Abstract
(S)-2-methyl-5-(prop-1-en-2-yl)cyclopent-1-ene-1-carbaldehyde (2) is a component of the essential oils of several plants and has also been used in many syntheses, usually of terpenes. Although the preparation of compound 2 has been reported several times, no side products have been reported previously. In this Short Note, we present the identification of one such side product. This new compound (3) arises from a vinylogous aldol autocondensation, occurring after the formation of 2, even when the reaction runs for a short time. However, it can diminish the yield of 2 when the reaction is allowed to continue for a longer time.
1. Introduction
Natural chiral building blocks are extremely useful for the construction of complex molecules, such as natural products and derivatives. Those that are easily prepared from natural sources have been especially employed in synthesis. Among them, compound 2, a terpenic molecule, has been used in different syntheses from its first preparation in 1964 [1] to the latest synthesis of diterpenes [2,3]. This molecule is also present in the essential oils of several plants [4,5] and is usually prepared from limonene. In an ongoing project, we needed to prepare 2, and thus, we resorted to the classical synthesis depicted in Scheme 1 [6]. Ozonolysis of (R)-limonene and intramolecular aldol reaction of the keto-aldehyde 1 using piperidine and acetic acid gave the expected compound 2 in 67.2% yield. The choice of reagents is aimed at avoiding the other possible intramolecular aldol reaction of the enolate of the ketone over the aldehyde [7].
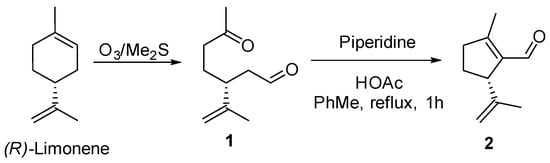
Scheme 1.
Classical synthesis of 2.
We repeated this process several times and observed that every time, a subproduct, slightly less polar than 2, was formed in a small but consistent amount.
We decided to study this side product to investigate the possible impact of its formation on the yield of the desired compound.
2. Results and Discussion
The new compound was isolated by column chromatography and studied by 1H-NMR at 600 MHz and 13C-NMR at 150 MHz. The NMR study was conducted in C6D6, since in CDCl3 the compound decomposed slowly. The 1H-NMR showed a simple and well-resolved spectrum (see Supporting Information) with one proton at 10.3 ppm, an AB system in the vinylic part, 2 sets of signals for terminal double bonds, two signals for allylic protons, three singlet methyl groups, and other signals accounting for a total of 26 protons. The 13C-NMR indicated the presence of 20 carbons, including 4 aliphatic and 2 vinylic CH2 carbons, 6 quaternary vinylic carbons, and the carbon of an aldehyde.
Overall, a composition of C20H26O was deduced from the NMR spectra, which implies an IHD of 8. Mass spectroscopy confirmed the suggested composition for compound 3, and UV indicates some conjugation on the molecule (λmax = 351 nm). Thus, the presence of five double bonds and two cycles was deduced.
A study including COSY, HSQC, HMBC, and ROESY spectra was undertaken, and structure 3 (Figure 1), an autocondensation product of 2, was proposed for the new compound 3.
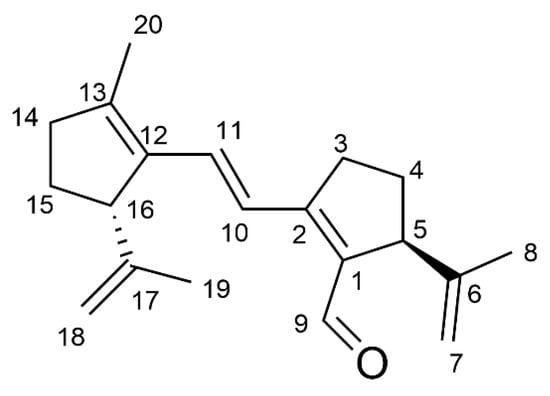
Figure 1.
Structure of compound 3. The numbering scheme for the NMR description is shown.
Figure 2 shows the more relevant correlations found for the COSY spectrum of compound 3.
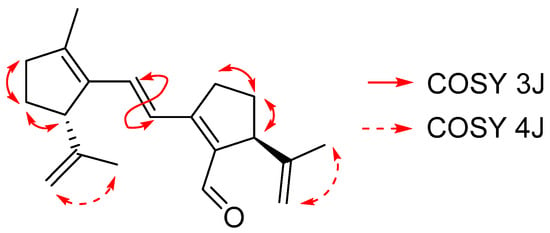
Figure 2.
