
Synthesis and Characterization of Hydrazine Bridge Cyclotriphosphazene Derivatives with Amide–Schiff Base Linkages Attached to Decyl and Hydroxy Terminal Groups



Abstract
1. Introduction
2. Results and Discussion
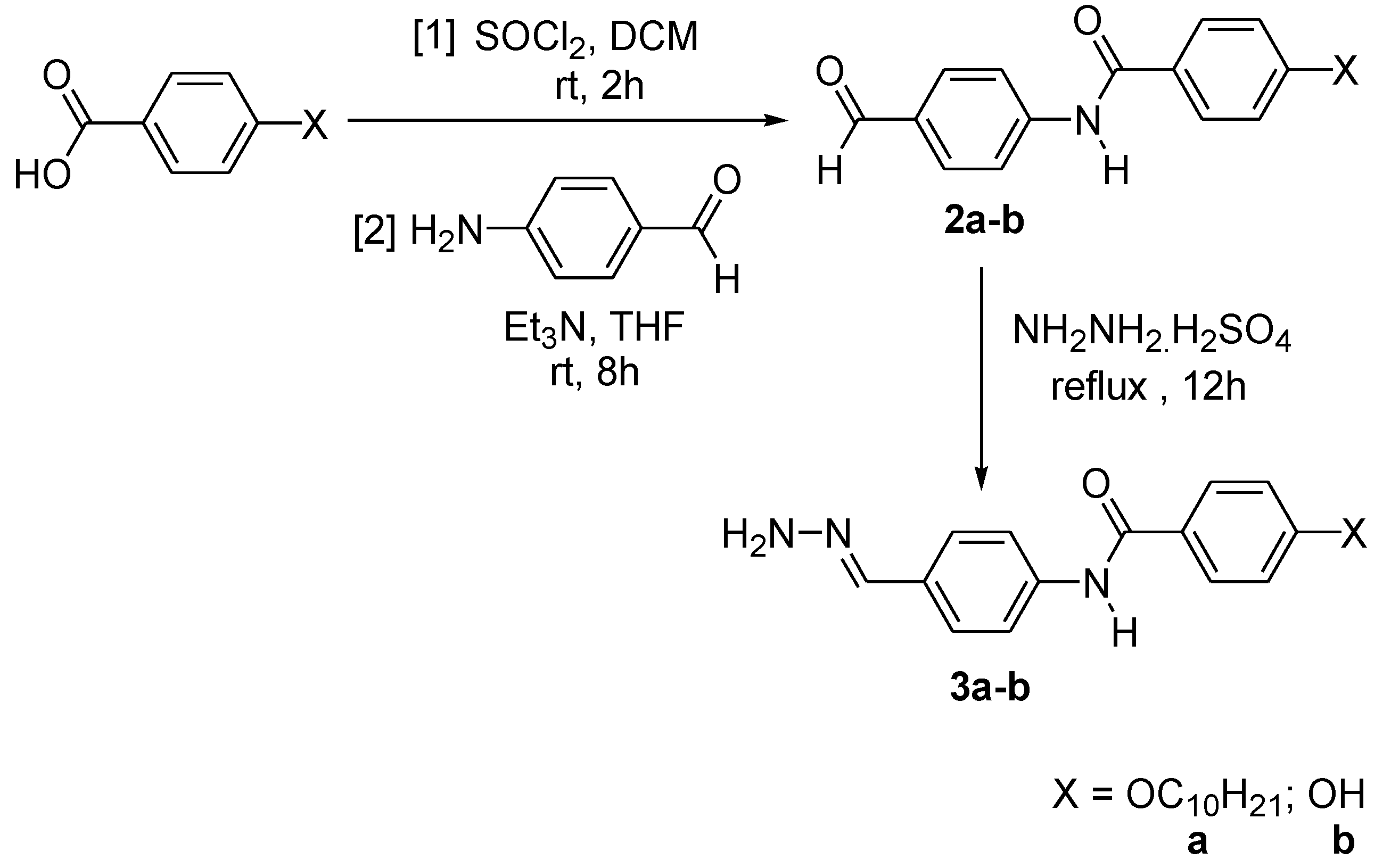
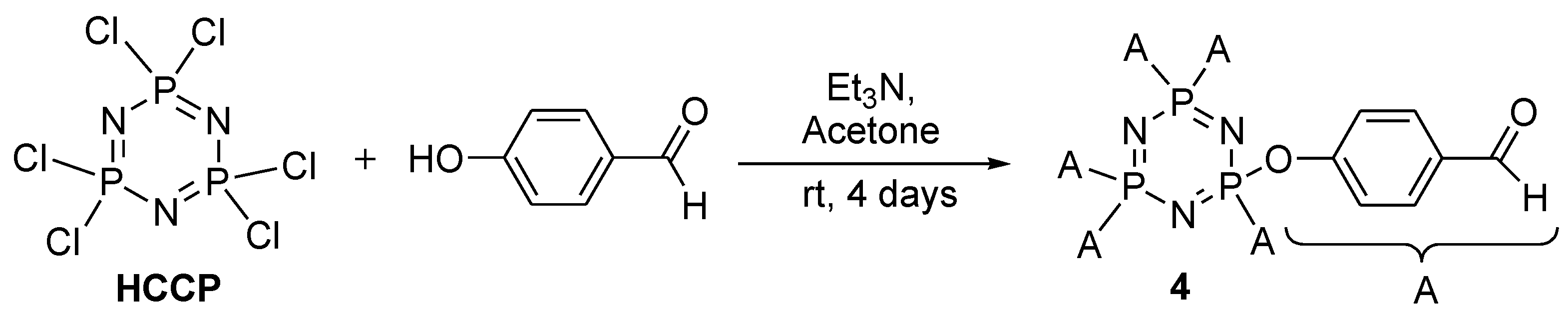
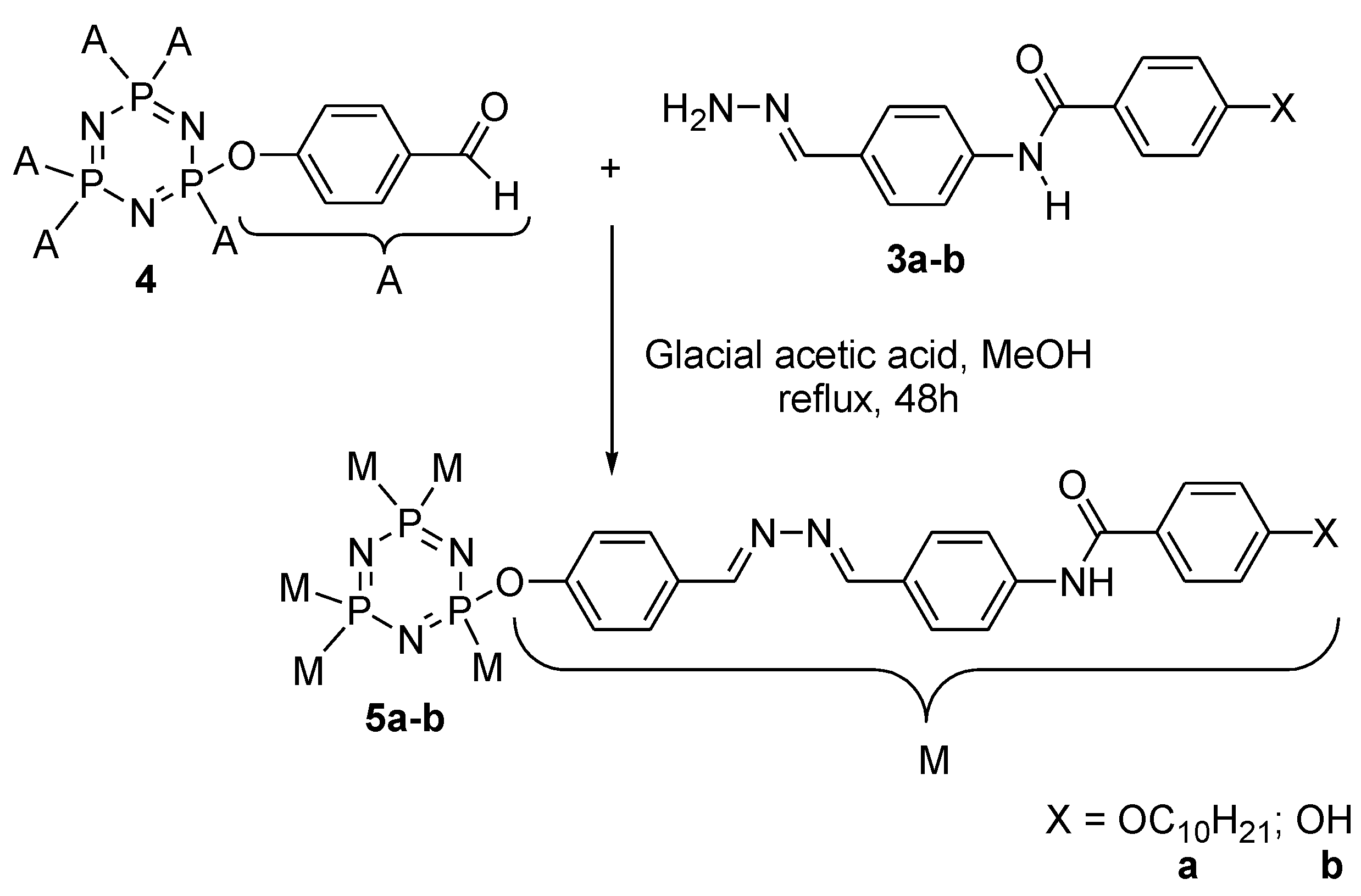
2.1. Synthesis Flow
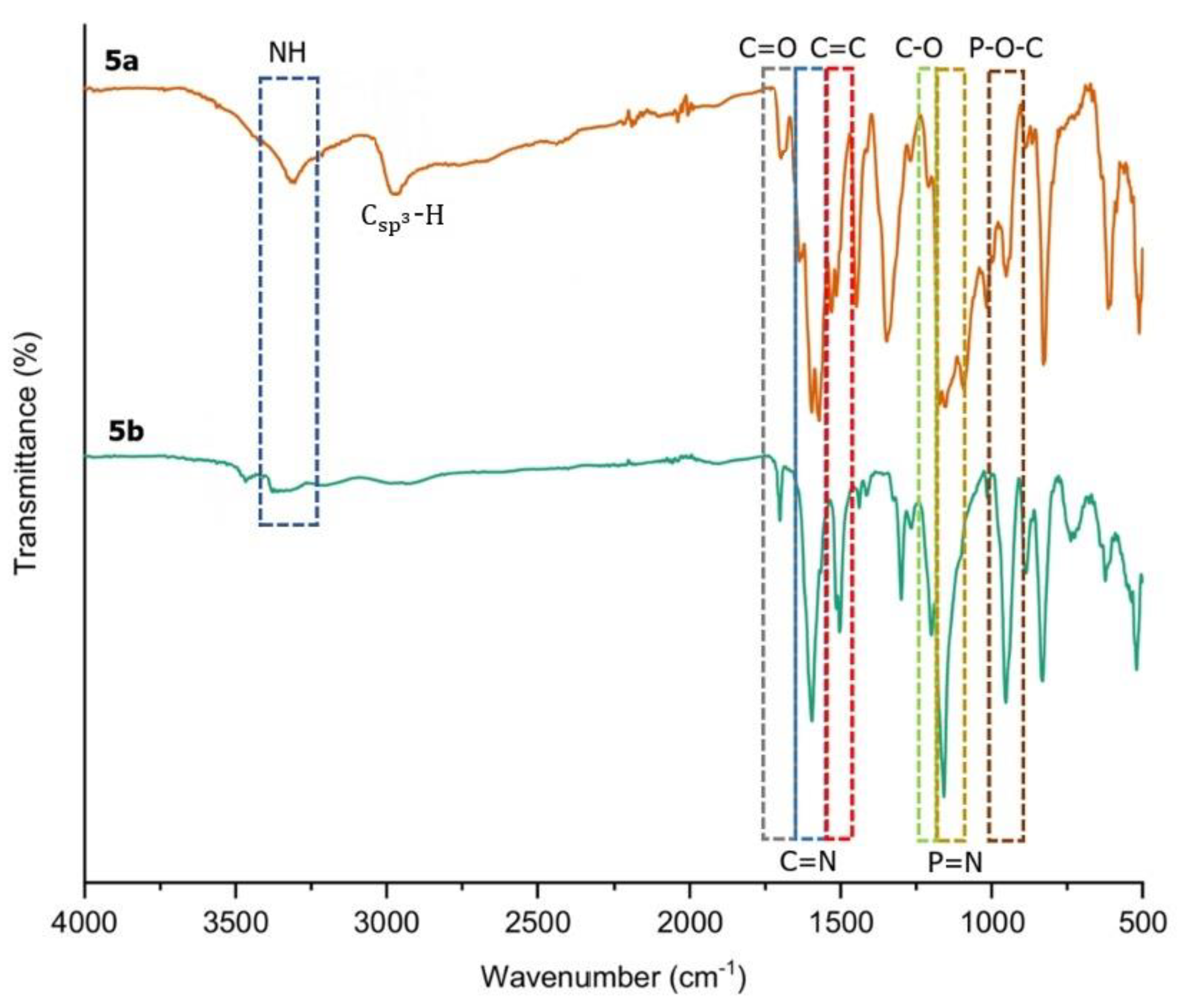
2.2. FTIR Spectral Discussion
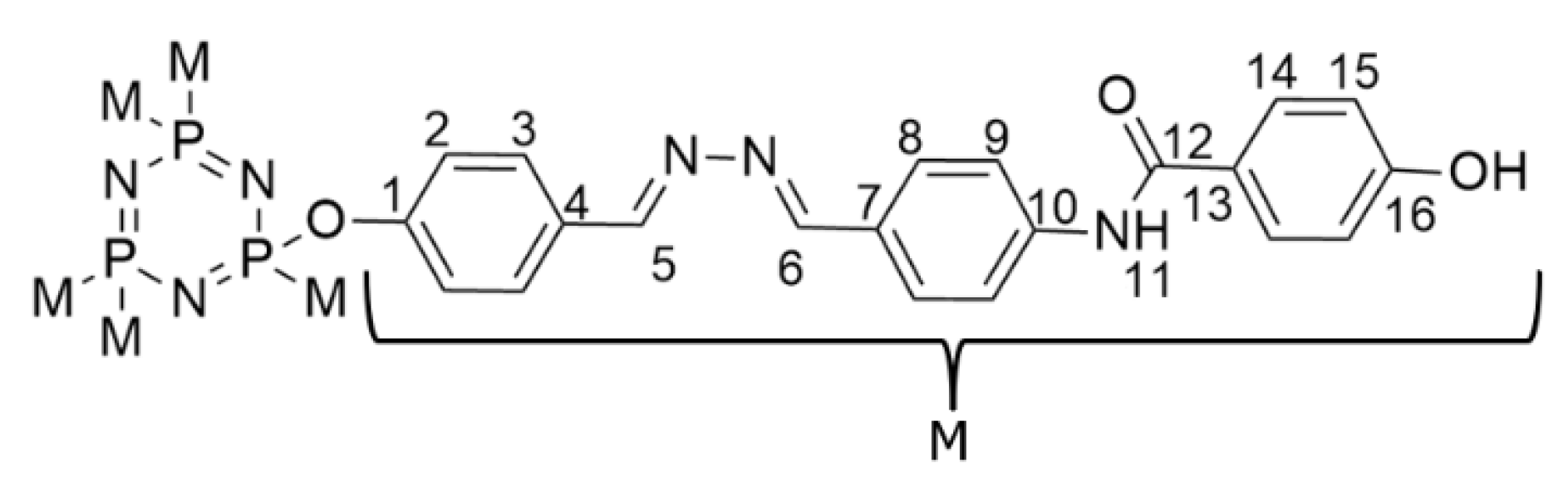
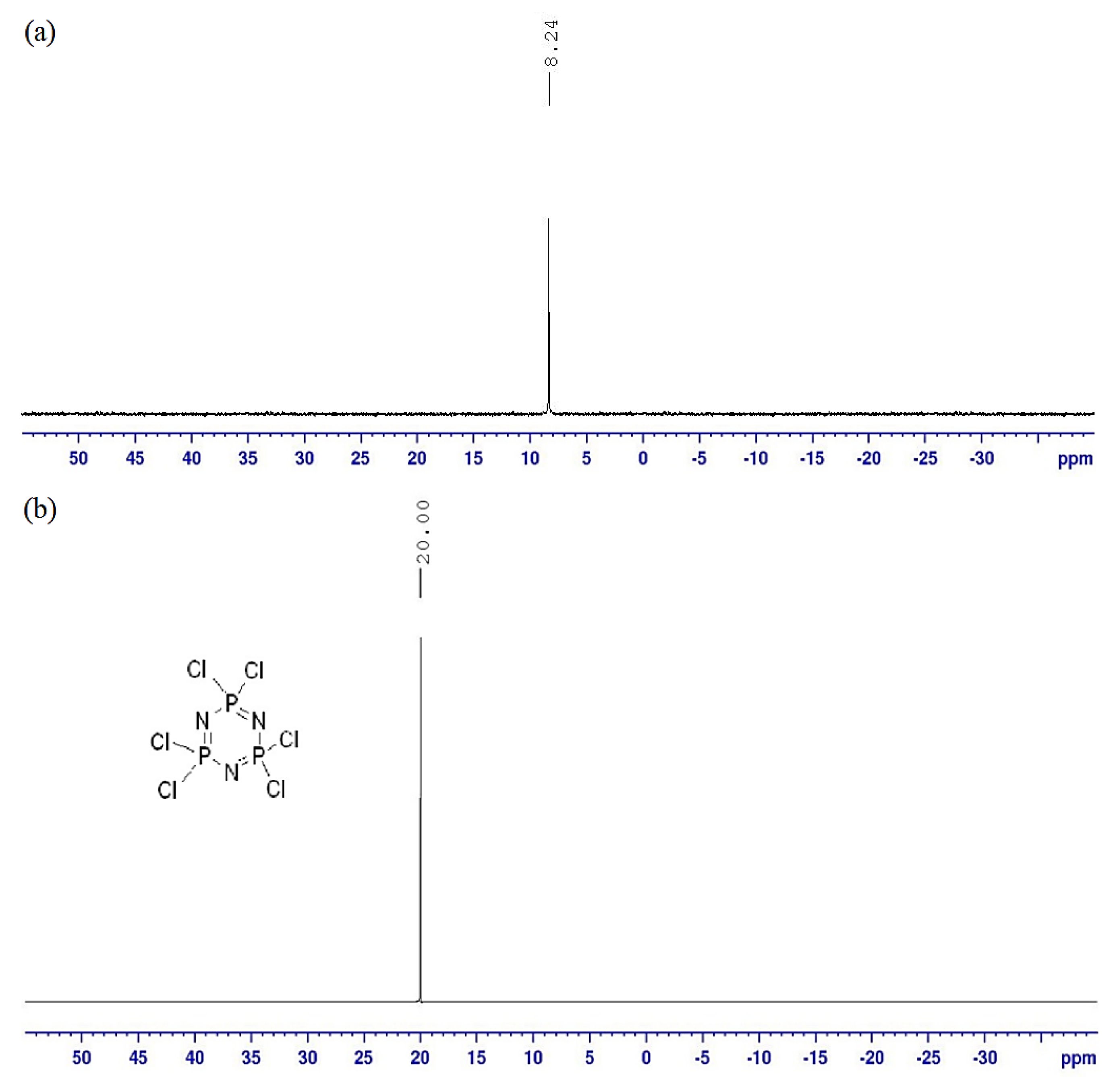
2.3. NMR Spectral Discussion
2.4. CHN Elemental Analysis
3. Materials and Methods
3.1. Chemicals
3.2. Instrumentation
3.3. Synthesis Method
3.3.1. Synthesis of 4-(Decyloxy)benzoic Acid, 1a
3.3.2. Synthesis of 4-{[4-(Substituted)benzoyl]amino}benzoic Acid, 2a–b
4-{[4-(Decyloxy)benzoyl]amino}benzoic Acid, 2a
4-{[4-(Hydroxy)benzoyl]amino}benzoic Acid, 2b
3.3.3. Synthesis of 4-[4-(Substituted-Hydrazonomethyl)phenyl]benzamide, 3a–b
4-[4-(Decyloxy-Hydrazonomethyl)phenyl]benzamide, 3a
4-[4-(Hydroxy-Hydrazonomethyl)phenyl]benzamide, 3b
3.3.4. Synthesis of Hexakis(4-Formlyphenoxy)cyclotriphosphazene, 4
3.3.5. Synthesis of Hexakis{4-((E)-((4-((E)-4-Substituted-Benzylidene)hydrazine-1-Ylidene) hydrazonomethyl)phenoxy}triphosphazene, 5a–b
Hexakis{4-((E)-((4-((E)-4-Decyloxy-Benzylidene)hydrazine-1-Ylidene)hydrazonomethyl) phenoxy}triphosphazene, 5a
Hexakis{4-((E)-((4-((E)-4-Hydroxy-Benzylidene)hydrazine-1-Ylidene)hydrazonomethyl) phenoxy}triphosphazene, 5b
