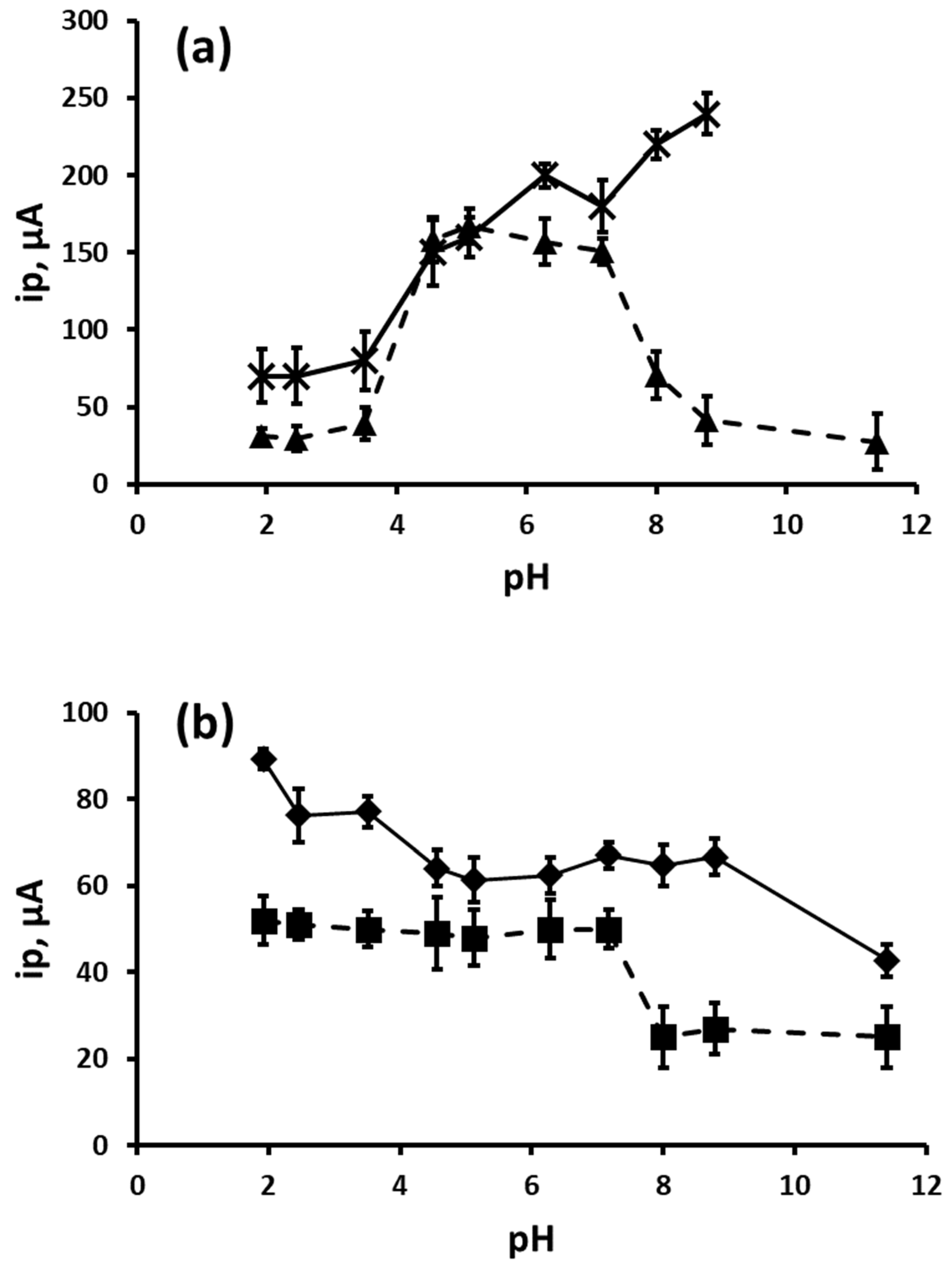
3.2. Effect of pH on Peak Potential and Peak Current
3.3. Calibration Plot, Limit of Detection and Precision
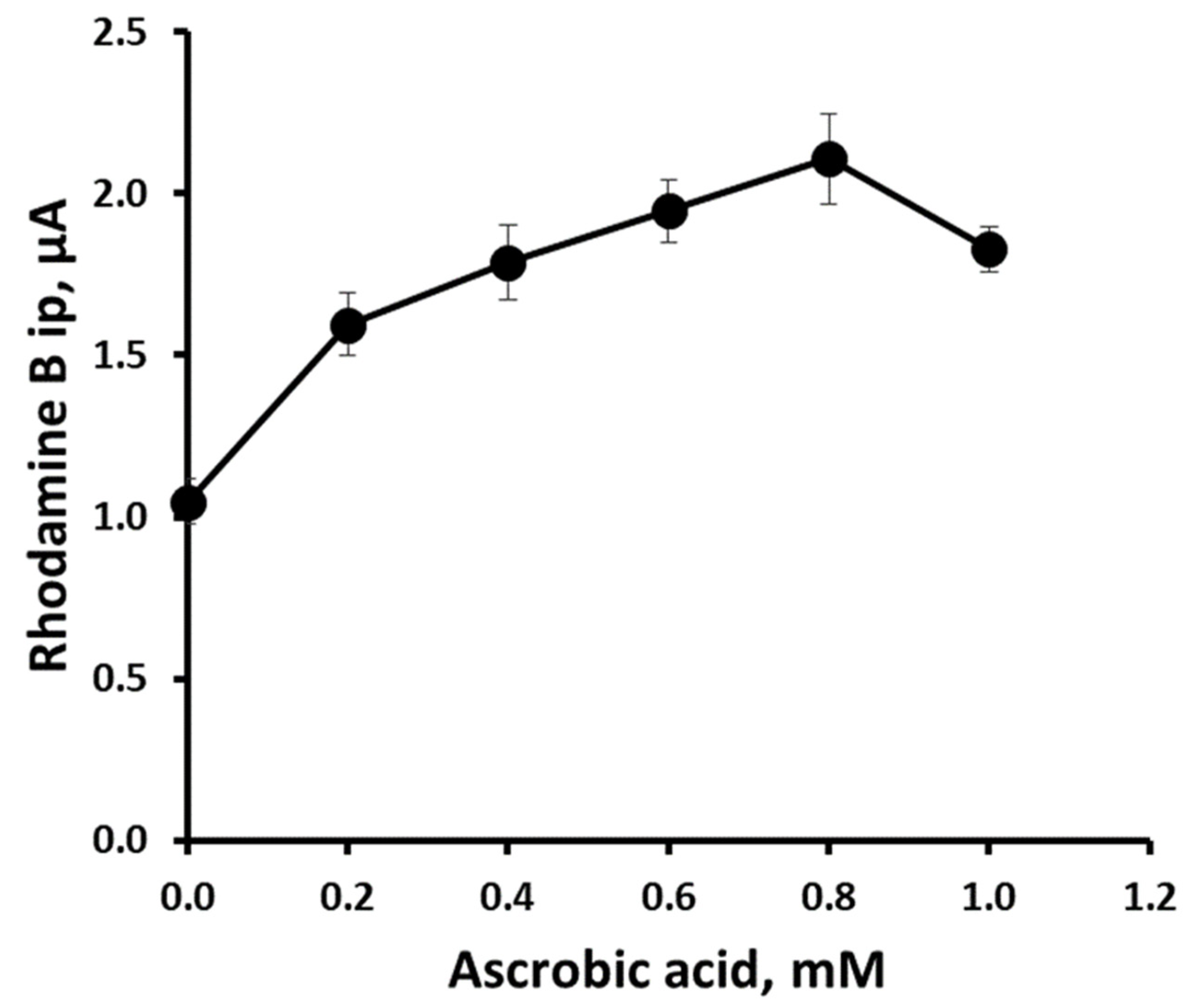
3.4. Effect of Ascorbic Acid on Rhodamine B Differential Pulse Voltammetric Response
3.5. Analytical Application
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pubchem, R.B. Use and Manufacturing. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Rhodamine-B#section=Use-and-Manufacturing&fullscreen=true (accessed on 22 April 2022).