Relevant correlations found in the COSY spectrum of compound 3.
The COSY spectrum indicates the protons of each cycle and the trans-double bond. The HMBC spectrum (Figure 3) confirmed the position of the quaternary carbons of 3.
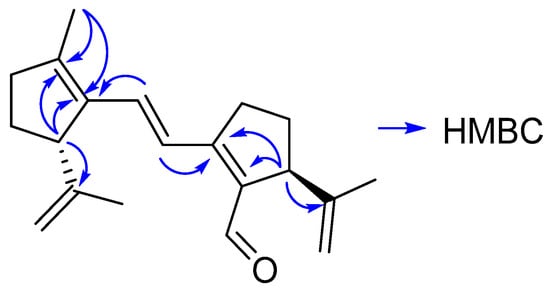
Figure 3.
Relevant correlations found in the HMBC spectrum of compound 3.
The ROESY spectrum (Figure 4) also corroborates the suggested structure of the molecule.
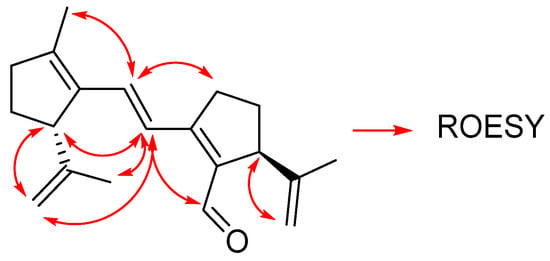
Figure 4.
Relevant correlations found in the ROESY spectrum of compound 3.
Compound 3 is thus the result of the reaction of two molecules of compound 2 under the cyclization conditions. To confirm this, a sample of 2 was subjected to the same conditions used for the cyclization of 1.
After 1 h of reaction, 3 was formed in the same proportion as found in the cyclization of 1, that is, around 7, with a large proportion of 2 being recovered unaltered. This confirms that 3 is formed by a vinylogous aldol condensation (Scheme 2).
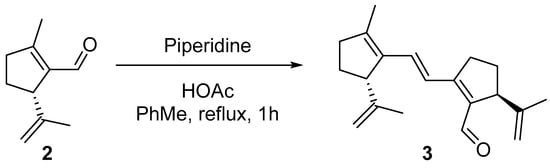
Scheme 2.
Preparation of 3 from 2.
This compound is formed in low yield under the usual reaction conditions, but can become relevant if the reaction is allowed to proceed for a longer time.
3. Materials and Methods
3.1. Chemicals and Instrumentation
All solvents and reagents were purified using standard techniques or used as supplied by commercial sources. Reactions under standard conditions were monitored by thin-layer chromatography (TLC) on Merck 60 F254 plates (Darmstadt, Germany). Silica gel (200–300 mesh) was used for flash column chromatography. NMR spectra were recorded in C6D6, at 600 MHz for 1H NMR and 150 MHz for 13C NMR on a Bruker Avance instrument (Karlsruhe, Germany). Chemical shifts are given in (δ) parts per million and coupling constants (J) are given in Hz. 1H- and 13C-spectra were referenced using the residual solvent signal as an internal standard (7.16 for 1H-NMR and 128.06 MHz for 13C-NMR). The data are reported as follows: s = singlet, m = multiplet, d = doublet. The numbers for protons and carbons refer to the numbering scheme shown in Figure 1. High-resolution mass spectral analysis (HRMS) data were obtained using a VION IMS Q-TOF (Waters, Milford, MA, USA). UV spectra were recorded using a Thermoscientific GENESYS™ 180 instrument (Madison, WI, USA). using CHCl3 as solvent.
3.2. Aldol Cyclization of 1
To a solution of 1 (1.0 g, 5.94 mmol) in toluene (10 mL) was added piperidine (0.2 mL) and acetic acid (0.2 mL), and the mixture was refluxed for 1 h. Then, water was added, and the reaction was extracted with EtOAc. The organic phase was washed with aqueous 10% HCl, decanted, and dried over anhydrous Na2SO4. After filtration, the solvent was removed, and the crude reaction mixture was purified by column chromatography (95:5 Hex: EtOAc), yielding a mixture of known compound 2 (0.6 g, 67.2%) and 3 (65 mg, 7.7%).