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jeevananthan, V.; Shanmugan, S. Halogen-free layered double hydroxide-cyclotriphosphazene carboxylate flame retardants: Effects of cyclotriphosphazene di, tetra and hexacarboxylate intercalation on layered double hydroxides against the combustible epoxy resin coated on wood substrates. RSC Adv. 2022, 12, 23322–23336. [Google Scholar] [CrossRef] [PubMed]
- Jamain, Z.; Azman, A.N.A.; Razali, N.A.; Makmud, M.Z.H. A Review on Mesophase and Physical Properties of Cyclotriphosphazene Derivatives with Schiff Base Linkage. Crystals 2022, 12, 1174. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, Q.; Hao, Y.; Lan, X.; Haurie, L.; Zheng, D.; Huang, G. Preparation of a cyclotriphosphazene microsphere bearing a phosphaphenanthrene structure towards fire-safety and mechanical enhancement for epoxy and its aramid fiber composite. Mater. Adv. 2024, 5, 2860–2871. [Google Scholar] [CrossRef]
- Ali, S.; Zuhra, Z.; Butler, I.S.; Dar, S.U.; Hameed, M.U.; Wu, D.; Zhang, L.; Wu, Z. High-throughput synthesis of cross-linked poly(cyclotriphosphazene-co-bis(aminomethyl)ferrocene) microspheres and their performance as a superparamagnetic, electrochemical, fluorescent and adsorbent material. Chem. Eng. J. 2017, 315, 448–458. [Google Scholar] [CrossRef]
- Zhou, B.; Yang, C.; Wu, F.; Deng, T.; Guo, S.; Zhu, G.; Jiang, Y.; Wang, Z. Cyclotriphosphazene-based flame-retardant polymer electrolytes for high performance sodium metal batteries. Chem. Eng. J. 2020, 450, 138385. [Google Scholar] [CrossRef]