- Abril, J.M.; Abdel-Aal, M.M.; Al-Gamal, S.A.; Abdel-Hay, F.M.; Zahar, H.M. Marine Radioactivity Studies in the Suez Canal, Part II: Field Experiments and a Modelling Study of Dispersion. Estuar. Coast. Shelf Sci. 2000, 50, 503–514. [Google Scholar] [CrossRef]
- Jones, C.; Moller, H.; Hamilton, W. A review of potential techniques for identifying individual stoats (Mustela erminea) visiting control or monitoring stations. N. Z. J. Zool. 2004, 31, 193–203. [Google Scholar] [CrossRef] [Green Version]
- Isoe, J.; Morita, K.; Kaneko, E. Determination of trace silica in highly purified water based on delayed quenching of Rhodamine B through nanoparticle formation with molybdosilicate. Analyst 2005, 130, 872–877. [Google Scholar] [CrossRef]
- Haddad, P.R. The application of ternary complexes to spectrofluorometric analysis. Talanta 1977, 24, 1–13. [Google Scholar] [CrossRef]
- Zhang, Z.X.; Sun, D.M.; Rong, Z.H. Catalytic kinetic spectrophotometric determination of trace ascorbic acid. Spectrosc. Spect. Anal. 2004, 24, 873–875. [Google Scholar]
- Varella, S.D.; Pozetti, G.L.; Vilegas, W.; Varanda, E.A. Mutagenic Activity of Sweepings and Pigments from a Household-Wax Factory Assayed with Salmonella typhimurium. Food Chem. Toxicol. 2004, 42, 2029–2035. [Google Scholar] [CrossRef] [PubMed]
- Nestmann, E.R.; Douglas, G.R.; Matula, T.I.; Grant, C.E.; Kowbel, D.J. Mutagenic activity of rhodamine dyes and their impurities as detected by mutation induction in Salmonella and DNA damage in Chinese hamster ovary cells. Cancer Res. 1979, 39, 4412–4417. [Google Scholar] [PubMed]
- Panciera, M. The toxicity of Rhodamine-B to eggs and larvae of Crassostrea virginica. Natl. Shellfieries Assoc. Proc. 1967, 58, 7–8. [Google Scholar]
- Cholnoky-Pfannkuche, K. Untersuchungen über Toxizität II Experimente mit Ulothrix tenerrima Kützing. Hydrobiologia 1969, 33, 223–236. [Google Scholar] [CrossRef]
- Sihombing, G. The toxic effect of Rhodamine B in rats. Paediatr. Indones. 1984, 24, 125–138. [Google Scholar] [PubMed]
- Dixit, S.; Pandey, R.C.; Das, M.; Khanna, S.K. Food quality surveillance on colours in eatables sold in rural markets of Uttar Pradesh. J. Food Sci. Technol. 1995, 32, 373–376. [Google Scholar]
- Skjolding, L.M.; Jørgensen, L.v.G.; Dyhr, K.S.; Köppl, C.J.; McKnight, U.S.; Bauer-Gottwein, P.; Mayer, P.; Bjerg, P.L.; Baun, A. Assessing the aquatic toxicity and environmental safety of tracer compounds Rhodamine B and Rhodamine WT. Water Res. 2021, 197, 117109. [Google Scholar] [CrossRef]
- Bernhard, M.; Cagnetti, P.; Zattera, A. Results with Rhodamine B as a pollutant tracer in shallow seawater. G. Fis. Sanit. Prot. Radiaz. 1972, 16, 71–80. [Google Scholar]
- Rochat, J.; Demenge, P.; Rerat, J.C. Toxicologic study of a fluorescent tracer: Rhodamine B. Toxicol. Eur. Res. 1978, 1, 23–26. [Google Scholar]
- Sørensen, B.L.; Wakeman, R.J. Filtration characterisation and specific surface area measurement of activated sludge by Rhodamine B adsorption. Water Res. 1996, 30, 115–121. [Google Scholar] [CrossRef]
- Chiang, T.-L.; Wang, Y.-C.; Ding, W.-H. Trace Determination of Rhodamine B and Rhodamine 6G Dyes in Aqueous Samples by Solid-phase Extraction and High-performance Liquid Chromatography Coupled with Fluorescence Detection. J. Chin. Chem. Soc. 2012, 59, 515–519. [Google Scholar] [CrossRef]