(S)-2-((E)-2-((S)-2-methyl-5-(prop-1-en-2-yl)cyclopent-1-en-1-yl)vinyl)-5-(prop-1-en-2-yl)cyclopent-1-ene-1-carbaldehyde 3.
Slight yellow oil, Rf = 0.46 (Hex: EtOAc, 95:5 v/v). = +102.9 (c 0.7, CHCl3). 1H NMR (600 MHz, C6D6) δ 10.32 (s, 1H, H9), 7.39 (d, J = 15.7 Hz, 1H, H10), 6.64 (d, J = 15.7 Hz, 1H, H11), 4.81 (s, 2H, H18a and H7a), 4.79 (s, 1H, H7b), 4.74 (s, 1H, H18b), 3.67 (d, J = 8.6 Hz, 1H, H5), 3.55 (d, J = 9.5 Hz, 1H, H16), 2.49 (m, 1H, H3a), 2.37–2.22 (m, 2H, H3b and H14a), 2.08 (m, 1H, H14b), 1.93 (m, 1H, H15a), 1.74 (m, 1H, H4a), 1.67 (s, 3H, Me8), 1.62 (s, 3H, Me20), 1.56 (s, 3H, Me19), 1.60–1.47 (m, 2H, H15b and H4b). 13C NMR (150 MHz, C6D6) δ 186.23 (C9), 157.61 (C2), 148.36 (C17), 147.27 (C6), 145.69 (C13), 139.85 (C1), 135.68 (C12), 130.18 (C11), 120.90 (C10), 111.06 (C18), 110.24 (C7), 53.98 (C16), 51.72 (C5), 38.61 (C14), 32.64 (C3), 29.12 (C15), 28.77 (C4), 20.97 (C8), 18.97 (C19), 14.31 (C20). HRMS (ESI+): Calcd for C20H26ONa [M + Na] 305,1881, found 305,1876.
Supplementary Materials
Figures containing the 1H-NMR, 13C-NMR, COSY, HSQC, HMBC, HRMS, and UV of compound 3.
Author Contributions
M.M.A. and J.A.P. conceived and designed the experiments; A.M.-G. and J.F.R.-C. performed the experiments. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
Data is contained within the article or Supplementary Materials.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviation
The following abbreviation are used in this manuscript:
| IHD | Index of Hydrogen Deficiency |
References
- Wolinsky, J.; Slabaugh, M.R.; Gibson, T. Synthesis of 4-(2-Methyl-5-isopropenyl-lcyclopenten-l-yl)butan-2-one. A By-Product in the Synthesis of Pseudoionone. J. Org. Chem. 1964, 29, 3740–3742. [Google Scholar] [CrossRef]
- Chen, B.; Yang, Y.; Zhang, X.; Xu, D.; Sun, Y.; Chen, Y.; Wang, L. Total Syntheses of Brassicicenes A, R, and T. Org. Lett. 2023, 25, 8570−8574. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-Q.; Xu, K.; Min, L.; Li, C.-C. Asymmetric Total Syntheses of Hypoestin A, Albolic Acid, and Ceroplastol II. J. Am. Chem. Soc. 2022, 144, 10162−10167. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.-T.; Lee, T.-C.; Valchanova, A.K.; Okamoto, T.; Liao, P.-C.; Kokubugata, G.; Okuyama, Y. Match of fruit-scented cone volatile composition with genetic boundary in Cycas revoluta and implications for fruit mimicry pollination. Plant Biol. 2025, 27, 725–739. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Fang, J.; Zhang, C.; Ma, W. Faster predicting the content of key non-volatile compounds in rosemary using electronic nose with multivariate algorithms. Food Control 2025, 168, 110886. [Google Scholar] [CrossRef]
- Srikrishna, A.; Babu, N.C. An enantiospecific formal total synthesis of (−)-aplysin and (−)-debromoaplysin. Tetrahedron Lett. 2001, 42, 4913–4914. [Google Scholar] [CrossRef]
- Wolinsky, J.; Barker, W. The Synthesis of 1-acetyl-4-Isopropenyl-1-Cyclopentene. J. Am. Chem. Soc. 1960, 82, 636. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).