- Ning, K.; Zhou, L.-L.; Zhao, B. A novel aminothiazole-based cyclotriphosphazene derivate towards epoxy resins for high flame retardancy and smoke suppression. Polym. Degrad. Stab. 2021, 190, 109651. [Google Scholar] [CrossRef]
- He, Y.-F.; Ning, K.; Zhang, C.-Y.; Shao, Z.-B.; Zhao, B. Intramolecular cooperation and biphasic flame retardant mode of action: Effectiveness of hexa(1,2,4-triazol-3-ylamine) cyclotriphosphazene in epoxy resin. Adv. Ind. Eng. Polym. Res. 2024, 7, 326–337. [Google Scholar] [CrossRef]
- Yang, R.; Wang, B.; Han, X.; Ma, B.; Li, J. Synthesis and characterization of flame retardant rigid polyurethane foam based on a reactive flame retardant containing phosphazene and cyclophosphonate. Polym. Degrad. Stab. 2017, 144, 62–69. [Google Scholar] [CrossRef]
- Wang, D.; Xu, X.; Qiu, Y.; Wang, J.; Meng, L. Cyclotriphosphazene based materials: Structure, functionalization and applications. Prog. Mater. Sci. 2024, 142, 101232. [Google Scholar]
- Jamain, Z.; Khairuddean, M.; Loh, M.L.; Manaff, N.L.A.; Makmud, M.Z.H. Synthesis and characterization of hexasubstituted cyclotriphosphazene derivatives with azo linking units. Malays. J. Chem. 2020, 22, 125–140. [Google Scholar]
- Dagdag, O.; Kim, H. Recent advances for poly(cyclotriphosphazene) functionalized graphene oxide composites: Synthesis, properties and applications. J. Ind. Eng. Chem. 2024, 136, 89–122. [Google Scholar] [CrossRef]