- International Atomic Energy Agency. Marine radioactivity studies in the Suez Canal. In Field Tracer Experiments; IAEA Mission Report; A. Plata (EGY/07/002-01): Vienna, Austria, 1995. [Google Scholar]
- Barek, J.; Zima, J. Eighty Years of Polarography—History and Future. Electroanalysis 2003, 15, 467–472. [Google Scholar] [CrossRef]
- Zittel, H.E.; Miller, F.J. A Glassy-Carbon Electrode for Voltammetry. Anal. Chem. 1965, 37, 200–203. [Google Scholar] [CrossRef]
- Adams, R.N. Carbon Paste Electrodes. Anal. Chem. 1958, 30, 1576. [Google Scholar] [CrossRef]
- Matthews, D.R.; Bown, E.; Watson, A.; Holman, R.R.; Steemson, J.; Hughs, S.; Scott, D. Pen-sized digital 30-second blood glucose meter. Lancet 1987, 1, 778–779. [Google Scholar] [CrossRef]
- Honeychurch, K.C.; Hart, J.P. Screen-printed electrochemical sensors for monitoring metal pollutants. TrAC 2003, 22, 456–469. [Google Scholar] [CrossRef]
- Hughes, G.; Westmacott, K.; Honeychurch, K.C.; Crew, A.; Pemberton, R.M.; Hart, J.P. Recent Advances in the Fabrication and Application of Screen-Printed Electrochemical (Bio)Sensors Based on Carbon Materials for Biomedical, Agri-Food and Environmental Analyses. Biosensors 2016, 6, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hart, J.P.; Crew, A.; Crouch, E.; Honeychurch, K.C.; Pemberton, R.M. Some Recent Designs and Developments of Screen-Printed Carbon Electrochemical Sensors/Biosensors for Biomedical, Environmental, and Industrial Analyses. Anal. Lett. 2004, 37, 789–830. [Google Scholar] [CrossRef]
- Hart, J.P.; Abass, A.-K.; Honeychurch, K.C.; Pemberton, R.M.; Ryan, S.L.; Wedge, R. Sensors/Biosensors, Based on Screen-printing Technology for Biomedical Applications. Indian J. Chem. A 2003, 42, 709–718. [Google Scholar]
- Honeychurch, K.C. Printed thick-film biosensors. In Printed Films Materials Science and Applications in Sensors Electronics and Photonics; Prudenziati, M., Hormadaly, J., Eds.; Woodhead Publishing: Sawston, UK, 2012; pp. 366–409. [Google Scholar] [CrossRef]
- Yu, L.; Mao, Y.; Qu, L. Simple Voltammetric Determination of Rhodamine B by Using the Glassy Carbon Electrode in Fruit Juice and Preserved Fruit. Food Anal. Methods 2013, 6, 1665–1670. [Google Scholar] [CrossRef]
- Kartika, A.E.; Setiyanto, H.; Manurung, R.V.; Jenie, S.-N.A.; Saraswaty, V. Silver Nanoparticles Coupled with Graphene Nanoplatelets Modified Screen-Printed Carbon Electrodes for Rhodamine B Detection in Food Products. ACS Omega 2021, 6, 31477–31484. [Google Scholar] [CrossRef]
- Benmassaoud, Y.; Murtada, K.; Salghi, R.; Zougagh, M.; Ríos, Á. Surface Polymers on Multiwalled Carbon Nanotubes for Selective Extraction and Electrochemical Determination of Rhodamine B in Food Samples. Molecules 2021, 26, 2670. [Google Scholar] [CrossRef]
- Zhu, X.; Wu, G.; Wang, C.; Zhang, D.; Yuan, X. A miniature and low-cost electrochemical system for sensitive determination of rhodamine B. Measurement 2018, 120, 206–212. [Google Scholar] [CrossRef]
- Harrison, I.R.; Quickenden, T.I. Effect of electrodic dye layers on electron transfer and power conversion efficiency in rhodamine photoelectrochemical cells. Phys. Chem. 1987, 91, 1481–1486. [Google Scholar] [CrossRef]