- Li, Y.; Wang, B.; Sui, X.; Xie, R.; Xu, H.; Zhang, L.; Zhong, Y.; Mao, Z. Durable flame retardant and antibacterial finishing on cotton fabrics with cyclotriphosphazene/polydopamine/silver nanoparticles hybrid coatings. Appl. Surf. Sci. 2018, 435, 1337–1343. [Google Scholar] [CrossRef]
- Allcock, H.R. The crucial role of inorganic ring chemistry in the development of new polymers. Phosphorus Sulfur Silicon Relat. Elem. 2004, 179, 661–671. [Google Scholar] [CrossRef]
- Saini, A.; Bhedi, D.; Dhanwant, K.; Thirumoorthi, R. Multi-azobenzene moieties on rigid cyclotriphosphazene core: Synthesis, structural characterization, electrochemistry, and photoisomerization study. J. Photochem. Photobiol. A 2024, 452, 115603. [Google Scholar] [CrossRef]
- Sönmez, E.B.; Ün, S.S.; Balcı, C.M.; Arslan, N.; Atilla, D.; Şeker, M.G.; İbişoğlu, H. New cyclotriphosphazenes with butylated oxyanisole motif; synthesis, characterization and biological properties. Polyhedron 2024, 563, 117252. [Google Scholar]
- Zhou, F.; Xi, W.; Qian, L.; Wang, J.; Qiu, Y.; Chen, Y. Hexaphenoxy cyclotriphosphazene/boron nitride high-efficiency charring system enhancing the flame retardancy and thermal conductivity of polycarbonate. Polym. Degrad. Stab. 2024, 219, 110601. [Google Scholar] [CrossRef]
- Doğan, S.; Tümay, S.O.; Balci, C.M.; Yeşïlot, S.; Besli, S. Synthesis of New Cyclotriphosphazene Derivatives Bearing Schiff Bases and Their Thermal and Absorbance Properties. Turk. J. Chem. 2020, 44, 31–47. [Google Scholar] [CrossRef] [PubMed]
- Waldin, N.A.; Jamain, Z. Synthesis and mechanical property of hexasubstituted cyclotriphosphazene derivatives attached to hydrazine-bridge linkage with high fire retardancy. J. Mol. Struc. 2023, 1284, 135330. [Google Scholar] [CrossRef]
- Davarci, D.; Doganci, S. Liquid Crystal Phosphazenes. J. Mol. Struc. 2022, 1269, 133819. [Google Scholar] [CrossRef]
- Ahmad, M.; Nawaz, T.; Hussain, I.; Chen, X.; Imran, M.; Hussain, R.; Assiri, M.A.; Ali, S.; Wu, Z. Phosphazene cyclomatrix network-based polymer: Chemistry, synthesis, and applications. ACS Omega 2022, 7, 28694–28707. [Google Scholar] [CrossRef]
- Mathews, L.D.; Capricho, J.C.; Peerzada, M.; Salim, N.V.; Parameswaranpillai, J.; Hameed, N. Recent progress and multifunctional applications of fire-retardant epoxy resins. Mater. Today Commun. 2022, 33, 104702. [Google Scholar] [CrossRef]
- Sulaiman, N.I.; Bakar, N.H.H.A.; Bakar, M.A. Effect of Al-Doping on Structural and Adsorption Properties of NiFe2O4 via Modified Sol–Gel Approach for CO2 Adsorption. Chem. Afr. 2024, 7, 2139–2154. [Google Scholar] [CrossRef]
- Palabıyık, D.; Mutlu Balcı, C.; Tümay, S.O.; Sengul, I.F.; Beşli, S. New Design of Cyclotriphosphazene Derivatives Bearing Carbazole Units: The Syntheses, Characterization, and Photophysical Properties. Inorg. Chim. Acta 2022, 539, 121022. [Google Scholar] [CrossRef]
- Elkhalgi, H.H.M.; Khandka, S.; Singh, U.B.; Pandey, K.L.; Dabrowski, R.; Dhar, R. Dielectric and electro-optical properties of a nematic liquid crystalline material with gold nanoparticles. Liq. Cryst. 2018, 45, 1795–1801. [Google Scholar] [CrossRef]