- Ngo, H.T.; Nguyen, V.T.; Manh, T.D.; Toan, T.T.T.; Triet, M.T.N.; Binh, N.T.; Hoan, N.T.V.; Thien, T.V.; Khieu, D.Q. Voltammetric Determination of Rhodamine B Using a ZIF-67/Reduced Graphene Oxide Modified Electrode. J. Nanomater. 2020, 2020, 4679061. [Google Scholar] [CrossRef]
- Golestaneh, M.; Ghoreishi, S.M. Sensitive Determination of Rhodamine B in Real Samples at the Surface of a Multi-walled Carbon Nanotubes Paste Electrode. Anal. Bioanal. Electrochem. 2020, 12, 81–92. [Google Scholar]
- Mchedlov-Petrossyan, N.O.; Kukhtik, V.I.; Bezugliy, V.D. Dissociation, tautomerism and electroreduction of xanthene and sulfonephthalein dyes in N, N-dimethylformamide and other solvents. J. Phys. Org. Chem. 2003, 16, 380–397. [Google Scholar] [CrossRef]
- Beitollahi, H.; Mohammadi, S.Z.; Safaeia, M.; Tajik, S. Applications of electrochemical sensors and biosensors based on modified screen-printed electrodes: A review. Anal. Methods 2020, 12, 1547–1560. [Google Scholar] [CrossRef]
- Masa, J.; Batchelor-McAuley, C.; Schuhmann, W.; Compton, R.G. Koutecky-Levich analysis applied to nanoparticle modified rotating disk electrodes: Electrocatalysis or misinterpretation. Nano. Res. 2014, 7, 71–78. [Google Scholar] [CrossRef]
- Honeychurch, K.C.; Hart, J.P.; Cowell, D.C. Voltammetric Behavior and Trace Determination of Lead at a Mercury-Free Screen-Printed Carbon Electrode. Electroanalysis 2000, 12, 171–177. [Google Scholar] [CrossRef]
- Honeychurch, K.C.; Crew, A.; Northall, H.; Radbourne, S.; Davies, O.; Newman, S.; Hart, J.P. The redox behaviour of diazepam (Valium®) using a disposable screen-printed sensor and its determination in drinks using a novel adsorptive stripping voltammetric assay. Talanta 2013, 116, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Honeychurch, K.C.; Brooks, J.; Hart, J.P. Development of a voltammetric assay, using screen-printed electrodes, for clonazepam and its application to beverage and serum samples. Talanta 2016, 147, 510–515. [Google Scholar] [CrossRef]
- Honeychurch, K.C.; Hart, J.P.; Cowell, D.C. Voltammetric studies of lead at a 1-(2-pyridylazo)-2-naphthol modified screen-printed carbon electrode and its trace determination in water by stripping voltammetry. Anal. Chim. Acta 2001, 431, 89–99. [Google Scholar] [CrossRef]
- Honeychurch, K.C.; Gilbert, L.; Hart, J.P. Electrocatalytic behaviour of citric acid at a cobalt phthalocyanine-modified screen-printed carbon electrode and its application in pharmaceutical and food analysis. Anal. Bioanal. Chem. 2010, 396, 3103–3111. [Google Scholar] [CrossRef]
- Thomas, T.; Mascarenhas, R.J.; Swamy, B.E.K. Poly(Rhodamine B) modified carbon paste electrode for the selective detection of dopamine. J. Mol. Liq. 2012, 174, 70–75. [Google Scholar] [CrossRef]
- Austin, J.M.; Harrison, I.R.; Quickenden, T.I. Electrochemical and Photoelectrochemical Properties of Rhodamine B. J. Phys. Chem. 1986, 90, 1839–1843. [Google Scholar] [CrossRef]
- Rajeev, J.; Nidhi, S.; Meenakshi, B. Electrochemical Degradation of Rhodamine B Dye in Textile and Paper Industries Effluent. J. Sci. Ind. Res. 2003, 62, 1138–1144. Available online: http://nopr.niscpr.res.in/handle/123456789/17640 (accessed on 30 May 2022).
- Aslam, Z.; Yousaf, I.; Zahir, A.; Akhtar, A. Adsorptive performance of MWCNTs for simultaneous cationic and anionic dyes removal; kinetics, thermodynamics, and isotherm study. Turk. J. Chem. 2021, 45, 1189–1200. [Google Scholar] [CrossRef] [PubMed]
- Stratton, S.G.; Taumoefolau, G.H.; Purnell, G.E.; Rasooly, M.; Czaplyski, W.L.; Harbron, E.J. Tuning the pKa of Fluorescent Rhodamine pH Probes through Substituent Effects. Chem. Eur. J. 2017, 23, 14064–14072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pletcher, D.; Greff, R.; Peat, R.; Peter, L.M.; Robinson, J. Potential Sweep Techniques and Cyclic Voltammetry. In Instrumental Methods in Electrochemistry; Pletcher, D., Greff, R., Peat, R., Peter, L.M., Robinson, J., Eds.; Woodhead Publishing: Sawston, UK, 2010; pp. 178–228. [Google Scholar] [CrossRef]
- Zhu, D.; Li, Q.; Honeychurch, K.C.; Piano, M.; Chen, G. Determination of Malachite Green in Aquaculture Water by Adsorptive Stripping Voltammetry. Anal. Lett. 2016, 49, 1436–1451. [Google Scholar] [CrossRef]
- Compton, R.G.; Mason, D.; Unwin, P.R. The reduction of fluorescein in aqueous solution (at pH 6). A new DISP2 reaction. J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases 1988, 84, 483–489. [Google Scholar] [CrossRef]
- Okada, M.; Nishio, I.; Takahashi, F.; Tatsumi, H.; Jin, J. Cathodic Electrochemiluminescence from Rhodamine B in Aqueous Media Using Peroxydisulfate as Co-reactant. Chem. Lett. 2021, 50, 1659–1661. [Google Scholar] [CrossRef]
- Bohrer, D.; Schwedt, G. Anodic-Stripping Voltammetric Determination of Thallium as [Tlbr4]-Rhodamine-B Complex. Fresenius J. Anal. Chem. 1998, 362, 224–229. [Google Scholar] [CrossRef]
- Hou, X.; Shen, W.; Huang, X.; Ai, Z.; Zhang, L. Ascorbic acid enhanced activation of oxygen by ferrous iron: A case of aerobic degradation of rhodamine B. J. Hazard Mater. 2016, 5, 67–74. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Du, Y.; Liu, H.; Ma, J. Ascorbic acid/Fe0 composites as an effective persulfate activator for improving the degradation of rhodamine B. RSC Adv. 2018, 8, 12791–12798. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.-L.; Liang, A.-H. Catalytic kinetic determination of trace amounts of ascorbic acid with single-sweep oscillopolarography. Anal. Chim. Acta 1993, 278, 53–58. [Google Scholar] [CrossRef]
- Pavan, F.A.; Ribeiro, E.S.; Gushikem, Y. Congo Red Immobilized on a Silica/Aniline Xerogel: Preparation and Application as an Amperometric Sensor for Ascorbic Acid. Electroanalysis 2005, 17, 625–629. [Google Scholar] [CrossRef]
- Wu, J.-M.; Zhang, T.-W. Photodegradation of Rhodamine B in water assisted by Titania films prepared through a novel procedure. J. Photochem. Photobiol. A 2004, 162, 171–177. [Google Scholar] [CrossRef]
- Shea, C.R.; Chen, N.; Wimberly, J.; Hasan, T. Rhodamine dyes as potential agents for photochemotherapy of cancer in human bladder carcinoma cells. Cancer Res. 1989, 49, 3961–3965. [Google Scholar]
- Ma, Y.; Yao, J.-N. Photodegradation of Rhodamine B catalyzed by TiO2 thin films. J. Photochem. Photobiol. A 1998, 116, 167–170. [Google Scholar] [CrossRef]
- Wu, T.X.; Liu, G.M.; Zhao, J.C.; Hidaka, H.; Serpone, N. Photoassisted degradation of dye pollutants. V. Self-photosensitized oxidative transformation of Rhodamine B under visible light irradiation in aqueous TiO2 dispersions. J. Phys. Chem. B 1998, 102, 5845–5851. [Google Scholar] [CrossRef]
- Watanabe, T.; Takizawa, T.; Honda, K. Photocatalysis through excitation of adsorbates. 1. Highly efficient N-deethylation of rhodamine B adsorbed to cadmium sulfide. J. Phys. Chem. 1977, 81, 1845–1851. [Google Scholar] [CrossRef]
- Wang, H.; Lu, L.; Zhu, S.; Li, Y.; Cai, W. The phototoxicity of xanthene derivatives against E. coli, Staphylococcus aureus, and Saccharomyces cerevisiae. Curr. Microbiol. 2006, 52, 1–5. [Google Scholar] [CrossRef]
- Mizuno, N.; Fujiwara, A.; Morita, E. Effect of dyes on the photodecomposition of pyridoxine and pyridoxamine. J. Pharm. Pharmacol. 1981, 33, 373–376. [Google Scholar] [CrossRef]