- Wang, J.; Liu, W.; Liu, H.; Wang, X.; Wu, D.; Zhang, S.; Shi, S.; Liu, W.; Wu, Z. Cyclotriphosphazene-based epoxy resins with excellent mechanical and flame retardant properties. Polymer 2022, 261, 125399. [Google Scholar] [CrossRef]
- Barberá, J.; Jiménez, J.; Laguna, A.; Oriol, L.; Pérez, S.; Serrano, J.L. Cyclotriphosphazene as a dendritic core for the preparation of columnar supermolecular liquid crystals. Chem. Mater. 2006, 18, 5437–5445. [Google Scholar] [CrossRef]
- Gioia, M.D.; Leggio, A.; Guarino, I.; Leotta, V.; Romio, E.; Liguori, A. A simple synthesis of anilines by LiAlH4/TiCl4 reduction of aromatic nitro compounds. Tetrahedron Lett. 2015, 56, 5341–5344. [Google Scholar] [CrossRef]
- Rong, Y.; Wentian, H.; Liang, X.; Yan, S.; Jinchun, L. Synthesis, mechanical and fire behaviours of rigid polyurethane foam with a reactive flame retardant containing phosphazene and phosphate. Polym. Degrad. Stab. 2015, 122, 102–109. [Google Scholar]
- Jamain, Z.; Khairuddean, M. Synthesis and Mesophase behaviour of benzylidene-based molecules containing two azomethine units. J. Phys. Conf. Ser. 2021, 1882, 012120. [Google Scholar] [CrossRef]
- Fang, Z.; Liu, Y.; Zhang, Y.; Cao, Z. Phosphorus-Containing Schiff Base Derivative Expanded Flame Retardant and Its Preparation. CN Patent No. CN102732041A, 29 May 2012. [Google Scholar]
- Jamain, Z.; Khairuddean, M.; Guan-Seng, T. Liquid-Crystal and Fire-Retardant Properties of New Hexasubstituted Cyclotriphosphazene Compounds with Two Schiff Base Linking Units. Molecules 2020, 25, 2122. [Google Scholar] [CrossRef]