- Kaminski, E.E.; Cohn, R.M.; Mcguire, J.L.; Carstensen, J.T. Light Stability of Norethindrone and Ethinyl Estradiol Formulated with FD&C Colorants. J. Pharm. Sci. 1979, 8, 368–370. [Google Scholar] [CrossRef]
- Baugh, R.; Calvert, R.T.; Fell, J.T. Stability of Phenylbutazone in Presence of Pharmaceutical Colors. J. Pharm. Sci. 1977, 66, 733–735. [Google Scholar] [CrossRef] [PubMed]
- Stanley, R.; Jebasingh, J.A.; Vidyavathy, S.M.; Stanley, K.P.; Ponmani, P.; Shekinah, M.E.; Vasanthi, J. Excellent Photocatalytic degradation of Methylene Blue, Rhodamine B and Methyl Orange dyes by Ag-ZnO nanocomposite under natural sunlight irradiation. Optik 2021, 231, 166518. [Google Scholar] [CrossRef]
- Maya, F.; Horstkotte, B.; Estela, J.M.; Cerdà, V. Lab in a syringe: Fully automated dispersive liquid–liquid microextraction with integrated spectrophotometric detection. Anal. Bioanal Chem. 2012, 404, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Hasi, W.-L.-J.; Lin, X.; Han, S.; Xiu-Tao Lou, X.-T.; Yang, F.; Lin, D.-Y.; Lu, Z.W. Rapid and sensitive SERS method for determination of Rhodamine B in chili powder with paper-based substrates. Anal. Methods 2015, 7, 5289–5294. [Google Scholar] [CrossRef]
- Dixit, S.; Purshottam, S.K.; Gupta, S.K.; Khanna, S.K.; Das, M. Usage pattern and exposure assessment of food colours in different age groups of consumers in the State of Uttar Pradesh. Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess. 2010, 27, 181–189. [Google Scholar] [CrossRef]
- Wang, C.C.; Masi, A.N.; Fernandez, L. On-line micellar-enhanced spectrofluorimetric determination of rhodamine dye in cosmetics. Talanta 2008, 75, 135–140. [Google Scholar] [CrossRef]
- Hofstraat, J.W.; Steendijk, M.; Vriezekolk, G.; Schreurs, W.; Broer, G.J.A.A.; Wijnstok, N. Determination of rhodamine WT in surface water by solid-phase extraction and HPLC with fluorescence detection. Water Res. 1991, 25, 883–890. [Google Scholar] [CrossRef]
- Soylak, M.; Unsal, Y.E.; Yilmaz, E.; Tuzen, M. Determination of rhodamine B in soft drink, waste water and lipstick samples after solid phase extraction. Food Chem. Toxicol. 2011, 49, 1796–1799. [Google Scholar] [CrossRef]
- Yilmaz, E.; Soylak, M. A novel and simple deep eutectic solvent based liquid phase microextraction method for rhodamine B in cosmetic products and water samples prior to its spectrophotometric determination. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2018, 202, 81–86. [Google Scholar] [CrossRef]
- Sun, J.; Gan, T.; Li, Y.; Shi, Z.; Liu, Y. Rapid and sensitive strategy for Rhodamine B detection using a novel electrochemical platform based on core–shell structured Cu@carbon sphere nanohybrid. J. Electroanal. Chem. 2014, 724, 87–94. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, L.; Wang, W.; Chen, Z. Sensitive Electrochemical Determination of Rhodamine B Based on a Silica-Pillared Zirconium Phosphate/Nafion Composite Modified Glassy Carbon Electrode. J. AOAC Int. 2016, 99, 760–765. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Linear Range µg/L | Limit of Detection µg/L | Technique | Comments | Reference |
|---|---|---|---|---|---|
| Wastewater and surface water | 50–1000 | 0.5 | High-performance liquid chromatography with fluorescence detection | Solid-phase extraction | [17] |
| Fruit juice and Preserved fruit | 4.78–956.1 | 2.93 | Differential pulse voltammetry at a glassy carbon electrode | Preserved fruits extracted in water with the aid of ultrasonication for 4 h, and then filtrated under vacuum | [28] |
| Chili powder and tomato sauce | 5–100 | 1.44 | Magnetic solid-phase extraction, with SPCE modified by multiwalled carbon nanotubes and a molecular imprinted polymer | Chili powder extracted with acetonitrile with the aid of ultrasonication. Centrifuged and mixed with NaCl and distilled water, stored at −20 °C for 2 h. Filtrated, under nitrogen, and reconstituted with distilled water at pH 5. Tomato sauce extract obtained via direct filtration | [30] |