- Rajkumar, P.; Selvaraj, S.; Anthoniammal, P.; Ram Kumar, A.; Kasthuri, K.; Kumaresan, S. Structural (monomer and dimer), spectroscopic (FT-IR, FT-Raman, UV–Vis and NMR) and solvent effect (polar and nonpolar) studies of 2-methoxy-4-vinyl phenol. Chem. Phys. Impact. 2023, 7, 100257. [Google Scholar] [CrossRef]
- Kara, Y.S.; Diran, D. Synthesis of novel 1,2,4-oxadiazine derivatives and the substituent effect study on 13C NMR spectra. J. Mol. Struct. 2024, 1310, 138309. [Google Scholar] [CrossRef]
- Kara, Y.S.; Yıldız, B. Synthesis and substituent effect study on 13C NMR chemical shifts of 4-(substitue-phenyl)-6-methyl-3-phenyl-4H-1,2,4-oxadiazin-5(6H)-one. J. Mol. Struct. 2022, 1250, 131787. [Google Scholar] [CrossRef]
- Kupka, T.; Dziuk, B.; Ejsmont, K.; Makieieva, N.; Fizer, L.; Monka, N.; Konechna, R.; Stadnytska, N.; Vasyliuk, S.; Lubenets, V. Impact of crystal and molecular structure of three novel thiosulfonate crystals on their vibrational and NMR parameters. J. Mol. Struct. 2024, 1313, 138642. [Google Scholar] [CrossRef]
- Jamain, Z.; Khairuddean, M.; Kamaruddin, K.; Rui, Y. Synthesis, structural elucidation and mesophase behavior of hexasubstituted cyclotriphosphazene molecules with amide linking unit. Malays. J. Chem. 2021, 23, 213–225. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | % Found (Calculated) | ||
|---|---|---|---|
| C (%) | H (%) | N (%) | |
| 1a | 73.29 (73.34) | 9.34 (9.41) | - |
| 2a | 75.50 (75.56) | 8.14 (8.19) | 3.62 (3.67) |
| 2b | 69.62 (69.70) | 4.53 (4.60) | 5.77 (5.81) |
| 3a | 72.81 (72.88) | 8.36 (8.41) | 10.57 (10.62) |
| 3b | 65.83 (65.87) | 5.10 (5.13) | 16.41 (16.46) |
| 4 | 58.47 (58.55) | 3.49 (3.51) | 4.84 (4.88) |
| 5a | 71.40 (71.45) | 6.93 (6.96) | 9.37 (9.41) |
| 5b | 66.18 (66.22) | 4.20 (4.23) | 12.84 (12.87) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohamad Fazli, F.J.; Jamain, Z. Synthesis and Characterization of Hydrazine Bridge Cyclotriphosphazene Derivatives with Amide–Schiff Base Linkages Attached to Decyl and Hydroxy Terminal Groups. Molbank 2024, 2024, M1934. https://doi.org/10.3390/M1934
Mohamad Fazli FJ, Jamain Z. Synthesis and Characterization of Hydrazine Bridge Cyclotriphosphazene Derivatives with Amide–Schiff Base Linkages Attached to Decyl and Hydroxy Terminal Groups. Molbank. 2024; 2024(4):M1934. https://doi.org/10.3390/M1934
Chicago/Turabian StyleMohamad Fazli, Fatin Junaidah, and Zuhair Jamain. 2024. "Synthesis and Characterization of Hydrazine Bridge Cyclotriphosphazene Derivatives with Amide–Schiff Base Linkages Attached to Decyl and Hydroxy Terminal Groups" Molbank 2024, no. 4: M1934. https://doi.org/10.3390/M1934
APA StyleMohamad Fazli, F. J., & Jamain, Z. (2024). Synthesis and Characterization of Hydrazine Bridge Cyclotriphosphazene Derivatives with Amide–Schiff Base Linkages Attached to Decyl and Hydroxy Terminal Groups. Molbank, 2024(4), M1934. https://doi.org/10.3390/M1934