| Tomato and chili sauces | 0.96–44.07 | 1.79 | Zeolite imidazolate framework-67/reduced graphene oxide modified glassy carbon electrode | Sample was sonicated in water by ultrasonication and stirring The Rhodamine B concentration was then determined using the optimized conditions | [33] |
| Water samples and hair colors | 48–720 | 9.6 | Multi-walled carbon nanotube carbon paste electrode | Hair color was sonicated in ethanol and diluted with PBS (pH = 3.0) and examined electrochemically | [34] |
| Water samples and soft drinks | 23–2000 | 7.0 | Spectrophotometric detection | Dispersive liquid–liquid microextraction | [67] |
| Chili powder | 1.0–10,000 µg/g | 1.0 µg/g | Surface-enhanced Raman spectroscopy | Extraction with acetonitrile via shaking, sonication and centrifuging | [68] |
| Bakery products, beverages and cooked foods | 600–5000 | 10 | High-performance liquid chromatography | Officially prescribed method of the Indian food regulatory authority | [69] |
| Cosmetics | 0.765–478.03 | 0.239 | Micellar-enhanced fluorimetry | Lipstick extracted in water by stirring at 333 K (ca. 60 °C) for 15 min | [70] |
| Surface water | -- | 0.000010 | High-performance liquid chromatography with fluorescence detection | Solid-phase extraction of 1 L of sample water reconstituted in a 1.0 mL of mobile phase | [71] |
| Soft drink, waste water and cosmetics | 250–3000 | 3.14 | Spectrophotometry at 556 nm | Solid-phase extraction | [72] |
| Cosmetic products and water samples | -- | 2.2 | Spectrophotometry at 550 nm | Deep eutectic solvent-based liquid-phase microextraction | [73] |
| Chili powder, tomato juice, soy sauce and pasta sauce | 144–1440 | 48 | Cu@CS nanohybrid-modified glassy carbon electrode | Tomato sauce extract was obtained by filtration. Chili powder was sonicated in acetonitrile and centrifuged. NaCl and water were added and the mixture frozen and centrifuged. The acetonitrile layer collected. Soy sauce was sonicated in a mixture of ethanol/water/acetic acid, and filtered. Pasta sauce was diluted in water and filtered. Once processed, extracts were made up in pH 6.5 PBS, and examined using the optimized procedure | [74] |
| Chili powder and preserved fruit | 5–2400 | 2.1 | Silica-pillared zirconium phosphate/Nafion composite modified glassy carbon electrode | Sample added to acetone–n-hexane solution and sonicated. Mixture then centrifuged and supernatant extracted with n-hexane and organic phase discarded following addition of water. An aliquot was then diluted in pH 5.0 BR buffer and investigated using the optimized conditions | [75] |
| Surface water | 60–4000 | 10 | Differential pulse voltammetry at an unmodified SPCE | Dilution with phosphate buffer and addition of ascorbic acid | This work |
| Original Concentration ng/mL | Added ng/mL | Found ng/mL | % Recovery | |
|---|---|---|---|---|
| 1 | ND | 96.0 | 90.0 | 93.8 |
| 2 | ND | 96.0 | 90.5 | 94.3 |
| 3 | ND | 96.0 | 89.9 | 93.6 |
| 4 | ND | 96.0 | 87.1 | 90.7 |
| 5 | ND | 96.0 | 95.3 | 99.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Honeychurch, K.C. Voltammetric Behaviour of Rhodamine B at a Screen-Printed Carbon Electrode and Its Trace Determination in Environmental Water Samples. Sensors 2022, 22, 4631. https://doi.org/10.3390/s22124631
Honeychurch KC. Voltammetric Behaviour of Rhodamine B at a Screen-Printed Carbon Electrode and Its Trace Determination in Environmental Water Samples. Sensors. 2022; 22(12):4631. https://doi.org/10.3390/s22124631
Chicago/Turabian StyleHoneychurch, Kevin C. 2022. "Voltammetric Behaviour of Rhodamine B at a Screen-Printed Carbon Electrode and Its Trace Determination in Environmental Water Samples" Sensors 22, no. 12: 4631. https://doi.org/10.3390/s22124631
APA StyleHoneychurch, K. C. (2022). Voltammetric Behaviour of Rhodamine B at a Screen-Printed Carbon Electrode and Its Trace Determination in Environmental Water Samples. Sensors, 22(12), 4631. https://doi.org/10.3390/s22124631